

Abstract
Structural variants (SVs) are a major source of human genomic variation; however, characterizing them at nucleotide resolution remains challenging. Here we assemble a library of breakpoints at nucleotide resolution from collating and standardizing ~2,000 published SVs. For each breakpoint, we infer its ancestral state (through comparison to primate genomes) and its mechanism of formation (e.g., non-allelic homologous recombination, NAHR). We characterize breakpoint sequences with respect to genomic landmarks, chromosomal location, sequence motifs and physical properties, finding that the occurrence of insertions and deletions is more balanced than previously reported and that NAHR-formed breakpoints are associated with relatively rigid, stable DNA helices. Finally, we demonstrate an approach, BreakSeq, for scanning the reads from short-read sequenced genomes against our breakpoint library to accurately identify previously overlooked SVs, which we then validate by PCR. As new data become available, we expect our BreakSeq approach will become more sensitive and facilitate rapid SV genotyping of personal genomes.
Introduction
Structural variation of large segments (>1kb), including copy-number variation (CNV) and unbalanced inversion events, is widespread in human genomes1–6 with ~20,000 SVs presently reported in the Database of Genomic Variants (DGV)2. These SVs considerably impact genomic variation by causing more nucleotide differences between individuals than single-nucleotide polymorphisms4–6 (SNPs). In several genomic loci, SV formation rates could even be orders of magnitude higher than single nucleotide substitution rates7, 8. In order to measure the influence on human phenotypes of common SVs (i.e., those present at substantial allele frequencies in populations) and de novo formed SVs, several studies have mapped SVs across individuals. They reported associations of SVs with normal traits and with a range of diseases including cancer, HIV, developmental disorders and autoimmune diseases9–14. While most SVs listed in DGV are presumably common, de novo SV formation is believed to occur constantly in the germline and several mutational mechanisms have been proposed15.
Nevertheless, so far our understanding of SVs and the way we analyze SV maps is limited by the fact that most recent surveys, such as those solely based on microarrays, have not revealed the precise start- and end-coordinates (i.e., breakpoints) of the SVs. This has hampered our understanding of the actual extent and effects of SVs in humans, as mapping at breakpoint resolution can reveal SVs that intersect with exons of genes or that lead to gene fusion events5, 16.
The lack of nucleotide-resolution maps has further prevented systematic deduction of the processes involved in SV formation, such as whether common SVs emerged initially as insertions or deletions at ancestral genomic loci. Instead, operational definitions have been applied for classifying common SVs into gains, losses, insertions and deletions either based on allele frequency measurements, or the ‘human reference genome’ (hereafter also referred as the reference genome) that was originally derived from a mixed pool of individuals17. Thus, inference of the ancestral state of an SV locus is crucial for relating SV surveys to primate genome evolution and population genetics.
In addition, the lack of data at breakpoint resolution has limited the number of SVs for which the likely mutational mechanisms of origin have been inferred. These mechanisms are thought to include (i) non-allelic homologous recombination (NAHR) involving homology-mediated recombination between paralogous sequence blocks; (ii) non-homologous recombination (NHR) associated with the repair of DNA double-strand breaks (i.e., non-homologous end-joining, NHEJ) or with the rescue of DNA replication-fork stalling events (i.e., fork-stalling and template switching18); (iii) variable number of tandem repeats (VNTRs) resulting from expansion or contraction of simple tandem repeat units; and (iv) transposable element insertions (TEIs) involving mostly long and short interspersed elements (LINEs and SINEs) and combinations thereof, along with other types of TEI-associated events (e.g., processed pseudogenes).
Finally, owing to the lack of resolution of most SV maps, junction sequences (the flanking sequences of breakpoints) have thus far not been exploited for testing the presence of SVs in a queried individual in a similar fashion to the way SNPs can be directly detected by oligonucleotide chips with probes designed for each polymorphism.
Recent advances in microarray technology and particularly large-scale DNA sequencing have paved the way for ‘high-resolution’ SV maps. To date, nearly two thousand SVs have been fine-mapped at breakpoint level and efforts such as the 1000 Genomes Project (http://1000genomes.org), which will soon sequence over a thousand human genomes, might in the near future report many more SVs at such resolution (Supplementary Fig. 1). Thus far however, no study has leveraged the potential of collectively analyzing breakpoint-level SV data.
Here we present a comprehensive analysis of a library of nearly two thousand breakpoint-level SVs assembled from eight recent surveys that involve individuals from three distinct populations. We demonstrate four uses of the breakpoint library—mapping structural variation at high resolution, revealing ancestral states of variants, inferring mechanisms of variant formation, and correlating the inferred mechanisms with DNA sequence features. We found several lines of evidence consistent with a non-uniform distribution of SV formation mechanisms and with locus-specific sequence properties, such as DNA helix stability, chromatin accessibility and the propensity for a DNA sequence to recombine, predisposing genomic regions to SV-mutational processes.
Results
Generation of a standardized SV breakpoint library
We compiled a set of breakpoints from eight published sources (Fig. 1). In accordance with the proposed operational definition by Feuk and coworkers19, we defined SVs to be deletions, insertions and inversions reported relative to the reference genome with a size of 1kb or larger. As our initial library encompassed SVs mapped using different types of evidence, sequencing technologies and genome assembly versions, an essential first step of our framework was ‘library standardization’. We therefore implemented a computational pipeline for generating a unified, non-redundant breakpoint library (Online Methods).
Figure 1.
