

Summary
There is both cellular and physiological evidence demonstrating that both Activins and Inhibins regulate osteoblastogenesis and osteoclastogenesis, and regulate bone mass in vivo. Although Activins and Inhibins were initially isolated from the gonad, Activins are also produced and stored in bone, whereas Inhibins exert their regulation on bone cell differentiation and metabolism via endocrine effects. The accumulating data provide evidence that reproductive hormones, distinct from classical sex steroids, are important regulators of bone mass and bone strength. Given the well described dominant antagonism of Inhibin over Activin, as well as over BMPs and TGFβ, the gonadally-derived Inhibins are important regulators of locally produced osteotrophic factors. Thus, the cycling Inhibins in females and diurnal changes in Inhibin B in males elicit temporal shifts in Inhibin levels (tone) that de-repress the pituitary. This fundamental action has the potential to de-repress locally stimulated changes in osteoblastogenesis and osteoclastogenesis, thereby altering bone metabolism.
Keywords: osteoblastogenesis, osteoclastogenesis, Activin, Inhibin, bone turnover
Introduction: Bone as an endocrine organ
It is widely accepted that estrogens play a critical role in the maintenance of bone homeostasis (Manolagas 2000) and that the cellular basis of bone loss in postmenopausal women results from de-repression of both osteoblast and osteoclast development by estrogen loss after ovarian failure. The pathophysiology of postmenopausal osteoporosis involves the overproduction of osteoclasts, relative to the increase in osteoblastogenesis, a process that, itself, facilitates the support of osteoclast development (Parfitt, et al. 1983; Martin and Ng 1994; Citron, et al. 1995). However, clinical indices of increased bone turnover (Ebeling, et al. 1996) and decreases in bone volume (Riggs, et al. 2004) are first detected in late premenopausal women with normal circulating estrogen levels (Riggs, et al. 2008), suggesting that other, non-steroidal, members of the hypothalamic-pituitary-gonadal hormone (HPG) axis may also regulate skeletal metabolism.
Ebeling and colleagues in 1996 were the first to report increases in serum markers of bone resorption in peri-menopausal women, which occurred in the absence of decreased estrogens (Ebeling, et al. 1996). Instead, the increases in serum N-telopeptide, a marker of bone resorption, were better predicted by increases in Follicle Stimulating Hormone (FSH) than serum estradiol (E2) (Ebeling, et al. 1996). Numerous clinical reproductive studies have demonstrated that the early peri-menopausal rise in FSH levels in women is attributed to a selective decrease in Inhibin B secretion that occurs in the presence of normal levels of estradiol (E2), Inhibin A, Gonadotropin Releasing Hormone (GnRH), and Luteinizing Hormone (LH) (Klein, et al. 1996; Welt, et al. 1999; Klein, et al. 2004). Because both Inhibin A and Inhibin B isoforms selectively inhibit pituitary FSH secretion, these data suggest that the increased FSH is due to the loss of feed-back inhibition of the pituitary by gonadal Inhibin B in peri-menopausal women. As the loss of gonadal function progresses, the well-established decreases in E2 accompany ever declining levels of both Inhibin B and Inhibin A, further increasing serum FSH (Klein, et al. 1996; Welt, et al. 1999; Klein, et al. 2004), markedly increasing bone loss in postmenopausal women. Thus, although the hormonal mechanisms responsible for bone loss in peri-menopausal women are clear, we hypothesized that the well characterized hormonal changes and the bone turnover changes at the peri-menopause reflects a role of Inhibins.
If correct (and the data described below suggest it is), this idea would support the emerging idea that the skeleton is an endocrine organ, regulated by gonadal hormones other than sex steroids, as well as other factors (Karsenty 2006; Takeda and Karsenty 2008; Yadav, et al. 2008). The clinical data and the in vitro and in vivo evidence supporting the concept that gonadal Inhibins are part of the normal repertoire of factors regulating bone metabolism will be discussed.
Since Inhibins antagonize Activin action in all known Inhibin target tissues (Vale, et al. 1994), and Activin A is known to be expressed in bone marrow cells (Shao, et al. 1992; Yamashita, et al. 1992; Tuuri, et al. 1994; Gaddy-Kurten, et al. 2002) the endocrine role of Inhibins in bone metabolism must be considered in the context of Activin expression in the skeleton. Moreover, the presence in the local bone microenvironment of the soluble Activin antagonist, Follistatin (Inoue, et al. 1994) suggests that net Activin action in the skeleton is influenced by both endocrine and paracrine factors. The signaling mechanisms responsible for action and antagonism between Inhibin and Activin have been elucidated in cell lines and primary pituitary cells (Vale, et al. 1994; Gaddy-Kurten, et al. 1995; Vale, et al. 2004; Bilezikjian, et al. 2006; Farnworth, et al. 2006) but are not well characterized in bone cells. This review will summarize these studies and provide insight into the potential mechanisms mediating Inhibin and Activin effects on bone metabolism.
Inhibins and Activins: Sources and effects on the HPG axis
Inhibin A and Inhibin B are heterodimeric proteins in the TGFβ superfamily composed of αβA or αβB subunits, respectively, that were originally isolated from ovarian follicular fluid (Mason, et al. 1985). Inhibins were originally identified and defined based on their ability to suppress pituitary follicle stimulating hormone (FSH) secretion (reviewed in (Vale, et al. 1994)). The suppression of FSH by the Inhibins is antagonized by the related homodimeric peptides, Activin A and Activin B. Activin A is a homodimer of βA subunits that is locally produced in the gonad and was also isolated from follicular fluid (Vale, et al. 1994), whereas Activin B is a homodimer of βB subunits that is locally produced in the pituitary (Corrigan, et al. 1991). Activins exert multi-level increases in pituitary FSH production (reviewed in (Gregory and Kaiser 2004)). Activins directly stimulates FSH biosynthesis and release from the gonadotroph cells of the pituitary gland (Corrigan, et al. 1991). In addition, Activin has been shown to up-regulate gonadotropin-releasing hormone (GnRH) receptor gene expression, leading to increased synthesis and release of both FSH and luteinizing hormone (LH) in response to GnRH. Finally, Activin can also regulate FSH (and LH) secretion by stimulation of GnRH release from GnRH neurons in the hypothalamus (Gregory and Kaiser 2004). Inhibins antagonize these effects by preventing Activin binding to the Type II Activin receptor, whereas locally produced Follistatin also suppresses FSH secretion via direct binding to and neutralization of Activin action (Vale, et al. 1994).
In addition to opposing effects on pituitary FSH production, Inhibins, Activins and Follistatin exert a variety of gonadal effects in both males and females (reviewed in (de Kretser, et al. 2002)). In females, these include a role for Activin in the development and maintenance of healthy estrogenic follicles and prevention of premature luteinization, whereas Follistatin opposes these Activin effects and promotes luteinization or atresia (Findlay 1994) (Figure 1). Inhibins are involved in LH-regulated steroid production, preovulatory follicle selection and maintenance prior to ovulation (Figure 1; reviewed in Welt 2002).
Figure 1. Skeletal effects of Inhibins, Activin, BMPs and Follistatin derived from bone and gonadal sources.
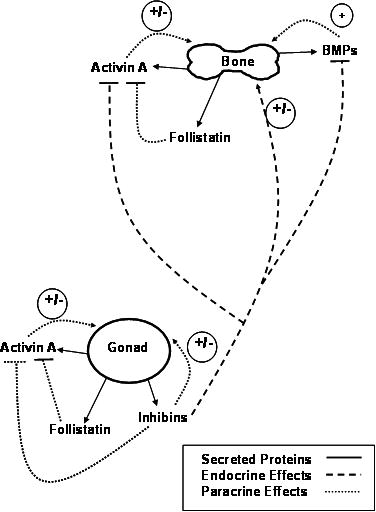
Skeletal effects of Activin A, its antagonist, Follistatin, and BMPs are paracrine effects (…….) from local production and sequestration of peptides in bone matrix. The stimulatory effects of Activin and BMP are blocked by the endocrine effects of Inhibins (- - - -) produced primarily by the gonads. The Inhibin antagonism of Activin and BMP action, demonstrating the importance of skeletal Inhibin “tone” that is associated with normal gonadal function. Activin A and Follistatin are also produced by the gonads, although their levels in serum are likely insufficient to exert endocrine effects on pituitary FSH production or contribute to the regulation of bone metabolism. See text for more details.
The gonadal Inhibin regulation of FSH production is not limited to females. In males, FSH secretion is suppressed by Inhibin B (de Kretser, et al. 2000), the principal form produced from adult Sertoli cells in the testis. Serum levels of Inhibin B show a clear diurnal variation that is closely linked to serum testosterone levels (Meachem, et al. 2001). Inhibin B levels are low in early development and remain low until puberty, when they rise, initially as a consequence of FSH stimulation and then as a result of the combined regulation by FSH and ongoing spermatogenesis (Meachem, et al. 2001). Serum Inhibin B levels are positively correlated with testicular volume and sperm counts (Kumanov, et al. 2005). In infertile patients, Inhibin B decreases and FSH increases, and the addition of Inhibin B to serum FSH improves prediction of impaired spermatogenesis (Abid, et al. 2008).