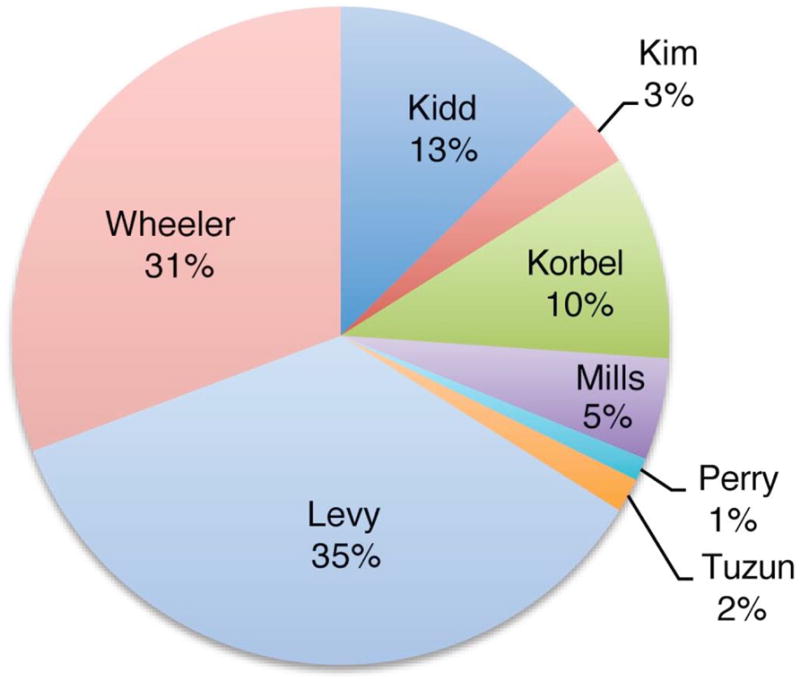
Composition of the SV breakpoint library. SVs in the library were based on different SV-mapping and breakpoint-sequencing strategies. A large fraction (44%) of the breakpoints were based on data generated using 454/Roche sequencing, including resequencing of an individual human genome (Wheeler21, 602 SVs) and sequencing of breakpoints in two individuals following high-resolution and massive paired-end mapping (Korbel5 and Kim16, 264 SVs). The remaining 56% of the breakpoints were identified using other approaches, including Sanger capillary sequencing of breakpoints identified by whole-genome shotgun sequencing and assembly of an individual human genome(Levy44, 694 SVs), fosmid-paired-end sequencing carried out in multiple individuals (Tuzun3 and Kidd6, 281 SVs), breakpoints mined from SNP discovery DNA resequencing traces(Mills45, 98 SVs), and tiling-array based comparative genomic hybridization followed by breakpoint sequencing (Perry25, 22 SVs). Fewer than five breakpoints were reported in two genomes sequenced using short 36 bp reads (Illumina/Solexa)22, 23, presumably owing to the complex DNA sequence patterns frequently associated with breakpoints5, 6, 25.
The pipeline yielded a non-redundant set of 1,889 SVs that were initially annotated as deletions (1,409), insertions (419) or inversions (61) relative to the reference genome (Supplementary Fig. 2). This set, which represents the most exhaustive compilation to date of SV breakpoints in phenotypically normal individuals, is available as Supplementary Table 1 and at http://sv.gersteinlab.org/breakseq. It also has been deposited into the BreakDB database20 (http://sv.gersteinlab.org/breakdb).
High-resolution mapping of SVs from short-read sequencing data
Personal genomics endeavors based on next-generation sequencing technology21–23 typically detect genomic variation by mapping relatively short sequencing reads directly onto the reference genome. Although many short indels (<1kb) can be accurately identified with such an approach, SVs >1kb are commonly missed, or not identified at nucleotide (that is, breakpoint-level) resolution. This is probably due to the difficulty in constructing accurate sequence alignments from short reads (e.g., 36mers), especially if they involve long sequence gaps or span breakpoints.
We thus devised an approach, BreakSeq, for detecting SVs by aligning raw reads directly onto SV breakpoint junctions of the alternative, non-reference, alleles contained in our library (Fig. 2a, Online Methods). Briefly, the genomic coordinates of each breakpoint in the standardized library are used to extract 30 bp of flanking sequence on the reference genome. These 30 bp flanking sequences are concatenated into 60 bp ‘junction sequences’. Thus, a deletion event is represented with a single junction sequence in the library (containing the sequence flanking its single breakpoint), while an insertion has both left and right junction sequences (containing the sequence flanking each of its two breakpoints). DNA reads from personal genomes are aligned against the junction sequences. Successful alignment requires a read to overlap a junction sequence by at least 10 bp on each side of the breakpoint. This approach is conceptually similar to using a library of exon splice junctions in transcriptome analyses, which leads to a considerably better coverage of alternatively spliced transcripts than restricting the analysis to reference genome sequences lacking splice junctions24.
Figure 2.



Mapping breakpoints using the library. (a) Overview of the BreakSeq approach. Breakpoints are used to generate junction sequences (upper)—the 30 bp of sequence flanking each side of the breakpoint is extracted to form a 60 bp of junction sequence. Then, DNA reads are aligned to the junction sequences (lower). Alignment results are interpreted as follows. In the case of insertions relative to the reference genome (left), sequences A and B represent the left and right breakpoint junction sequences of the non-reference SV allele, respectively. In the case of deletions (right), sequence C represents the junction sequence of the non-reference SV allele. Solid lines with arrows, successful alignments. Dashed lines with crosses, no proper alignment. (b) Representative PCR validation of detected SVs in NA12891. Primers flanking each SV were used to amplify41 different genomic regions(see Supplementary Table 3 for genomic coordinates and primer sequences). Expected band sizes for the reference and non-reference SV alleles are given at the top of each lane. The difference in size of the products for the reference and non-reference alleles confirmed the presence of the SVs for all loci except 6, 13 (confirmed by LongAmp Taq in a separate experiment), 21, 25 and 36. M1 is a 100bp marker and M2 is a 1kb marker. (c) A subset of SVs, which were confirmed by sequencing, was analyzed in nine additional genomic DNA samples (HapMap individuals with ancestry in Europe) to test for SV frequency within the CEPH population. An asterisk indicates that the SV is present polymorphically.