On the other hand, Activin A has its highest concentrations in the immediate postnatal period during which it is involved in the developmental regulation of both germ cells and Sertoli cells and its action is modulated by Follistatin (de Kretser, et al. 2004). Activin A levels are considerably lower in the adult testis, but its production in Sertoli cells is stimulated by Interleukin-1 and inhibited by FSH (de Kretser, Buzzard et al. 2004). In Sertoli cells, local low level Activin production is thought to be involved in autocrine/paracrine actions within the seminiferous epithelium (de Kretser, Buzzard et al. 2004). Interestingly, since castration does not decrease circulating Activin A levels (Activin A is almost exclusively bound to Follistatin in the serum (de Kretser, et al. 2000)), the testis is not the major source of circulating Activin A in males (de Kretser, et al. 2000). Finally, the local production of both Activin and Follistatin in the pituitary is subject to both autocrine and endocrine regulation by gonadal testosterone and hypothalamic GnRH (de Kretser, et al. 2000; Gregory and Kaiser 2004). Analogous to females, the net level of FSH production and secretion in males is dependent upon the balance between local processes, and the circulating feedback exerted by Inhibin B and gonadal steroids (de Kretser, et al. 2000).
With reproductive aging in women, Inhibin B levels decline, along with the follicular pool, and the dynamics of the normal menstrual cycle becomes disturbed (Hurwitz and Santoro 2004). The loss of Inhibin suppression of pituitary FSH secretion appears to be the initiating endocrine event that leads to menstrual cycle shortening as well as to some of the hormonal unpredictability of the peri-menopausal years (Klein, et al. 1996; Burger, et al. 2002; Robertson and Burger 2002). Changes in Inhibin B levels may also be related to the decline in fertility that occurs with age (Danforth, et al. 1998). Similarly, Inhibin B is an excellent marker for gonadal competence in men, with the age-related decline in Inhibin B levels reflecting decreased gonadal reserves as it does in women (Hurwitz and Santoro 2004).
The changes in Inhibin B and FSH at the onset of menopause are accompanied by sustained Activin A and age-dependent decreases in Follistatin (Reame, et al. 2007), which is consistent with an enhanced Activin bioavailability at the menopause. In adult men, Activin A is present in the circulation bound to Follistatin (Muttukrishna, et al. 2001). However, as in females, Activin bioavailability may be increased in men with advancing age, as Activin levels appear to be elevated, whereas Follistatin levels do not appear to change greatly with age (Hurwitz and Santoro 2004). Collectively, the age-related fluctuations in Inhibins, Activin and Follistatin influence reproductive capacity as well as the sequelae of reproductive changes that are a feature of chronological aging in both men and women (Hurwitz and Santoro 2004).
Activins and Inhibins and the skeleton
Inhibins and Activin A also exert opposing effects on several cells of the hematopoietic lineage, including erythroid, (Yu, et al. 1987) megakaryocyte, (Fujimoto, et al. 1991; Okafuji, et al. 1995), granulocyte-macrophage lineage cells (Broxmeyer, et al. 1988; Scher, et al. 1990), as well as cells of the monocyte/macrophage lineage (Yamada, et al. 1992; Fuller, et al. 2000; Gaddy-Kurten, et al. 2002; Perrien, et al. 2006). Activin βA subunit mRNA is locally produced in bone marrow; (Yu, et al. 1994) and, like TGFβ (Bonewald and Mundy 1990) and bone morphogenetic proteins (BMPs) (Wozney 1992), Activin A protein is abundantly localized in the bone matrix (Ogawa, et al. 1992). In addition, we have shown that Activin A is an endogenous growth factor produced during ex vivo bone marrow cell osteoblastogenesis (Gaddy-Kurten, et al. 2002). Although expression of the Inhibin α-subunit (required for Inhibin dimer formation) is very low in both human and rat bone marrow (Meunier, et al. 1988; Yu, Shao et al. 1994), [125I]-Inhibin A rapidly accumulates in the bone marrow of 25 day-old rats (within 10 min of i.v. injection) and is retained for at least an hour (Woodruff, Krummen et al. 1993). These results are consistent with the idea that the effects of Inhibin on marrow cell hematopoiesis (Yu, et al. 1987; Broxmeyer, et al. 1988; Fujimoto, Kawakita et al. 1991) are attributable to endocrine Inhibin derived from gonadal sources (Meunier, et al. 1988). These data provided the basis for the hypothesis that gonadal Inhibins also regulate osteoclast differentiation (Gaddy-Kurten, et al. 2002), as well as cells in the mesenchymal lineage. Collectively, the accumulating data indicate that gonadal Inhibins contribute to the normal endocrine repertoire responsible for the regulation of bone cell differentiation, turnover, and metabolism (Perrien, et al. 2006; Perrien, et al. 2007).
Inhibin/Activin/Follistatin signaling in the pituitary provides a model paradigm for endocrine effects of this system in bone (Ying 1988). The emerging theme over the past two decades is that both endocrine-derived Inhibin and locally produced Follistatin can act coordinately as extracellular regulators of Activin action to regulate the timing, duration, and amplitude of pituitary FSH secretion (Welt, et al. 2002; Bilezikjian, et al. 2004). Autocrine production of bone morphogenetic proteins (BMP) 6 and 7 in gonadotrophs has been demonstrated, as has BMP 6 and 7 stimulation of FSH biosynthesis and secretion (Huang, et al. 2001). These data provide compelling evidence reinforcing the obvious parallels between the pituitary and bone Inhibin/Activin/FSH axis. In both regulatory systems, FSH secretion and bone cell differentiation can be regulated by the combination of endocrine-derived Inhibins, and locally produced Activins, Follistatin, and BMPs (Wozney 1992).
Interestingly, a recent report suggests that FSH may directly stimulate bone resorption via effects on osteoclast formation (Sun, et al. 2006). FSH receptor expression was demonstrated in both human and murine osteoclast precursors, and in vitro FSH treatment of these precursors was found to activate several signaling mechanisms in osteoclasts and their precursors, namely phosphorylation of ERK, Akt and IkBα (Sun, et al. 2006). These data were proposed to support the concept that increases in circulating FSH during the menopause transition may have a direct role in stimulating bone resorption (Gao, et al. 2007).
The evaluation of transgenic mice deficient in either the FSH-β ligand or FSH receptor (FORKO) has provided additional insight into the contribution of FSH to bone turnover (Table 1). As previously indicated, key endocrine products of FSH-stimulation of the HPG axis are gonadal estrogens and Inhibins. Thus, while LH-stimulation of androgen production is maintained in FSHβ-deficient and FORKO mice, estrogen levels are low (Danilovich, et al. 2000; Gao, et al. 2007). FORKO mice developed age-dependent declines in bone mineral density and trabecular bone volume of the lumbar spine and femur, which could be partly reversed by ovarian transplantation (Gao, et al. 2007). Bilateral ovariectomy reduced elevated circulating testosterone levels in FORKO mice and decreased bone mass to levels indistinguishable from those in ovariectomized wild-type controls (Gao, et al. 2007). Responses to androgen receptor blockade and aromatase inhibitor administration indicated that ovarian secretory products, such as estrogen, and peripheral conversion of ovarian androgen to estrogen can alter bone homeostasis independent of any bone resorptive action of FSH (Gao, et al. 2007). However, caution should be used in interpreting these data, as the independent contribution of Inhibin depletion in these ovariectomized transgenic models of altered FSH signaling has not been determined. To date, the lack of commercially available murine Inhibin assays have precluded the measurement of serum Inhibin levels in the absence of FSH signaling. Thus, the individual contribution of Inhibins and gonadal steroids to any FSH-independent regulation of bone mass in these FORKO and FSHβ-deficient mice is unknown.
Table 1. Skeletal parameters altered by disruption of FSH in the HPG axis.
| Genotype | Reproductive effects | Skeletal effects | Following OVX | Reference |
|---|---|---|---|---|
| F ORKO | Immature ovary development, thread-like uteri | Spinal kyphosis, vertebral compression (4 mo) | Decreased BMD, BV/TV (spine, femur) | 2, 3, 4 |
| Decreased circulating estrogen | Decreased BMD, Tb BV, ObS/BS, MAR, BFR at 3-4 | Increased OcS/BS | ||
| Increased FSH, LH | mo. (spine, femur) | Decreased testosterone | ||
| 10-fold increase in testosterone at 3-4 mo. | Decreased ex vivo osteoclastogenesis | OVX effect is mimicked by androgen receptor blockade or aromatase inhibition | ||
| Inhibin – not determined | ||||
| FSHβ +/- | Normal ovaries and uteri | Decreased osteoclast number | Not determined | 1, 3 |
| Normal estrogen | Decreased bone resorption | |||
| 50% decreased FSH | Increased Tb BV, Tb N (spine, femur) | |||
| Inhibin, testosterone, LH – not determined | No changes in serum osteocalcin, MAR | |||
| FSHβ -/- | Atrophic ovaries, Thread-like uteri | No bone loss | Not determined | 1, 3 |
| Decreased estrogen | Increased BMD (femur) | |||
| Decreased FSH; Increased LH | Increased BV/TV, Tb N | |||
| Inhibin, testosterone – not determined | ||||
References: 1. Kumar, 1997; 2. Danilovich, 2000, 2002; 3. Sun, 2006; 4. Gao, 2007
BMD, bone mineral density; Tb BV, trabecular bone volume; ObS/BS, osteoblast Surface/bone surface; MAR, mineral apposition rate; BFR, bone formation rate; BV/TV, bone volume/total volume; OcS/BS, osteoclast surface/bone surface; Tb N, trabecular number
Activin effects on osteoblastogenesis
Several investigators have explored the effects of Activin A on osteoblast development, using multiple in vitro models. Both primary cells and osteoblastic cell lines of murine, rat and human origin have been used. Results demonstrate opposing effects of Activin on osteoblastogenesis depending upon the model systems and species used. Thus, the evidence will be presented separately for each model.
Calvarial cells
The demonstration that the genetic deletion of the Activin betaA gene (inhβa) results in the disruption of whisker, palate and tooth development (Matzuk, et al. 1995), suggested that locally produced Activin A may stimulate osteoblastogenesis in vivo, particularly at sites of intramembranous bone formation. Many initial studies supported this concept, using the well described fetal rat calvarial cell (FRC) culture system of osteoblast precursors, which can be stimulated to undergo complete osteoblast development and maturation (Rowe and Kream 1982).
Using dispersed cells from collagenase digestions 3, 4 and 5 of FRCs, stimulatory effects of Activin A on DNA synthesis as well as both collagen and non-collagen protein synthesis were observed (Centrella, et al. 1991). These authors were the first to suggest a potential endocrine role of Activin A on bone cells from that derived from blood or gonadal tumors overexpressing Activin A (Centrella, et al. 1991). However, subsequent FRC studies using only the third collagenase digestion demonstrated an inhibitory effect of Activin A on osteoblast differentiation (Ikenoue, et al. 1999). Interestingly, this suppression of osteoblast development was strongest when Activin A treatment was initiated early during osteoblastogenesis (during the first 5 days of culture). The effects of Activin A during FRC osteoblast cultures are similar to those observed for TGFβ (Ikenoue, et al. 1999), and suggest the importance of cell context (cells obtained from the third collagenase digestion are different than those pooled from the third, fourth and fifth digests) and that exposure time to the growth factor is critical for eliciting effects on osteoblastogenesis.
Bone marrow-derived cells
Additional data supporting the idea that Activin A stimulates osteoblastogenesis in a context-dependent manner has been demonstrated in studies of murine ex vivo bone marrow cell cultures. These ex vivo cultures of whole adult murine bone marrow cells are comprised of both mesenchymal and hematopoietic progenitors (Gaddy-Kurten, et al. 2002). Activin A is endogenously produced in the cultures over time and stimulates osteoblast differentiation (Gaddy-Kurten, et al. 2002). Activin A (beta A subunit) is produced and secreted during the second half of culture (days 10-28), and its stimulatory effects on later osteoblast differentiation and mineralization (as indicated by bone nodule formation, CFU-OB, on day 28) can be blocked by either the soluble Activin antagonist, Follistatin, or by Inhibins (Gaddy-Kurten, et al. 2002). The cellular source of endogenous Activin A production in these cultures are the stromal mesenchymal progenitors, as both mouse and human bone marrow cells have been demonstrated to produce bioactive Activin A (Yamashita, et al. 1992; Yu, et al. 1994; Uchimaru, et al. 1995; Dolter, et al. 1998; Abe, et al. 2002).
Interestingly, during ex vivo bone marrow cell osteoblastogenesis, Follistatin had no effect on osteoblast recruitment (as indicated by alkaline phosphatase positive CFU-F formation on day 10), consistent with a lack of Activin A action (Gaddy-Kurten, et al. 2002). Moreover, treatment with exogenous Activin A substituted for the stimulatory effects of endogenous BMPs on both early osteoblast precursor recruitment and subsequent maturation and mineralization (Gaddy-Kurten, et al. 2002).
These robust stimulatory effects of Activin A on osteoblast recruitment and mineralization in primary, heterogeneous adult murine cultures are in stark contrast to recent observations using homogeneous cultures of human osteoblast precursors (Eijken, et al. 2007). Activin A treatment altered matrix protein production, and suppressed mineralization of SV40 transformed human osteoblastic cells, normal human osteoblast preparations, and human mesenchymal stem cells (Eijken, et al. 2007).
These opposing effects of Activin A on osteoblastogenesis are somewhat difficult to reconcile. Although the differences may simply reflect species specific differences between mouse and human cell cultures, it is interesting to note that the stimulatory actions of Activin A on osteoblastogenesis have most often been observed in cells cultured in a heterogeneous context (Centrella, et al. 1991; Ikenoue, et al. 1999; Gaddy-Kurten, et al. 2002). The multicellular context is consistent with other reports demonstrating the stimulatory ectopic bone formation effects of daily periosteal injections of Activin A in newborn rat calvaria (Oue, et al. 1994). Activin treatment has also been shown to stimulate bone mass and strength (Sakai, et al. 2000), and prevent ovariectomy-induced bone loss by preserving bone volume and mineralized surface (Sakai, et al 1999). The effects strongly support a stimulatory role for Activin A on osteoblast activity, rather than the inhibition of osteoclast function.
The inhibitory effects of Activin A on osteoblastogenesis are more often observed in cultures of homogeneous murine and human osteoblast precursors, cell lines, and in single enzymatic digested fractions of FRC. Thus, it is tempting to speculate that the microenvironmental context determines the net effect of Activin A. Indeed, other cell types in the bone marrow, including lymphoid, erythroid and granulocyte-macrophage precursors (Broxmeyer, et al. 1988; Broxmeyer, et al. 1991; Abe, et al. 2002) as well as monocytes (Yamashita, et al. 1993) have been shown to respond to Activin A. Some of these cells, such as monocytes, respond to Activin A by producing additional cytokines, such as Interleukin-1alpha and TNFalpha (Yamashita, et al. 1993), which themselves can influence osteoblast and/or osteoclast development. These data lend support to the idea that in heterogeneous cultures, the contributions of other cell types may contribute to the overall stimulatory effects of Activin A on osteoblast development, and that such effects would be lacking in purified populations of cells.
Activin and osteoclastogenesis
One of the consequences of stimulating osteoblastogenesis is the production of critical pro-osteoclastogenic molecules, such as the receptor activator of NFkB ligand (RANKL) and mCSF, thereby providing the foundation for the coupling of osteoblastogenesis and osteoclastogenesis (Manolagas 2000; Horowitz, et al. 2001; Teitelbaum and Ross 2003; Martin, et al. 2006). Although Activin A has been shown to exert both stimulatory and suppressive effects on in vitro osteoblast development, Activin A has consistent pro-osteoclastogenic effects in vitro. Initial studies demonstrated Activin A stimulation of TRAP positive multinucleated cell (TRAP+ MNC) formation in cultures of murine bone marrow macrophages as well as neonatal mouse calvarial organ cultures supported by 1,25-dihydroxyvitamin D3 or PTH (Sakai, et al. 1993). These findings were later confirmed in whole murine bone marrow (Gaddy-Kurten, et al. 2002) and in a murine macrophage cell line (RAW 264.7) (Koseki, et al. 2002). Activin A also enhances RANKL-stimulated osteoclastogenesis (Fuller, et al. 2000), but most importantly, Activin A is one of the few factors demonstrated to stimulate TRAP+ MNC development independent of RANKL (Sugatani, et al. 2003).
Mechanistically, the general pathway of Activin signaling has been well characterized (Vale, et al. 2004). Activin binds to its Type II receptor serine kinase (ActRIIA or ActRIIB), which recruits and phosphorylates the Type I receptor (ALK4). ALK4 then phosphorylates pathway-restricted Smad transcription factors (Smad2 or Smad3) which interact with the common mediator Smad4, translocate to the nucleus, and activate gene transcription. However, it appears that in murine bone marrow macrophage cultures, Activin A treatment rapidly activates IκB-α through phosphorylation (within 5-15 minutes) and induces nuclear translocation of phosphorylated-NFκB (Sugatani, et al. 2003), a critical step downstream of RANKL signaling that is essential for osteoclastogenesis (Xing, et al. 2002). This rapid effect on NFκB activation suggests an alternative Activin signaling mechanism that is likely independent of Smad-dependent gene transcription, (Derynck and Zhang 2003). Activin A also enhances RANK expression in osteoclast precursors, further stimulating osteoclastogenic differentiation, but has had no observed effects on osteoclast survival via activation of Akt, BAD, and mTOR (Sugatani, et al. 2003). Finally, Activin A has been immunolocalized in actively resorbing multinucleated osteoclasts in vivo, as well as in osteoclasts differentiated in vitro (Hosoi, et al. 1996).
Activin A is secreted in its bioactive form, and its secretion from osteoclasts, as well as its liberation from bone matrix during osteoclastic resorption has been reported (Sakai, Eto et al. 2000). These data suggests that local Activin A may support sustained osteoclastogenesis and/or exert stimulatory effects in the bone marrow microenvironment to stimulate (or inhibit) the many marrow cell types described above.
Activin effects in vivo
Similar to the paradoxical in vitro effects of Activin A on osteoblastogenesis, conflicting skeletal effects of Activin A in vivo have also been reported. Stimulatory effects of Activin A treatment on bone mass have been observed in several systems. Local injection over the periosteum of neonatal calvaria increased bone formation (Oue, et al. 1994), and direct injection into rat fibula fracture sites enhanced both callus and bone formation (Sakai, et al. 1999). In addition, intramuscular injection of Activin A in ovariectomized rats maintained bone mass and strength (Sakai, Fujita et al. 2000).
Interestingly, a soluble extracellular domain of the Type IIA Activin receptor (ActRIIA ECD) was generated and demonstrated an ability to attenuate FSH secretion in response to exogenous Activin A or endogenous Activin B in cultured pituitary cells, thereby suppressing Activin action (Donaldson, et al. 1999). This ActRIIA ECD retains the ability to bind both Activin A and Inhibin A with high affinity, based upon its binding capacity to the shared βA subunit of both Activin A and Inhibin A (Donaldson, et al. 1999). Treatment in vivo with this same ActRIIA ECD was shown recently to increase bone formation, bone mass, and bone strength in both normal and ovariectomized mice (Pearsall, et al. 2008). Consistent with the in vitro data demonstrating Activin suppression of mineralization (Eijken, et al. 2007), the cellular basis for the effects of the ActRIIA ECD in vivo appears to result from stimulatory effects on cells of the osteoblastic lineage, rather than suppression of osteoclast parameters (Pearsall, et al. 2008).