To demonstrate the utility of our approach for mapping personal SVs at high resolution, we mapped short reads from three personal genomes sequenced with Illumina/Solexa technology. These included two previously published genomes22, 23 from individuals of Nigerian (Yoruba from Ibadan, YRI) and Han Chinese (HCH) origins. The third genome was from a HapMap individual of European ancestry (CEPH) that was sequenced recently in the pilot phase of the 1000 Genomes Project (http://www.1000genomes.org). To prioritize the SV calls generated by BreakSeq, we developed a scoring system based on supportive read-matches (the number of reads that map to a breakpoint; Online Methods) and distinguished low-support SV calls (with 1 to 4 supportive read-matches) from high-support SV calls. For the HCH, CEPH (NA12891) and YRI (NA18507) genomes, we identified 158, 219 and 179 SVs, respectively (Supplementary Table 2). Several SVs were shared among the three, suggesting that they may represent common alleles. For example, among the high-support calls, we found that 57 SVs were shared between the YRI and HCH genomes, 62 between the YRI and NA12891 genomes, 52 between the HCH and NA12891 genomes, and 42 were common to all three genomes.
To validate these results, we used PCR to test 24 insertion and 33 deletion calls predicted in NA12891 relative to the reference (Supplementary Table 3). Specifically, PCR amplification of predicted non-reference SV alleles5 was used as a means for validation. In 48 cases the predicted SVs were validated, and in one case the reaction was inconclusive (Fig. 2b and Supplementary Fig. 3). Furthermore, seven reactions neither revealed the reference allele nor the predicted SV allele. (This primer failure rate can be explained by repetitive and GC-rich sequences that occur in association with SVs.) Finally, in a single case only the reference allele was found, suggesting either a false positive prediction or the inability to amplify the event band of a predicted size of 7.5kb.
We then sequenced 12 of the PCR-validated amplicons with Sanger capillary sequencing and confirmed the predicted breakpoint in all—that is, the Sanger-sequenced junction was identical to that in the library, with few single base-pair differences (presumable SNPs). We also analyzed a panel including 9 unrelated CEPH individuals for the presence of 6 of the sequenced SVs and found that most SVs (4) were present polymorphically, whereas the remaining SVs likely represent rare alleles (Fig. 2c and Supplementary Table 3). All together, 48 out of 57 predicted SVs (84.2%) were confirmed successfully, and the validation rate was estimated at 98% (48 out of 49) based on the PCR reactions that could be scored, demonstrating high specificity. Notably, as about half of our validated SVs were low-support SV calls, our validations demonstrate that accurate calls are generated both at high- and low-support levels. This suggests that BreakSeq may perform reasonably even in conjunction with low-coverage sequencing projects.
Inferring ancestral states of SV loci by comparing breakpoint junctions to primate genomes
Global SV surveys have been so far reporting SV events such as insertions and deletions using operational definitions—that is, comparisons with the human reference genome or allele frequency measurements. However, we reasoned that a systematic assessment of SV formation requires an unambiguous discrimination of SV event types—that is, one minimally affected by ascertainment biases. Since the human reference genome presumably contains a mixture of common and rare SV alleles, it can serve only as a provisional reference for classifying SVs as insertions or deletions. Likewise, allele frequency measurements are of limited use in the context of classifying SVs into ‘gains’ and ‘losses’, as they may be affected by population-specific allele frequencies. In fact, ancestral state assignments facilitate systematic surveys of SVs in the context of studies focusing on human genome evolution, SV formational processes as well as minor/major allele assignment (as the ancestral allele often corresponds to the major one).
We therefore devised a framework that automatically assigns ancestral states of SV genomic loci based on a comparison of SV breakpoint junction sequence with the corresponding syntenic segments from the chimpanzee, orangutan and macaque genomes. Our approach (Fig 3a, Online Methods) involves extracting ±500bp flanking sequences around each breakpoint junction, combining them into putative ancestral regions (stretches resembling the allele present in the reference genome and stretches resembling the alternative allele), and then comparing the regions with syntenic primate genome sequences to deduce the most likely ancestral state. We defined SV loci as ‘rectifiable’ if unambiguous high-quality alignments to putative ancestral regions could be constructed for the loci in any primate genomes.
Figure 3.


Ancestral state classification. (a) Junction sequences are aligned onto syntenic regions of a non-human primate genome to infer SV ancestral states. For rectifying an SV insertion event (from deletion) according to ancestral state (left), sequences A and B represent the junction sequences of the reference SV allele, where as sequence C represents the junction sequence of the non-reference SV allele. For rectifying an SV deletion event (from deletion) according to ancestral state(right), sequence C represents the junction sequence of the reference SV allele and sequences A and B represent the junction sequences of the non-reference SV allele. Solid lines with arrows indicate successful alignments and dashed lines with crosses indicate no proper alignment. (b) Results of classifying SVs as insertions or deletions according to ancestral state. An SV event is defined as ‘rectifiable’ (indicated by darker color) if unambiguous high-quality alignments to putative ancestral regions could be constructed for the loci in any primate genomes (regardless of whether the classification is changed according to the ancestral state), and as ‘unrectifiable’ (represented by lighter color) if not.