Interestingly, inhibition of Activin signaling via the ActRIIA ECD is mechanistically equivalent to either administration of the soluble Activin antagonist, Follistatin, or the opposing growth factor Inhibin (Donaldson, et al. 1999). Mechanistically, Inhibin binding to the TGFβ type III receptor betaglycan (BG) sequesters the ActRIIA into a non-signaling complex and prevents Activin binding to endogenous receptors (Lewis, et al. 2000; Wiater, et al. 2006). The findings with ActRIIA ECD on murine bone mass in vivo (Pearsall, et al. 2008) are similar to those reported with Inhibin transgenic mice, where continuous Inhibin expression in vivo increased bone mass and strength, via increases in osteoblast differentiation and activity ((Perrien, et al. 2007), described in more detail below).
Inhibin Effects on Osteoblastogenesis and Osteoclastogenesis
As has been demonstrated with many Activin target tissues, Inhibins exert dominant and opposing effects to Activin on both osteoblast and osteoclast development (Gaddy-Kurten, et al. 2002). Both Inhibin A and Inhibin B isoforms suppressed osteoblast and osteoclast development in murine bone marrow cultures (Gaddy-Kurten, et al. 2002), human mesenchymal stem cells (hMSC), and peripheral blood mononuclear cells (PBMC), respectively (Perrien, et al. 2006). Inhibin treatment blocked recruitment of cells into the osteoblastic lineage (AP expression), as well as later differentiation (mineralized matrix formation). As has been demonstrated in other Activin and Inhibin responsive tissues, Inhibins blocked Activin A stimulation of osteoblastogenesis and osteoclastogenesis in ex vivo murine bone marrow cultures (Gaddy-Kurten, et al. 2002). Surprisingly however, Inhibins also blocked the well-described stimulatory effects of bone morphogenetic proteins (BMPs) on both osteoblast and osteoclast formation, regardless of whether the BMP was generated endogenously in the culture or provided by exogenous administration (Gaddy-Kurten, et al. 2002). These data were the first to demonstrate the antagonism/blockade of the stimulatory effects of BMPs by Inhibins in any tissue, a finding that was later confirmed in pituitary and other cell types (Wiater and Vale 2003). The demonstration that Inhibins block the stimulatory effects of both BMP and Activin support the concept that bone is an endocrine target of Inhibin. Moreover, it suggests that regulation of osteoblast and osteoclast differentiation by gonadal Inhibins can be dominant over the local paracrine/autocrine effects of BMPs, Activin A, and their soluble antagonists (chordin, follistatin, and gremlin) also expressed in bone (reviewed in (Rosen 2006)).
Inhibins antagonize Activin and BMP actions by sequestering their type II receptors in high-affinity complexes with betaglycan (BG), a co-receptor that Inhibin shares with TGFβ (also known as TGFβRIII, (Esparza-Lopez, et al. 2001; Wiater and Vale 2003; Vale, et al. 2004; Farnworth, et al. 2006; Kirkbride, et al. 2008)). Inhibin binding to BG facilitates interaction with other members of the Type II TGFβ receptor superfamily, including ActRIIA, ActRIIB, BMPRII and TβRII (Esparza-Lopez, et al. 2001; Wiater and Vale 2003; Vale, et al. 2004; Farnworth, et al. 2006; Kirkbride, et al. 2008).
BG expression in cells of the osteoblastic lineage and in bone marrow cells has been well characterized (Centrella, et al. 1991; Centrella, et al. 1994; McCarthy, et al. 2007). BG expression on the surface of osteoblast precursors has been shown to limit TGFβ signaling via TβRII, and in this way delay TGFβ-dependent activation of osteoblasts (Centrella, et al. 1991; Centrella, et al. 1994; McCarthy, et al. 2007). Inhibin suppression of osteoblastogenesis is likely similar, as Inhibin has been shown to utilize a similar mechanism via BG binding in other cell types (Esparza-Lopez, et al. 2001). In fact, competition for BG binding between Inhibins and TGFβ has been determined to occur via the uromodulin-like core domain in the BG molecule (Esparza-Lopez, et al. 2001).
More recently, BG was shown to also serve as a co-receptor for BMPs, including BMP-2, BMP-4, and BMP-7 (Kirkbride, et al. 2008), which stimulate bone cell differentiation and bone formation. Binding of BG to BMP ligands occurs with similar kinetics and ligand binding domains as previously identified for TGFβ and Inhibin (Wiater and Vale 2003). Whereas, Inhibin-BG complexes bind to Type II receptors to block Activin and BMP signaling (Lewis, et al. 2000), BMP binding to BG enhances ligand binding to the BMP Type I receptors and amplifies downstream BMP signaling (Kirkbride, et al. 2008),
Collectively, the competition for ligand binding to BG between Inhibins, BMPs and TGFβ, and the competition between Inhibin and Activin for binding to the Type II Activin receptors, provides potential mechanisms by which Inhibins exert their dominant suppressive Inhibin effects over the stimulatory actions of BMPs or Activin in both mouse (Gaddy-Kurten, et al. 1998) and human (Perrien, et al. 2006) cells.
Correlation of Inhibins with bone turnover in humans
The demonstration that Inhibins exert direct effects on osteoblastogenesis and osteoclastogenesis suggested that Inhibins may have a clinically relevant role in bone turnover. Decreases in serum Inhibin B are the first indicators of loss of ovarian function at the onset of menopause, causing de-repression of pituitary FSH secretion (Klein, et al. 1996; Welt, et al. 1999; Klein, et al. 2004). Moreover, serum FSH was demonstrated to be a better predictor than estradiol of the earliest menopause-related increases in bone resorption (Ebeling, et al. 1996). Subsequently, Inhibin B levels were also correlated with bone mass and bone turnover in regularly menstruating women aged 35-50 years, whereas neither FSH nor estradiol levels in these women were significantly associated with bone mass (Vural, et al. 2005).
These important findings were confirmed and expanded in a cross-sectional study of 188 women, not on oral contraceptives or hormone replacement, across the menopause transition from 21-85 yo (Perrien, et al. 2006). Both serum Inhibin A and Inhibin B were measured in addition to FSH and bioavailable estradiol, and these hormone levels were correlated with indices of bone turnover. The data revealed significant inverse correlations between serum Inhibin A and Inhibin B levels with markers of bone formation and bone resorption, in both pre-menopausal and peri-menopausal women. Interestingly, FSH was not significantly correlated with any bone formation markers in either pre- or post-menopausal women, but was significantly correlated with bone resorption markers in post-menopausal women (Perrien, et al. 2006). Importantly, in multivariate analysis serum Inhibin A was the best endocrine predictor of changes in markers of both bone formation and resorption in pre-and post-menopausal women, exceeding the predictive potential of FSH and bioavailable estradiol (Perrien, et al. 2006). Collectively, these clinical data suggest that decreases in cycling Inhibin levels associated with diminished ovarian function contribute to the initial bone loss observed during the peri-menopausal period, regardless of any demonstrable changes in sex steroids or FSH (Perrien, et al. 2006).
Furthermore, it is likely that decreases in Inhibin levels, in the absence of changes in estradiol, that occur throughout reproductive life are associated with the reported increases in bone turnover and observed decreases in volumetric BMD in these women (Riggs, et al. 2008). High resolution peripheral QCT assessment of women before and after the menopause demonstrated that young adult women (and men) already have marked diminished volumetric BMD in the trabecular bone of the distal radius, distal tibia and lumbar spine, whereas cortical bone loss begins in midlife (Riggs, et al. 2008). As expected, the cortical bone loss in post-menopausal women (and older men over age 75) was associated with lower levels of bioavailable estradiol and testosterone and with higher levels of FSH and bone turnover markers (Riggs, et al. 2008). However, the early trabecular bone loss did not consistently correlate with putative causal factors, except for a trend with IGF-related variables at the distal tibia in women Indeed, this has been identified in men in an earlier cross-sectional study by the same group (Khosla S, et al. 2006) The authors concluded that the early-onset, substantial trabecular bone loss in both sexes during sex steroid sufficiency is incompletely unexplained, and indicates that current paradigms on the pathogenesis of osteoporosis are incomplete (Riggs, et al. 2008). Unfortunately, Inhibins were not assessed in this study. However, it is interesting to speculate that declines in cycling gonadally-derived serum Inhibins may lead to subtle increases in Activin/BMP tone in the local bone microenvironment, enhancing bone cell differentiation, and bone turnover, resulting in bone loss in these younger subjects.
Inhibin effects in vivo
The effects of Inhibins on the regulation of bone mass have been less well studied. This is in part due to the limited quantities and costs of commercially available recombinant Inhibins. However, transgenic mouse models have been used to inducibly express recombinant human Inhibin A at in intact and gonadectomized mice (Perrien, et al. 2007). Continuous systemic exposure to Inhibin A for 4 weeks was strongly anabolic in intact adult mice at multiple skeletal sites, including the tibia, spine and humerus in both sexes (Perrien, et al. 2007). Moreover, Inhibin A overexpression prevented orchidectomy-induced bone loss at the tibia and spine, and prevented loss of vertebral bone strength due to gonadectomy (Perrien, et al. 2007).