Overall, ancestral states of 1,281 (70%) out of 1,828 SV indel events could be assigned. For the vast majority of these (1,142), the chimpanzee genome contributed to the ancestral state assignment. For an an additional 139 cases located in hard-to-align regions in the chimpanzee genome (e.g., sequence assembly gaps), the ancestral state was inferred based on aligning junctions to the orangutan and macaque genomes. After ancestral state assignment, 665 SVs (36%) were classified as insertions and 1,163 (64%) as deletions. Furthermore 925 out of the 1,281 events were consistently rectifiable in at least two genomes. Of those, 420 were consistently rectifiable in all three genomes, with an approximate balance between insertions (212) and deletions (208) (Fig. 3b). We note that this balance differs substantially from earlier provisional SV classifications, which were strongly biased towards deletions, probably owing to the difficulty of many SV detection approaches in identifying insertions relative to the reference genome.
Inferring mechanisms of SV formation
Breakpoint junction sequences can also be used to deduce the molecular mechanisms of origin for SVs5, 6, 25. To systematically classify SVs in our library, we evaluated previously reported signatures of particular formation mechanisms (such as VNTR, TEI, NAHR and NHR) with a computational pipeline (Fig. 4a, Online Methods). TEIs can be identified by the underlying genomic signatures of transposable elements; VNTRs by underlying tandem repeats and low-complexity DNA; NAHRs by the extended stretches of high sequence identity at the breakpoint junctions; and NHRs by events lacking the former patterns. Parameters of the pipeline were chosen so as to yield results comparable to those achieved manually; in this regard, we confirmed the applicability of the chosen parameters by performing a sensitivity analysis (Online Methods and Supplementary Fig. 4).
Figure 4.






Inferring mechanisms of SV formation. (a) Pipeline for classifying SV-formation mechanisms. TE, transposable element. TSD, target site duplication. (b) Mechanisms of formation inferred for SVs in the library (larger circle on right). For NAHR (red) and MTEI/STEI (green), darker wedges represent high-confidence classification subsets, and lighter wedges are extended subsets. STEI is further subdivided in the left circle according to the fraction of previously reported L1insertions26, novel L1 insertions and processed pseudogene insertions in our dataset. (c) SV-indel distribution for all rectifiable events, broken down by formation mechanism. (d) Distribution of inter-vs. intra-chromosomal events for all consistently rectifiable insertions, broken down by formation mechanism. (e) Distances of putative ancestral loci to insertion sites for all consistently rectifiable intra-chromosomal insertions, showing that intra-chromosomal NAHR insertions usually involve nearby sequences, whereas TEIs and NHR-associated insertions usually involve distant sequences. (f) Genome-wide view of insertion trace. The outermost circle represents chromosomal ideograms; the second circle represents SV formational mechanisms of 1,554 events in a stacked histogram. The lines in the innermost circle indicate the origin of the insertion sequences in the human genome for all 321 consistently rectifiable insertions.
We found, consistent with earlier findings based on considerably smaller datasets5, 25, that NHR events constitute the most abundant mechanism of SV formation in the genome (Fig. 4b). Our analyses inferred NHR as the formation mechanism for nearly half of all SVs in our set (45%), whereas 28% involved NAHRs, 21% involved TEIs, 5% involved VNTRs, and 2% were ambiguous (the full list of events is available in Supplementary Table 1). Although VNTRs have the ability to contract and expand over kilobase-ranges, most of the 92 VNTRs identified in this study involved simple repeat units <1kb in size. We thus reasoned that they do not fall strictly into the stringent SV definition given above and excluded VNTRs from most of the remaining analyses below. Additionally, for NAHR and TEI mechanisms, we focused on the high-confidence sets in the analyses unless indicated otherwise (Online Methods).
We then analyzed SV formation mechanisms of 1,281 rectifiable SV-indel events. As discussed above, SVs were provisionally mostly reported as deletions owing to ascertainment biases5, 16, 21, regardless of the respective formation mechanisms. For example, despite the fact that retrotransposons are thought to move within the genome by a ‘copy-and-paste process’ involving reverse transcription of RNA intermediates and insertion of full-length or fragmented mobile elements26, most TEIs were previously annotated as deletions. Nevertheless, our ancestral state analysis revised the actual locus origin for a considerable number of SVs, and helped to resolve this apparent contradiction.
Our results show that nearly all transposable-element associated SVs for which ancestral states could be assigned were categorized as insertions (98%). Through manual inspection, we found that the remaining TE-associated deletions can be reasonably explained as NHR-mediated SV-deletions in regions of concentrated transposon annotations, which are difficult to be distinguished from retrotranspositions. This shows that using the class name TEI (“Transposable Element Insertion”) was justified in retrospect, and that our ancestral analysis pipeline is able to produce results consistent with prior knowledge on the formation mechanism of TEI. On the other hand, even after classification by ancestral states, NAHR and NHR events were mostly annotated as deletions (Fig. 4c), which may be due to biases of these formation mechanisms towards deletions (as previously reported for NAHR7) or due to biases in SV detection methods towards ascertaining deletions in ancestral loci.
Further analysis of TEI events showed that they involved LINEs, SINEs, LTR-elements, composite retrotransposons and processed pseudogenes. Our results show that LINE-1s (L1s) represent the most abundant class at the given size range (>1kb) as expected27, with 71% of the TEIs mediated by LINE/L1 transposable elements. Although many transposable elements in the human genome have lost their ability to retrotranspose autonomously, several full-length elements, including 147 L1s, are still implicated in recent or ongoing retrotransposition activity26. Interestingly, our results suggest the possible recent activity in the human population of at least 84 L1 elements, which were reported by our pipeline as ‘full-length’ with poly-A tracts and target site duplications. To the best of our knowledge, 38 of these putative active mobile elements have not yet been implicated with recent L1 activity (Fig. 4b; Supplementary Table 1; Supplementary Fig. 5). The remaining TEIs include three potential processed pseudogenes that were identified on the basis of their spliced primary transcripts, poly-A tracts and target site duplications (Fig. 4b and Supplementary Table 4).