Dynamic histomorphometry revealed that mineral apposition and bone formation rates were increased by Inhibin A overexpression. Similarly, the mechanistic basis of Inhibin A anabolism involved the stimulation of both osteoblast differentiation and activity, as ex vivo marrow cultures from Inhibin A overexpressing mice had enhanced capacity for osteoblast recruitment (alkaline phosphatase expression) and differentiation (mineralization). It should be noted that the anabolic effect of Inhibin A was observed in transgenic mice continuously expressing Inhibin A (Perrien, et al. 2007). Whether Inhibin A is anabolic when delivered or expressed in an intermittent (cyclic) administration regiment remains to be determined. Unlike other endocrine regulators of bone turnover, the lack of Inhibin A effects on bone resorption and osteoclast number suggest an Inhibin A induced imbalance between bone resorption and bone formation. The primarily osteoblastic effect observed is reminiscent to the mechanistic explanation for the bone anabolism reported for the ActRIIA ECD (Pearsall, et al. 2008), and may implicate a common or related mechanism of action.
Conclusions
Collectively, significant evidence has accumulated demonstrating in vitro and in vivo effects of both Activins and Inhibins on osteoblast and osteoclast development (summarized in Table 2). The data provide overwhelming evidence to suggest that reproductive hormones other than sex steroids play an important role in regulating both bone mass and bone strength. However, the specific roles that Inhibins and Activins play in regulating bone metabolism remains far from clear. Activins have well-demonstrated but conflicting effects on in vitro osteoblastogenesis that appear to be dependent on the in vitro test system (Centrella, et al. 1991; Ikenoue, et al. 1999; Eijken, et al. 2007), but consistent stimulatory effects on osteoclastogenesis in vitro (Sakai, et al. 1993) (Gaddy-Kurten, et al. 1998; Fuller, et al. 2000; Koseki, et al. 2002). Similarly, Activins have demonstrated stimulatory effects on bone formation and strength in vivo (Oue, et al. 1994; Sakai, et al. 1999; Sakai, et al. 2000); effects that are difficult to reconcile with recent findings that antagonism of Activin signaling via ActRIIA ECD is anabolic to the skeleton (Pearsall, et al. 2008) and suppresses bone turnover (Ruckle, et al. 2009).
Table 2. Summary of Inhibin and Activin Effects on Bone Cells.
| Model | Activin A effects | Inhibin A/B effects | References |
|---|---|---|---|
| In vitro models | |||
| Fetal rat Calvaria (FRC, digests 3-5) | ↑ OB | NR | (Centrella, 1991) |
| Fetal rat Calvaria (FRC, digest 3) | ↓ mineralization | NR | (Ikenoue, 1999). |
| Murine bone Marrow cultures | ↑ OB AP+ CFU-F, CFU-OB, ↑ TRAP+ OCL | ↓ AP+ CFU-F, CFU-OB, ↓ TRAP+ OCL | (Gaddy-Kurten, 2002) |
| Human PBMCs | ↑TRAP+ OCL | ↓TRAP+ OCL | (Eijken, 2007; Perrien, 2006; Fuller, 2000) |
| Human mesenchymal stem cells | ↓AP+ cells, Mineralization | ↓ AP+ cells, Mineralization | (Eijken, 2007; Perrien, 2006) |
| Human fetal osteoblasts | ↓AP+cells, Mineralization | NR | (Eijken, 2007) |
| In vivo models | |||
| Local injection over neonatal calvaria | ↑ bone formation (rat) | NR | (Oue, 1994) |
| Systemic administration in intact rodents | ↑ bone formation (rat) | ↑ bone formation (mouse) | (Sakai, 2000; Perrien, 2007) |
| Systemic administration in gonadectomorized rodents | ↑ bone formation (rat) | ↑ bone formation (mouse) | (Sakai, 2000; Perrien, 2007) |
| Local injection at fracture site | ↑ bone formation (rat) | NR | (Sakai, 1999) |
| Clinical correlations | |||
| Bone formation marker levels | NR | Serum Inhibins inversely correlated | (Perrien, 2006) |
| Bone resorption marker levels | NR | Serum Inhibins inversely correlated | (Perrien, 2006; Vural, 2005) |
| Bone mass | NR | Serum Inhibins inversely correlated | (Vural, 2005) |
PBMC, peripheral blood mononuclear cells; OB, osteoblastogenesis; OCL, osteoclastogenesis; AP+, alkaline phosphatase positive; CFU-F, colony forming unit-fibroblast; TRAP+, tartrate resistant acid phosphatase positive; NR, Not Reported
Inhibins have consistent suppressive effects on both osteoblast and osteoclast differentiation in vitro and these effects antagonize the local effects of Activins, TGFβs and BMPs (Gaddy-Kurten, et al. 2002; Perrien, et al. 2006). The suppressive effects of Inhibin are consistent with the concept that normal cyclic levels of Inhibins suppress bone cell differentiation and thereby, bone turnover. Clinical studies demonstrating that decreases in Inhibins are good predictors of increases in bone turnover markers across the menopause transition (Vural, et al. 2005; Perrien, et al. 2006) support this contention. As such, these findings suggest that like the pituitary, the skeleton is an Inhibin target organ. Moreover, as gonadal Inhibin secretion diminishes, the bone (like the pituitary) becomes de-repressed, allowing local Activins, TGFbetas and BMPs to stimulate bone cell differentiation unopposed (Figure 1), contributing to increased bone turnover and subsequent bone loss. In addition, the inverse correlations of Inhibins with bone turnover are consistent with Inhibins playing a potential role in the recently reported decreases in trabecular volumetric BMD in pre- and peri-menopausal women, and adult men (Riggs, et al. 2008).
Decreases in Inhibins at the peri-menopause leads to de-repression of pituitary FSH secretion and increased serum FSH (Danforth, et al. 1998; Klein and Soules 1998; Burger 1999; Muttukrishna, et al. 2000). FSH levels continue to increase into the post-menopausal period, as estradiol levels begin to decline, leading to the well reported elevations in bone resorption and further bone loss (Burger 1999; Riggs 2002).
The clinical data suggesting a role for cycling Inhibins (Vural, et al. 2005; Perrien, et al. 2006) are not easily reconciled with the anabolic effects of continuous Inhibin A exposure on the skeleton (Perrien, et al. 2007). However, the data suggest a bimodal mechanism of Inhibin action, similar to other bone active hormones. The best described precedent for bimodal hormone action on the skeleton is parathyroid hormone (PTH).
The well-described catabolic action of continuous versus strongly anabolic activity of intermittent PTH administration have been known for more than 6 decades (Ingalls, et al. 1943; Reeve, et al. 1980; Hock and Gera 1992; Lotinun, et al. 2002; Locklin, et al. 2003; Neer, et al. 2001). The primary target of Inhibin A anabolism appears to be cells in the osteoblastic lineage (Perrien, et al. 2007); however, the extent to which these are direct effects of continuous Inhibin A on bone cells is entirely unclear.
Additional studies are required to determine if Inhibins are also anabolic when administered in an intermittent fashion. Certainly, it is reasonable to speculate that the anabolic effects of continuous Inhibin A are targeting the same cells that ActRIIA ECD administration targets to achieve similar skeletal and cellular effects (Pearsall, et al. 2008). A direct comparison of ActRIIA ECD and Inhibin effects in vivo and in vitro is required to determine the extent to which ActRIIA ECD and Inhibin A are exploiting the same signaling pathways to block Activin action, and/or if these Inhibin A and ActRIIA ECD effects target the same cell type(s).
What controls the effect of Inhibin or Activin in a given situation? While the answer is unclear, it appears that the multiple effects of Inhibins and Activins may be regulated in both an exposure time- and context-dependent fashion. The integrated effects of Activin and Inhibin on a given cell type in vitro may reflect the microenvironment, the presence of local opposing hormones (Follistatin, BMPs, TGFβ), and the length of the exposure time in an in vivo setting. Moreover, the concept of Activin tone, while historically referred to solely in the context of Follistatin, should be amended to include Inhibin in the context of bone cell differentiation and metabolism. Given the dominant antagonism of Inhibin over Activin, as well as over BMPs and TGFβ (Gaddy-Kurten, et al. 2002; Wiater and Vale 2003; Farnworth, et al. 2006), it would appear that the rate limiting “tone” is that of the gonadally derived Inhibins, which have the capacity to antagonize the other locally produced stimulatory factors. Thus, cycling Inhibins in females and diurnal changes in Inhibin B in males would allow temporal shifts in Inhibin secretion to regulate locally stimulated increases in osteoblastogenesis and osteoclastogenesis.
Finally, there is ample evidence that bone is a reproductive endocrine target organ whose metabolism is not only regulated by sex steroids. Inhibins are a component of the normal reproductive endocrine repertoire that can regulate bone volume and strength in both the axial and appendicular skeleton, via regulation of bone cell differentiation. The ability of gonadal Inhibins to suppress bone cell differentiation and bone turnover (Perrien, et al. 2006) in the presence of locally produced Activins, TGFβs and BMPs (Gaddy-Kurten, et al. 2002; Perrien, et al. 2006) demonstrates that the bone loss associated with gonadectomy is not solely due to the loss of gonadal steroids. In fact, gonadectomy-induced bone loss results from the loss of sex steroids, gonadal Inhibins, and likely other undetermined factors from the HPG axis that affect bone. This concept is supported by data demonstrating the anabolic effect of exogenous transgenic Inhibin A expression in mice (Perrien, et al. 2007). Moreover, the onset of the loss of ovarian function at the menopause transition that occurs independent of changes in estradiol (Klein and Soules 1998; Burger 1999; Muttukrishna, et al. 2000) but with decreases in serum Inhibins, is temporally correlated with changes in bone turnover and decreases in volumetric BMD (Vural, et al. 2005; Riggs, et al. 2008).
Although the independent contributions of the non-sex steroid hormones in the HPG axis, including Inhibins, Activins and Follistatin, have yet to be completely characterized, it is abundantly clear that the continued use of the term “sex-steroid deficiency” to describe the reproductive hormone deficiencies that occur following gonadectomy that contribute to changes in bone mass and metabolism should be changed to “loss of ovarian function”.