We then focused on SVs associated with NAHR and NHR. Because these SVs mostly involve deletions relative to ancestral sequence, we reasoned that they might represent a particularly interesting class of SVs with potential impact on conserved DNA sequence. In fact, we found that 41% and 33% of the NAHR and NHR-based deletions, respectively, intersect with annotated exons from RefSeq genes (Online Methods) and thus may have a functional impact. On the other hand, insertions generated by NAHR or NHR have thus far received little attention, presumably due to difficulties in tracing these. Therefore, we extended our analysis to infer the most likely loci of origin of the inserted DNA sequences for 427 consistently rectifiable insertions (Online Methods). We found that NAHR-insertions usually involve nearby sequence stretches stemming from the same chromosome as would be expected from the NAHR duplication mechanism. On the contrary, TEIs were found to originate randomly from inter-chromosomal locations in the genome, probably due to the nature of retrotransposition of RNA intermediates. Furthermore, NHR-based insertions commonly involve both intra- and inter-chromosomal rearrangements (Fig. 4d–f).
Insights into SV formational biases
Finally, we analyzed the relationship between mechanisms of SV formation and sequence features located near to the breakpoints (including chromosomal landmarks, recombination hotspots, repeat sequences, GC content, short DNA motifs and microhomology regions). Briefly, we extracted the DNA sequences flanking both sides of each breakpoint junction. In the case of insertions, junction sequences included flanking DNA reconstructed from the inserted sequence. We also generated two random background sets, one by randomly picking sequences from the reference genome (global background), and the other by randomly picking DNA sequences from the local sequence context specific to each mechanistic class (local background). We then identified sequence features in the flanking regions of each breakpoint and calculated their enrichment with P-values based on randomization tests (Online Methods). We also tested for significant differences between SV formation mechanisms with respect to each feature using a Wilcoxon rank sum test (Fig. 5a and Supplementary Fig. 6).
Figure 5.



Analysis of breakpoint features. (a) Distance to chromosomal landmarks. Brackets indicate significantly different classes (P-value <0.05in Wilcoxon rank sum test after multiple hypothesis test correction by the Holm method). NAHR events are found to be significantly closer to telomeres and human-chimpanzee synteny block boundaries than the other mechanistic classes; VNTRs are significantly enriched in centromeric and pericentromeric regions. (b) DNA flexibility (dashed lines and left y-axis) and helix stability (solid lines and right y-axis) around NAHR and NHR breakpoints. (c) Distribution of NHR events with different lengths of microhomologies at the breakpoints. Microhomologies are significantly enriched in NHR breakpoints compared to a random background (KS test P-value=2.43E-11).
Chromosomal landmarks and structure
We first correlated SVs with chromosomal landmarks and found that NAHR events are significantly (P≤1E-05) more proximal to telomeres and human-chimp synteny block boundaries than the other mechanistic classes. Moreover, we observed that VNTRs are significantly (P≤1E-10) enriched in centromeric and pericentromeric regions, as expected (Fig. 5a). These results demonstrate a non-uniform distribution of SV formation mechanisms in the human genome (Fig. 4f).
Recombination hotspots
We also correlated SVs with recombination hotspots28 and observed that they are significantly enriched for NAHR events (1.5-fold enrichment; P-value=2.96E-03). Recombination hotspots are typically enriched for segmental duplications29, which may act as mediators for NAHR during meiotic recombination. We further observed biases towards recombination hotspots for TEIs (Supplementary Table 5), but not for NHR-mediated events. Whereas the accumulation of TEIs might in part be due to the formation of such elements by NAHR-mediated recombination involving interspersed repeat sequence, the lack of an enrichment for NHR indicates that DNA double-strand breaks occurring during recombination might be insufficient for initiating double-strand repair mediated by nonhomologous end-joining.
Repeat sequence
We next assessed associations between SV formation mechanisms and common repeat elements in the genome. For example, NAHR events have previously been reported to be associated with various types of genomic DNA repeats, in particular segmental duplications5, 6, 16. Following classification of NAHR events by our pipeline, we confirmed that significant (P~0) associations with segmental duplications are present both for NAHR-insertions (3.9-fold) and NAHR-deletions (7.4-fold). Furthermore, we found NAHR significantly (P~0) associated with the SINE/Alu class of mobile elements. On the other hand, LINE elements (both the L1 and L2 classes) were significantly (P≤1E-03) depleted among the NAHR events in our set whereas NHR events did not show significant enrichment (or depletion, except marginally for L2) with genomic repeat-structure (Supplementary Table 5).
GC-content and signatures of DNA fragility
We then analyzed various features related to the physical properties of DNA at SV breakpoint junctions. In contrast to NHR, NAHR events were found to be biased towards GC-rich regions (Supplementary Table 5). A possible explanation for this bias is the known GC-richness of recombination hotspots30, which we found to be significantly (P=2.96E-03) enriched for NAHR events. Further, our results may indicate SV formation biases owing to DNA duplex stability. We thus extended our analyses by two additional features: DNA helix stability predicted by calculating the average of the dissociation free energy of each overlapping dinucleotide31, and DNA flexibility based on the calculation of the average of the twist angle among each overlapping dinucleotide32. Our results indicate that in contrast to NAHR, NHR events are associated with high DNA flexibility and low helix stability, both of which are believed to be markers of fragility33. This is possibly due to sequence-specific biases for SV formation (Supplementary Table 5). We went on to characterize the change of these fragility marker signatures in a region of ±500bp around the breakpoint by smoothing the signal with a 50bp sliding window. Interestingly, we observed that the strength of the marker signatures was most extreme at or very close to the SV breakpoints (Fig. 5b).
DNA sequence motifs
We reasoned that our comprehensive breakpoint junction library may enable us to identify simple DNA sequence motifs associated with SV breakpoints. Thus, we used the MEME tool34 to carry out a comprehensive search for DNA motifs (6–12 nt, Online Methods) and found a significant enrichment (2.1-fold; P-value~0) of the dinucleotide repeat (TG)6 near breakpoints of NHR events, a sequence motif that fits with their relatively neutral GC content as shown above. We further analyzed all the NHR breakpoint sequences and found that the maximum consecutive occurrence of the TG-dinucleotide was 26. The MEME search did not reveal significantly enriched sequence motifs near NAHR or TEI events. Nevertheless, we used the MAST tool34 to search for the DNA sequence motif ‘CCNCCNTNNCCNC’ that recently was reported to be associated with chromosomal recombination hotspots35, and found a significant enrichment (1.5-fold; P-value~0) of the motif near NAHR-associated SVs, but not near NHR- or TEI-associated SVs.
Microhomology at breakpoints
Previous studies have observed the occurrence of stretches of short repeating sequences of 2 to ~10 bp (i.e., microhomologies) at the breakpoints of NHR events18, 36. We used our breakpoint junction library to scan NHR breakpoints for microhomology stretches of different lengths, and observed statistical enrichment relative to a random background (1.4-fold on average; KS test P-value=2.43E-11; Fig. 5c and Supplementary Table 6) as expected. This suggests a strong association of microhomology stretches with SV formation by NHEJ36 or fork-stalling and template switching18.
Discussion
In this study we presented a comprehensive library of 1,889 non-redundant SVs identified by breakpoint-resolution mapping in eight studies. Our approach, BreakSeq, leverages a breakpoint junction library for SV detection. While other computational approaches for SV detection (such as paired-end mapping (PEM)5, 37, DNA read depth analysis38–40 and split-read alignment analysis41) remain essential for identifying previously unknown SVs (a process that typically involves targeted PCR and sequencing), our approach serves as a reliable tool for rapidly identifying specific SV alleles in personal genomics data. Specifically, by mining personal genomes for sequences present in the breakpoint junction library, BreakSeq leverages alternative, non-reference genomic sequence data to rapidly detect previously described SVs that short-read based personal genomics surveys commonly fail to ascertain. As such, BreakSeq enables a step towards overcoming reference biases, which is the favoring in ascertainment of SV alleles present in the human reference genome sequence.
We foresee that BreakSeq will further gain in utility as datasets grow (e.g., when SV calls from the 1000 Genomes Project are published). As our approach has a linear time complexity (Online Methods), it is easily extendable to larger datasets. In this regard, the size of our junction library currently comprises 0.004% of the reference genome in terms of nucleotide bases, and even a 100-fold increase of its size (>0.2 million SVs; ~10 times of DGV) will result in a dataset considerably smaller than the reference genome. Thus, applying BreakSeq in personal genomics studies adds negligible computing efforts (compared to SNP genotyping) and at the same time dramatically improves SV calling. The library will be updated regularly to serve the personal genomics community in enabling precise SV detection with various next-generation sequencing platforms.
In addition to enabling accurate SV mapping, our junction library allows characterizing SV ancestral states. While the ancestral states of SNPs and small indels have been inferred according to ancestral alignments in earlier studies42, 43, we here report systematic ancestral state inference for SVs. When applying our new classification approach to 1,281 SVs, we found that overall there is a balance of insertions and deletions, unlike most currently published SV sets that display a considerable bias towards deletions. It should be noted that the non-human primate genomes used in our ancestral state inference correspond to single animals, which certainly do not represent idealized ancestral genomes. Nonetheless, here we reasonably assume that SV loci can be classified at high confidence when ancestral states can be consistently inferred across three distinct primates.
Furthermore, we have developed a computational pipeline for classifying SVs according to their formation mechanisms, and for analyzing various DNA sequence characteristics of the affected genomic loci. Together with the ancestral state analysis, this allowed us to analyze SV formation processes with respect to likely ancestral loci, an analysis that revealed some insights into SV formation. For example, our analyses suggest that the physical properties of the underlying DNA sequence influence locus-specific propensities for different SV formation mechanisms. We observed that NAHR-based SVs are associated with a relatively high GC content and with recombination hotspots, indicating that double-strand breaks (DSBs) occurring specifically during meiotic recombination contribute to NAHR-associated SV formation. On the other hand, NHR breakpoint regions appear to have lower DNA stability and higher flexibility, features that may increase the chance of DSBs in general. Overall, our analysis reveals formational biases underlying SV formation and conforms to the fact that NAHR is driven by recombination between repeat sequences, whereas NHR is likely driven by DNA repair and replication errors.
By applying BreakSeq on a large scale, we envisage that it could be used for genotyping and determining SV allele frequencies. In fact, it should be possible to put each of the breakpoint sequences in our library directly onto a commercially available ‘SNP chip’, which could be used to precisely assess SV genotypes simultaneously with all of the SNPs in an individual. (This should add only a small number of probes to the approximately 1M probes already on the commercial chips.)