Acknowledgments
Our studies of the action of activin and inhibin in bone have been supported by NIH grants R01-DK54044 and R21-DK74024 (DG) and F31-DK079362 (KMN), The Porter Physiology Developmental Fellowship (KMN), the NASA Graduate Student Research Program (DSP), and the Carl L. Nelson Chair of Orthopaedic Creativity (LJS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe M, Shintani Y, et al. Potent induction of activin A secretion from monocytes and bone marrow stromal fibroblasts by cognate interaction with activated T cells. J Leukoc Biol. 2002;72(2):347–52. [PubMed] [Google Scholar]
- Abid S, Maitra A, et al. Clinical and laboratory evaluation of idiopathic male infertility in a secondary referral center in India. J Clin Lab Anal. 2008;22(1):29–38. doi: 10.1002/jcla.20216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilezikjian LM, Blount AL, et al. Pituitary actions of ligands of the TGF-beta family: activins and inhibins. Reproduction. 2006;132(2):207–15. doi: 10.1530/rep.1.01073. [DOI] [PubMed] [Google Scholar]
- Bilezikjian LM, Blount AL, et al. Autocrine/paracrine regulation of pituitary function by activin, inhibin and follistatin. Mol Cell Endocrinol. 2004;225(1-2):29–36. doi: 10.1016/j.mce.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Bonewald LF, Mundy GR. Role of transforming growth factor-beta in bone remodeling. Clin Orthop. 1990;(250):261–76. [PubMed] [Google Scholar]
- Broxmeyer HE, Hangoc G, et al. Effects in vivo of purified recombinant human activin and erythropoietin in mice. Int J Hematol. 1991;54(6):447–54. [PubMed] [Google Scholar]
- Broxmeyer HE, Lu L, et al. Selective and indirect modulation of human multipotential and erythroid hematopoietic progenitor cell proliferation by recombinant human activin and inhibin. Proceedings of the National Academy of Sciences of the United States of America. 1988;85:9052–9056. doi: 10.1073/pnas.85.23.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger HG. The endocrinology of the menopause. J Steroid Biochem Mol Biol. 1999;69(1-6):31–5. doi: 10.1016/s0960-0760(98)00145-9. [DOI] [PubMed] [Google Scholar]
- Burger HG, Dudley EC, et al. Hormonal changes in the menopause transition. Recent Prog Horm Res. 2002;57:257–75. doi: 10.1210/rp.57.1.257. [DOI] [PubMed] [Google Scholar]
- Centrella M, Horowitz MC, et al. Transforming growth factor-beta gene family members and bone. Endocr Rev. 1994;15(1):27–39. doi: 10.1210/edrv-15-1-27. [DOI] [PubMed] [Google Scholar]
- Centrella M, McCarthy TL, et al. Activin-A binding and biochemical effects in osteoblast-enriched cultures from fetal-rat parietal bone. Mol Cell Biol. 1991;11(1):250–8. doi: 10.1128/mcb.11.1.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centrella M, McCarthy TL, et al. Glucocorticoid regulation of transforming growth factor beta 1 activity and binding in osteoblast-enriched cultures from fetal rat bone. Mol Cell Biol. 1991;11(9):4490–6. doi: 10.1128/mcb.11.9.4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citron JT, Ettinger B, et al. Spinal bone mineral loss in estrogen-replete, calcium-replete premenopausal women. Osteoporos Int. 1995;5(4):228–33. doi: 10.1007/BF01774011. [DOI] [PubMed] [Google Scholar]
- Corrigan AZ, Bilezikjian LM, et al. Evidence for an autocrine role of activin B within rat anterior pituitary cultures. Endocrinology. 1991;128(3):1682–4. doi: 10.1210/endo-128-3-1682. [DOI] [PubMed] [Google Scholar]
- Danforth DR, Arbogast LK, et al. Dimeric inhibin: a direct marker of ovarian aging. Fertil Steril. 1998;70(1):119–23. doi: 10.1016/s0015-0282(98)00127-7. [DOI] [PubMed] [Google Scholar]
- Danilovich N, Babu PS, et al. Estrogen deficiency, obesity, and skeletal abnormalities in follicle-stimulating hormone receptor knockout (FORKO) female mice. Endocrinology. 2000;141(11):4295–308. doi: 10.1210/endo.141.11.7765. [DOI] [PubMed] [Google Scholar]
- de Kretser DM, Buzzard JJ, et al. The role of activin, follistatin and inhibin in testicular physiology. Mol Cell Endocrinol. 2004;225(1-2):57–64. doi: 10.1016/j.mce.2004.07.008. [DOI] [PubMed] [Google Scholar]
- de Kretser DM, Hedger MP, et al. Inhibins, activins and follistatin in reproduction. Hum Reprod Update. 2002;8(6):529–41. doi: 10.1093/humupd/8.6.529. [DOI] [PubMed] [Google Scholar]
- de Kretser DM, Meinhardt A, et al. The roles of inhibin and related peptides in gonadal function. Mol Cell Endocrinol. 2000;161(1-2):43–6. doi: 10.1016/s0303-7207(99)00222-1. [DOI] [PubMed] [Google Scholar]
- Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425(6958):577–84. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- Dolter KE, Palyash JC, et al. Analysis of activin A gene expression in human bone marrow stromal cells. J Cell Biochem. 1998;70(1):8–21. doi: 10.1002/(sici)1097-4644(19980701)70:1<8::aid-jcb2>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Donaldson CJ, Vaughan JM, et al. Activin and inhibin binding to the soluble extracellular domain of activin receptor II. Endocrinology. 1999;140(4):1760–6. doi: 10.1210/endo.140.4.6665. [DOI] [PubMed] [Google Scholar]
- Ebeling PR, Atley LM, et al. Bone turnover markers and bone density across the menopausal transition. J Clin Endocrinol Metab. 1996;81(9):3366–71. doi: 10.1210/jcem.81.9.8784098. [DOI] [PubMed] [Google Scholar]
- Eijken M, Swagemakers S, et al. The activin A-follistatin system: potent regulator of human extracellular matrix mineralization. Faseb J. 2007;21(11):2949–60. doi: 10.1096/fj.07-8080com. [DOI] [PubMed] [Google Scholar]
- Esparza-Lopez J, Montiel JL, et al. Ligand binding and functional properties of betaglycan, a co-receptor of the transforming growth factor-beta superfamily. Specialized binding regions for transforming growth factor-beta and inhibin A. J Biol Chem. 2001;276(18):14588–96. doi: 10.1074/jbc.M008866200. [DOI] [PubMed] [Google Scholar]
- Farnworth PG, Stanton PG, et al. Inhibins differentially antagonize activin and bone morphogenetic protein action in a mouse adrenocortical cell line. Endocrinology. 2006;147(7):3462–71. doi: 10.1210/en.2006-0023. [DOI] [PubMed] [Google Scholar]
- Findlay JK. Peripheral and local regulators of folliculogenesis. Reprod Fertil Dev. 1994;6(2):127–39. doi: 10.1071/rd9940127. [DOI] [PubMed] [Google Scholar]
- Fujimoto K, Kawakita M, et al. Purification of megakaryocyte differentiation activity from a human fibrous histiocytoma cell line: N-terminal sequence homology with activin A. Biochem Biophys Res Commun. 1991;174(3):1163–8. doi: 10.1016/0006-291x(91)91543-l. [DOI] [PubMed] [Google Scholar]
- Fuller K, Bayley KE, et al. Activin A is an essential cofactor for osteoclast induction. Biochem Biophys Res Commun. 2000;268(1):2–7. doi: 10.1006/bbrc.2000.2075. [DOI] [PubMed] [Google Scholar]
- Gaddy-Kurten D, Coker JK, et al. Inhibin suppresses and activin stimulates osteoblastogenesis and osteoclastogenesis in murine bone marrow cultures. Endocrinology. 2002;143(1):74–83. doi: 10.1210/endo.143.1.8580. [DOI] [PubMed] [Google Scholar]
- Gaddy-Kurten D, Coker JK, et al. Activin substitutes for the BMP2/4 requirement for, and the noggin inhibition of, osteoblastogenesis and osteoclastogenesis in adult murine bone marrow cultures. Bone. 1998;23(5S):S166. [Google Scholar]
- Gaddy-Kurten D, Tsuchida K, et al. Activins and the receptor serine kinase superfamily. Recent Prog Horm Res. 1995;50:109–29. doi: 10.1016/b978-0-12-571150-0.50010-x. [DOI] [PubMed] [Google Scholar]
- Gao J, Tiwari-Pandey R, et al. Altered ovarian function affects skeletal homeostasis independent of the action of follicle-stimulating hormone. Endocrinology. 2007;148(6):2613–21. doi: 10.1210/en.2006-1404. [DOI] [PubMed] [Google Scholar]
- Gregory SJ, Kaiser UB. Regulation of gonadotropins by inhibin and activin. Semin Reprod Med. 2004;22(3):253–67. doi: 10.1055/s-2004-831901. [DOI] [PubMed] [Google Scholar]
- Hock JM, Gera I. Effects of continuous and intermittent administration and inhibition of resorption on the anabolic response of bone to parathyroid hormone. Journal of Bone and Mineral Research. 1992;7:65–72. doi: 10.1002/jbmr.5650070110. [DOI] [PubMed] [Google Scholar]
- Horowitz MC, Xi Y, et al. Control of osteoclastogenesis and bone resorption by members of the TNF family of receptors and ligands. Cytokine Growth Factor Rev. 2001;12(1):9–18. doi: 10.1016/s1359-6101(00)00030-7. [DOI] [PubMed] [Google Scholar]
- Hosoi T, Inoue S, et al. Immunohistochemical detection of activin A in osteoclasts. Gerontology. 1996;42 1:20–24. doi: 10.1159/000213821. [DOI] [PubMed] [Google Scholar]
- Huang HJ, Wu JC, et al. A novel role for bone morphogenetic proteins in the synthesis of follicle-stimulating hormone. Endocrinology. 2001;142(6):2275–83. doi: 10.1210/endo.142.6.8159. [DOI] [PubMed] [Google Scholar]
- Hurwitz JM, Santoro N. Inhibins, activins, and follistatin in the aging female and male. Semin Reprod Med. 2004;22(3):209–17. doi: 10.1055/s-2004-831896. [DOI] [PubMed] [Google Scholar]
- Ikenoue T, Jingushi S, et al. Inhibitory effects of activin-A on osteoblast differentiation during cultures of fetal rat calvarial cells. J Cell Biochem. 1999;75(2):206–14. doi: 10.1002/(sici)1097-4644(19991101)75:2<206::aid-jcb3>3.3.co;2-k. [DOI] [PubMed] [Google Scholar]
- Ingalls TH, Donaldson G, et al. The locus of action of the parathyroid hormone: Experimental studies with parathyroid extract on normal and nephrectomized rats. J Clin Invest. 1943;22(4):603–8. doi: 10.1172/JCI101432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue S, Nomura S, et al. Localization of follistatin, an activin-binding protein, in bone tissues. Calcif Tissue Int. 1994;55(5):395–7. doi: 10.1007/BF00299321. [DOI] [PubMed] [Google Scholar]
- Karsenty G. Convergence between bone and energy homeostases: leptin regulation of bone mass. Cell Metab. 2006;4(5):341–8. doi: 10.1016/j.cmet.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Khosla S, Riggs BL, et al. Effects of sex and age on bone microstructure at the ultradistal radius: A population-based noninvasive in vivo assessment. J Bone Miner Res. 2006;21:124–131. doi: 10.1359/JBMR.050916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkbride KC, Townsend TA, et al. Bone morphogenetic proteins signal through the transforming growth factor-beta type III receptor. J Biol Chem. 2008 doi: 10.1074/jbc.M704883200. [DOI] [PubMed] [Google Scholar]
- Klein NA, Battaglia DE, et al. Reproductive aging: accelerated ovarian follicular development associated with a monotropic follicle-stimulating hormone rise in normal older women. J Clin Endocrinol Metab. 1996;81(3):1038–45. doi: 10.1210/jcem.81.3.8772573. [DOI] [PubMed] [Google Scholar]
- Klein NA, Houmard BS, et al. Age-related analysis of inhibin a, inhibin B, and activin a relative to the intercycle monotropic follicle-stimulating hormone rise in normal ovulatory women. J Clin Endocrinol Metab. 2004;89(6):2977–81. doi: 10.1210/jc.2003-031515. [DOI] [PubMed] [Google Scholar]
- Klein NA, Soules MR. Endocrine changes of the perimenopause. Clin Obstet Gynecol. 1998;41(4):912–20. doi: 10.1097/00003081-199812000-00017. [DOI] [PubMed] [Google Scholar]
- Koseki T, Gao Y, et al. Role of TGF-beta family in osteoclastogenesis induced by RANKL. Cell Signal. 2002;14(1):31–6. doi: 10.1016/s0898-6568(01)00221-2. [DOI] [PubMed] [Google Scholar]
- Kumanov P, Nandipati KC, et al. Significance of inhibin in reproductive pathophysiology and current clinical applications. Reprod Biomed Online. 2005;10(6):786–812. doi: 10.1016/s1472-6483(10)61124-8. [DOI] [PubMed] [Google Scholar]
- Lewis KA, Gray PC, et al. Betaglycan binds inhibin and can mediate functional antagonism of activin signalling. Nature. 2000;404(6776):411–4. doi: 10.1038/35006129. [DOI] [PubMed] [Google Scholar]
- Locklin RM, Khosla S, et al. Mediators of the biphasic responses of bone to intermittent and continuously administered parathyroid hormone. J Cell Biochem. 2003;89(1):180–90. doi: 10.1002/jcb.10490. [DOI] [PubMed] [Google Scholar]
- Lotinun S, Sibonga JD, et al. Differential effects of intermittent and continuous administration of parathyroid hormone on bone histomorphometry and gene expression. Endocrine. 2002;17(1):29–36. doi: 10.1385/ENDO:17:1:29. [DOI] [PubMed] [Google Scholar]
- Manolagas SC. Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev. 2000;21(2):115–37. doi: 10.1210/edrv.21.2.0395. [DOI] [PubMed] [Google Scholar]
- Martin TJ, Ng KW. Mechanisms by which cells of the osteoblast lineage control osteoclast formation and activity. Journal of Cellular Biochemistry. 1994;56:357–366. doi: 10.1002/jcb.240560312. [DOI] [PubMed] [Google Scholar]
- Martin TJ, Quinn JM, et al. Mechanisms involved in skeletal anabolic therapies. Ann N Y Acad Sci. 2006;1068:458–70. doi: 10.1196/annals.1346.043. [DOI] [PubMed] [Google Scholar]
- Mason AJ, Hayflick JS, et al. Complementary DNA sequences of ovarian follicular fluid inhibin show precursor structure and homology with transforming growth factor-beta. Nature. 1985;318(6047):659–63. doi: 10.1038/318659a0. [DOI] [PubMed] [Google Scholar]
- Matzuk MM, Kumar TR, et al. Functional analysis of activins during mammalian development. Nature. 1995;374(6520):354–6. doi: 10.1038/374354a0. [DOI] [PubMed] [Google Scholar]
- McCarthy TL, Pham TH, et al. Prostaglandin E2 increases transforming growth factor-beta type III receptor expression through CCAAT enhancer-binding protein delta in osteoblasts. Mol Endocrinol. 2007;21(11):2713–24. doi: 10.1210/me.2007-0210. [DOI] [PubMed] [Google Scholar]
- Meachem SJ, Nieschlag E, et al. Inhibin B in male reproduction: pathophysiology and clinical relevance. Eur J Endocrinol. 2001;145(5):561–71. doi: 10.1530/eje.0.1450561. [DOI] [PubMed] [Google Scholar]
- Meunier H, Rivier C, et al. Gonadal and extragonadal expression of inhibin alpha, beta A, and beta B subunits in various tissues predicts diverse functions. Proc Natl Acad Sci U S A. 1988;85(1):247–51. doi: 10.1073/pnas.85.1.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muttukrishna S, Child T, et al. Serum concentrations of dimeric inhibins, activin A, gonadotrophins and ovarian steroids during the menstrual cycle in older women. Hum Reprod. 2000;15(3):549–56. doi: 10.1093/humrep/15.3.549. [DOI] [PubMed] [Google Scholar]
- Muttukrishna S, Farouk A, et al. Serum activin A and follistatin in disorders of spermatogenesis in men. Eur J Endocrinol. 2001;144(4):425–9. doi: 10.1530/eje.0.1440425. [DOI] [PubMed] [Google Scholar]
- Neer RM, Arnaud CD, et al. Effect of parathyroid hormone (1-34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med. 2001;344(19):1434–41. doi: 10.1056/NEJM200105103441904. [DOI] [PubMed] [Google Scholar]
- Ogawa Y, Schmidt DK, et al. Bovine bone activin enhances bone morphogenetic protein-induced ectopic bone formation. J Biol Chem. 1992;267(20):14233–7. [PubMed] [Google Scholar]
- Okafuji K, Kaku K, et al. Effects of activin A/erythroid differentiation factor on erythroid and megakaryocytic differentiations of mouse erythroleukemia (Friend) cells: evidence for two distinct modes of cell response. Exp Hematol. 1995;23(3):210–6. [PubMed] [Google Scholar]
- Oue Y, Kanatani H, et al. Effect of local injection of activin A on bone formation in newborn rats. Bone. 1994;15:361–366. doi: 10.1016/8756-3282(94)90301-8. [DOI] [PubMed] [Google Scholar]
- Parfitt AM, Mathews CH, et al. Relationships between surface, volume, and thickness of iliac trabecular bone in aging and in osteoporosis. Implications for the microanatomic and cellular mechanisms of bone loss. J Clin Invest. 1983;72(4):1396–409. doi: 10.1172/JCI111096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearsall RS, Canalis E, et al. A soluble activin type IIA receptor induces bone formation and improves skeletal integrity. Proc Natl Acad Sci U S A. 2008;105(19):7082–7. doi: 10.1073/pnas.0711263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrien DS, Achenbach SJ, et al. Bone turnover across the menopause transition: correlations with inhibins and follicle-stimulating hormone. J Clin Endocrinol Metab. 2006;91(5):1848–54. doi: 10.1210/jc.2005-2423. [DOI] [PubMed] [Google Scholar]
- Perrien DS, Akel NS, et al. Inhibin A is an endocrine stimulator of bone mass and strength. Endocrinology. 2007;148(4):1654–65. doi: 10.1210/en.2006-0848. [DOI] [PubMed] [Google Scholar]