Lastly, we note that as our approach depends on the current SV lists, it is inevitably affected by their existing biases owing to presently applied technologies. Likely biases include the difficulty in mapping insertions relative to the reference genome and in ascertaining SVs in repetitive regions, e.g. segmentally duplicated sequences. We anticipate that in the near future, as technologies advance in terms of read lengths, inherent biases against repeat-rich sequences will be further reduced and the mapping of SVs onto our junction library will further improve, making it essentially comparable to SNP-genotyping. In this regard, as thousands of human genomes will be sequenced in the coming years, there will be a huge demand for a reliable and accurate SV-mapping and SV-genotyping.
Online Methods
Data preparation
Our initial breakpoint library altogether represented 1,961 structural variants (SVs) identified at high precision based on NCBI build 36 of the human genome. It was compiled from 8 different published sources based on paired-end mapping5, 16, fosmid-paired-end sequencing3, 6, Sanger capillary sequencing44, resequencing of an individual human genome using second generation sequencing21, DNA resequencing traces for SNP discovery projects (support by at least two reads was required for an SV to be included in our dataset)45, and high-resolution array-based comparative genomic hybridization (aCGH)25. For the 253 SVs identified through fosmid-paired-end sequencing3, 6, 387 published sequenced clones originally used to identify SVs in NCBI build 35 were realigned to the NCBI build 36 human genome before inclusion in the library. A split-read analysis was then carried out using Blat to infer the breakpoints of the events. For the 98 SVs from resequencing traces45, the liftover tool available at the UCSC Genome Browser (http://genome.ucsc.edu/) was used to convert the breakpoint coordinates from human NCBI build 35 to build 36. All SVs in our analysis were between 1kb and 1Mb in length (i.e., we removed events >1Mb, reasoning that they may be lower in confidence). After accounting for redundancy, our standardized breakpoint library consisted of 1,889 SVs that were used in all subsequent calculations and analyses.
SV mechanism classification pipeline
Four major steps were involved in our procedure to classify SV formation mechanisms. First, SVs were examined for extensive coverage by tandem repeats and regions of low complexity (here, low-complexity DNA refers to micro-satellite DNA, poly-purine/poly-pyrimidine stretches, and regions of extremely high AT or GC content, as defined by the RepeatMasker program; www.repeatmasker.org) to identify instances of expansion or contraction of VNTRs. Second, ±100bp flanking sequences derived from both breakpoint junctions were aligned against each other to scan for blocks of extensive homology. SVs were classified as “high-confidence NAHR” if the homologous blocks had a minimum sequence identity of 85%, a minimum length of 50bp for the identical sequences, a maximum offset of 20bp between the homologous blocks, correct orientations, and covered the breakpoints. SVs displaying at least three but not all of the above criteria were classified as “extended NAHR”. Third, SVs aligning to known interspersed mobile elements carrying the common diagnostic features of corresponding transposable elements, i.e., target site duplications and poly-A tracts26, were classified as “high-confidence TEIs”. Events missing one or more of the diagnostic features were classified as “extended TEIs”. TEIs were furthered categorized into “Single Transposable Element Insertions” (STEIs) if a single element was involved and “Multiple Transposable Element Insertions” (MTEIs) if multiple elements appeared to be involved. Furthermore, full-length TEIs were discriminated from transposable element fragments and transposable element subfamilies were also recorded. Through identification of spliced protein-coding gene sequences and TEI-diagnostic features, processed pseudogenes likely inserted via a TEI-associated mechanism were identified. Finally, SVs lacking signatures of any of the above diagnostic sequence features were classified as NHR events.
Sensitivity analysis for the SV mechanism classification
Sensitivity analysis was performed on five key parameters used in the mechanism classification pipeline (Supplementary Fig. 4). Classification results were examined as each parameter was varied over a large range while fixing the other parameters at default values. First, the cutoff for the length of homologous blocks in the flanking sequences alignment for classifying NAHR events (NAHRhomolen) was varied from 10 to 150bp with a step size of 10bp. Second, the cutoff for the percentage identity of homologous blocks in the flanking sequences alignment for classifying NAHR events (NAHRpct) was varied from 70 to 100% with a step size of 1%. Third, the cutoff for the coverage of VNTR regions in the SV was varied from 0 to 100% with a step size of 5%. Fourth, the window size used to examine the consistency of the transposable element boundary with a breakpoint for classifying STEI and MTEI events (TEIwin) was varied from 10 to 400bp with a step size of 10bp. Finally, the gap size used to examine whether adjacent transposable elements can be joined for classifying MTEI events (TEIgap) was varied from 0 to 300bp with a step size of 10bp. Default values for NAHRhomolen, NAHRpct, VNTRcutoff, TEIwin and TEIgap used in the pipeline were 50, 85, 50, 200, and 150 respectively.
Analysis of ancestral state
For a “deletion” relative to the reference genome, a ±500bp flanking sequence at each breakpoint was extracted to obtain two sequences of 1,000bp representing both the left (“A”) and right (“B”) breakpoint junction sequences. Then a 1,000bp junction sequence at the breakpoint of the alternative allele, representing 500bp upstream and downstream of the left and right breakpoints, respectively (“C”), was also extracted. If C aligned onto a non-human primate genome (i.e., a potential ancestral genomic locus) at high-quality and with better length and sequence identity (represented by the Blat score) than A and B, then the event was rectified as an insertion relative to the ancestral genome. Conversely, for an “insertion” relative to the reference genome, the A, B (alternative allele) and C (reference allele) junction sequences of the event were extracted. If A and B both displayed an alignment better than C onto a non-human primate genome, the event was rectified as a deletion relative to the ancestral genome.