- Reame NE, Lukacs JL, et al. Differential effects of aging on activin A and its binding protein, follistatin, across the menopause transition. Fertil Steril. 2007;88(4):1003–5. doi: 10.1016/j.fertnstert.2006.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve J, Meunier PJ, et al. Anabolic effect of human parathyroid hormone fragment on trabecular bone in involutional osteoporosis: a multicentre trial. Jr Br Med J. 1980;280(6228):1340–4. doi: 10.1136/bmj.280.6228.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs BL. Endocrine causes of age-related bone loss and osteoporosis. Novartis Found Symp. 2002;242:247–59. discussion 260-4. [PubMed] [Google Scholar]
- Riggs BL, Melton IiiLJ, 3rd, et al. Population-based study of age and sex differences in bone volumetric density, size, geometry, and structure at different skeletal sites. J Bone Miner Res. 2004;19(12):1945–54. doi: 10.1359/JBMR.040916. [DOI] [PubMed] [Google Scholar]
- Riggs BL, Melton LJ, et al. A population-based assessment of rates of bone loss at multiple skeletal sites: evidence for substantial trabecular bone loss in young adult women and men. J Bone Miner Res. 2008;23(2):205–14. doi: 10.1359/JBMR.071020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson DM, Burger HG. Reproductive hormones: ageing and the perimenopause. Acta Obstet Gynecol Scand. 2002;81(7):612–6. doi: 10.1034/j.1600-0412.2002.810706.x. [DOI] [PubMed] [Google Scholar]
- Rosen V. BMP and BMP inhibitors in bone. Ann N Y Acad Sci. 2006;1068:19–25. doi: 10.1196/annals.1346.005. [DOI] [PubMed] [Google Scholar]
- Rowe DW, Kream BE. Regulation of collagen synthesis in fetal rat calvaria by 1,25-dihydroxyvitamin D3. J Biol Chem. 1982;257(14):8009–15. [PubMed] [Google Scholar]
- Ruckle J, Jacobs M, et al. Single-dose, randomized, double-blind, placebo-controlled study of ACE-011 (ActRIIA-IgG1) in postmenopausal women. J Bone Miner Res. 2009;24(4):744–52. doi: 10.1359/jbmr.081208. [DOI] [PubMed] [Google Scholar]
- Sakai R, Eto Y, et al. Activin release from bone coupled to bone resorption in organ culture of neonatal mouse calvaria. Bone. 2000;26(3):235–40. doi: 10.1016/s8756-3282(99)00268-9. [DOI] [PubMed] [Google Scholar]
- Sakai R, Eto Y, et al. Activin enhances osteoclast-like cell formation in vitro. Biochem Biophys Res Commun. 1993;195:39–46. doi: 10.1006/bbrc.1993.2006. [DOI] [PubMed] [Google Scholar]
- Sakai R, Fujita S, et al. Activin increases bone mass and mechanical strength of lumbar vertebrae in aged ovariectomized rats. Bone. 2000;27(1):91–6. doi: 10.1016/s8756-3282(00)00307-0. [DOI] [PubMed] [Google Scholar]
- Sakai R, Miwa K, et al. Local administration of activin promotes fracture healing in the rat fibula fracture model. Bone. 1999;25(2):191–6. doi: 10.1016/s8756-3282(99)00152-0. [DOI] [PubMed] [Google Scholar]
- Scher W, Eto Y, et al. Phorbol ester-treated human acute myeloid leukemia cells secrete G-CSF, GM-CSF and erythroid differentiation factor into serum-free media in primary culture. Biochim Biophys Acta. 1990;1055(3):278–86. doi: 10.1016/0167-4889(90)90044-e. [DOI] [PubMed] [Google Scholar]
- Shao L, Frigon NL, et al. Regulation of production of activin A in human marrow stromal cells and monocytes. Experimental Hematology. 1992;20:1235–1242. [PubMed] [Google Scholar]
- Sugatani T, Alvarez UM, et al. Activin A stimulates IkappaB-alpha/NFkappaB and RANK expression for osteoclast differentiation, but not AKT survival pathway in osteoclast precursors. J Cell Biochem. 2003;90(1):59–67. doi: 10.1002/jcb.10613. [DOI] [PubMed] [Google Scholar]
- Sun L, Peng Y, et al. FSH directly regulates bone mass. Cell. 2006;125(2):247–60. doi: 10.1016/j.cell.2006.01.051. [DOI] [PubMed] [Google Scholar]
- Takeda S, Karsenty G. Molecular bases of the sympathetic regulation of bone mass. Bone. 2008;42(5):837–40. doi: 10.1016/j.bone.2008.01.005. [DOI] [PubMed] [Google Scholar]
- Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function. Nat Rev Genet. 2003;4(8):638–49. doi: 10.1038/nrg1122. [DOI] [PubMed] [Google Scholar]
- Tuuri T, Eramaa M, et al. The tissue distribution of activin beta A- and beta B-subunit and follistatin messenger ribonucleic acids suggests multiple sites of action for the activin-follistatin system during human development. J Clin Endocrinol Metab. 1994;78(6):1521–4. doi: 10.1210/jcem.78.6.8200957. [DOI] [PubMed] [Google Scholar]
- Uchimaru K, Motokura T, et al. Bone marrow stromal cells produce and respond to activin A: Interactions with basic fibroblast growth factor and platelet-derived growth factor. Experimental Hematology. 1995;23:613–618. [PubMed] [Google Scholar]
- Vale W, Bilezikjian LM, et al. Reproductive and other roles of inhibins and activins. In: Knobil E, Neil JD, editors. The Physiology of Reproduction. New York: Raven Press; 1994. pp. 1861–1878. [Google Scholar]
- Vale W, Wiater E, et al. Activins and inhibins and their signaling. Ann N Y Acad Sci. 2004;1038:142–7. doi: 10.1196/annals.1315.023. [DOI] [PubMed] [Google Scholar]
- Vural F, Vural B, et al. Ovarian aging and bone metabolism in menstruating women aged 35-50 years. Maturitas. 2005;52(2):147–53. doi: 10.1016/j.maturitas.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Welt C, Sidis Y, et al. Activins, inhibins, and follistatins: from endocrinology to signaling. A paradigm for the new millennium. Exp Biol Med (Maywood) 2002;227(9):724–52. doi: 10.1177/153537020222700905. [DOI] [PubMed] [Google Scholar]
- Welt CK. The physiology and pathophysiology of inhibin, activin and follistatin in female reproduction. Curr Opin Obstet Gynecol. 2002;14(3):317–23. doi: 10.1097/00001703-200206000-00012. [DOI] [PubMed] [Google Scholar]
- Welt CK, McNicholl DJ, et al. Female reproductive aging is marked by decreased secretion of dimeric inhibin. J Clin Endocrinol Metab. 1999;84(1):105–11. doi: 10.1210/jcem.84.1.5381. [DOI] [PubMed] [Google Scholar]
- Wiater E, Harrison CA, et al. Identification of distinct inhibin and transforming growth factor beta-binding sites on betaglycan: functional separation of betaglycan co-receptor actions. J Biol Chem. 2006;281(25):17011–22. doi: 10.1074/jbc.M601459200. [DOI] [PubMed] [Google Scholar]
- Wiater E, Vale W. Inhibin is an antagonist of bone morphogenetic protein signaling. J Biol Chem. 2003;278(10):7934–41. doi: 10.1074/jbc.M209710200. [DOI] [PubMed] [Google Scholar]
- Woodruff TK, Krummen L, et al. In situ ligand binding of recombinant human [125I] activin-A and recombinant human [125I]inhibin-A to the adult rat ovary. Endocrinology. 1993;133(6):2998–3006. doi: 10.1210/endo.133.6.8243328. [DOI] [PubMed] [Google Scholar]
- Wozney JM. The bone morphogenetic protein family and osteogenesis. Mol Reprod Dev. 1992;32(2):160–7. doi: 10.1002/mrd.1080320212. [DOI] [PubMed] [Google Scholar]
- Xing L, Bushnell TP, et al. NF-kappaB p50 and p52 expression is not required for RANK-expressing osteoclast progenitor formation but is essential for RANK- and cytokine-mediated osteoclastogenesis. J Bone Miner Res. 2002;17(7):1200–10. doi: 10.1359/jbmr.2002.17.7.1200. [DOI] [PubMed] [Google Scholar]
- Yadav VK, Ryu JH, et al. Lrp5 controls bone formation by inhibiting serotonin synthesis in the duodenum. Cell. 2008;135(5):825–37. doi: 10.1016/j.cell.2008.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada R, Suzuki T, et al. Induction of differentiation of the human promyelocytic cell line HL-60 by activin/EDF. Biochem Biophys Res Commun. 1992;187(1):79–85. doi: 10.1016/s0006-291x(05)81461-5. [DOI] [PubMed] [Google Scholar]
- Yamashita N, Nakajima T, et al. Effects of activin A on IgE synthesis and cytokine production by human peripheral mononuclear cells. Clin Exp Immunol. 1993;94(1):214–9. doi: 10.1111/j.1365-2249.1993.tb06003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Takahashi S, et al. Expression of activin A/erythroid differentiation factor in murine bone marrow stromal cells. Blood. 1992;79(2):304–7. [PubMed] [Google Scholar]
- Ying SY. Inhibins, activins, and follistatins: gonadal proteins modulating the secretion of follicle-stimulating hormone. Endocr Rev. 1988;9(2):267–93. doi: 10.1210/edrv-9-2-267. [DOI] [PubMed] [Google Scholar]
- Yu AW, Shao LE, et al. Detection of functional and dimeric activin A in human marrow microenvironment. Implications for the modulation of erythropoiesis. Ann N Y Acad Sci. 1994;718:285–98. doi: 10.1111/j.1749-6632.1994.tb55727.x. discussion 298-9. [DOI] [PubMed] [Google Scholar]
- Yu J, Shao LE, et al. Importance of FSH-releasing protein and inhibin in erythrodifferentiation. Nature. 1987;330(6150):765–7. doi: 10.1038/330765a0. [DOI] [PubMed] [Google Scholar]