All the alignments were performed using Blat on the Chimpanzee (panTro2), Macaque (rheMac2), and Orangutan (ponAbe2) genomes, the sequences of which were downloaded from the UCSC genome browser (http://genome.ucsc.edu/). The Net alignments46, 47 from UCSC were also downloaded and the top level was chosen to verify that the alignment of the junction sequences were in the syntenic regions of the corresponding SVs. Since all the primate ancestral genomes are highly similar, the alignment identity and coverage were required to be >90%. Furthermore, the length ratio of target vs. query was required not to exceed a deviation of 10%.
SVs were classified as “rectifiable” if unambiguous high-quality alignments to putative ancestral regions could be constructed in any non-human primate genome. Particularly, an SV was classified as “rectified” if its state was changed from its original to another after the analysis (from deletion to insertion, or vice versa). The state of each SV was then assigned based on the closest non-human primate genome (e.g. from chimpanzee to orangutan and to macaque) in which a corresponding syntenic region existed. SVs were considered as “consistently rectifiable” if they were rectifiable for the same state with no inconsistent ancestral assignment inferred.
Insertion trace
After rectification based on the ancestral state analysis, all insertions that were consistently rectifiable were aligned onto to the human reference genome to scan for the presumable origin of the inserted sequences. Since the inserted sequence of an event rectified from a deletion is already present in the reference genome, any alignments overlapping with >50% of the SV region were discarded and the next best match was chosen. Blat alignments tracing inserted sequences were required to have a sequence identity >90%.
Enrichment calculation
To calculate the enrichment and p-value for each feature and repeat association with breakpoints, a non-parametric randomization test based on sampling was employed. For the observed samples, the exact coordinates of the breakpoints were taken for location-dependent computation and sequences flanking the breakpoints were extracted for sequence-dependent computation. A random global background was generated by randomly sampling a set of coordinates, or sequences with the same length, of the same amount from the reference genome (build 36). Similarly, a local background was generated by randomly sampling in a 10kb window at the breakpoints. The sampling was repeated 1,000 times with replacement and the observed statistic of the breakpoints was tested against the sampling distribution based on the whole genome. The enrichment value was calculated by comparing the observed statistic over the mean of the statistics of the samplings. Then, the p-value of the enrichment was calculated by counting the number of samplings that yielded a statistic as extreme as, or more extreme than, the observed one. The enrichment was reported as significant for any P-value < 0.05.
Correlation of chromosomal landmarks
Distance to telomeres was calculated from the midpoint of an SV to the end of the chromosome in the same arm. Distances to centromeres and pericentromeric gaps were calculated from the midpoint of an SV to the closest centromeric or pericentromeric gap boundary on the same chromosome. Distance to the closest synteny block boundary was calculated by computing the distance from each breakpoint to the closest synteny block boundary and then taking the average for the two breakpoints. Synteny block boundaries were taken from the human-chimpanzee Net alignment file46, 47 available at the UCSC genome browser (http://genome.ucsc.edu/) and the ‘gap’ type was excluded from the analysis. A Wilcoxon rank sum test was then performed to compare the distance measurements of different formation mechanisms in a pair-wise fashion, followed by a correction for multiple hypothesis testing using the Holm method.
Feature computation
We considered the following features at SV breakpoints in our analysis: GC content, helix stability, and DNA flexibility. All features were computed for sequences 50bp around the breakpoints or randomly extracted from the genome. GC content was calculated by computing the percentage of Guanine and Cytosine nucleotides over the given length of the sequence. Helix stability of the DNA duplex was predicted by calculating the average of the dissociation free energy of each overlapping dinucleotide31. Similarly, DNA flexibility was estimated by calculating the average of the twist angle among all overlapping dinucleotides32. To observe the change of the DNA flexibility and helix stability around a breakpoint, values at each nucleotide were smoothed using a sliding window of 50bp, which was slid across an interval of 1kb centered on the breakpoint.
Repeat association
The association of repeat elements and pseudogenes was calculated by intersecting the relevant datasets. Each element was overlapped with a breakpoint and the average number of overlapping elements for all the input breakpoints was calculated. Repeat elements in the human genome build 36 were downloaded from the RepeatMasker track of the UCSC Genome Browser (March 2006 assembly). Only the elements annotated with repeat classes SINE and LINE were included in this analysis. In total, there were 1,783,897 SINE elements and 1,407,547 LINE elements of which 1,193,509 were Alu elements and 927,909 were L1 elements respectively. For the pseudogene analysis, we used PseudoPipe48 to identify pseudogenes in the genome based on the protein annotations in the Ensembl database (release 48). This analysis involved 2,454 duplicated pseudogenes and 10,999 processed pseudogenes.
Motif discovery
MEME was used to discover sequence motifs near SV breakpoints and to generate position weight matrices (PWMs) for significantly enriched motifs. The input data to MEME were sequences of 200bp centered on the breakpoints. Motif width was allowed to range from 6bp to 12bp. For SVs classified as NAHR-mediated we also looked for an over-representation of a previously described sequence motif specific to recombination-hotspots35. The recombination-hotspot motif was converted into a PWM by considering the average genomic frequencies of the four bases ACGT (0.295, 0.205, 0.205, 0.295) and by adding pseudocounts of 1. After identifying the motifs, MAST was applied to search for a motif match in the original set and the global background set. The p-value cutoff for each motif match was P<0.0001 and a randomization test was performed as described above to calculate the enrichment P-values for each motif.
Microhomology enrichment analysis
The lengths of the microhomology sequences at the breakpoints of NHR-mediated events were compared with the local background and a theoretical distribution. The theoretical expectation was calculated by assuming independence between genomic positions and a uniform distribution of the four nucleotides (ATCG) in the genome. The formula P×(1−P)2×(i+1) was used to calculate the probability of observing homology of a specific length, where i is the length of homology and P is the probability of observing the same pair of nucleotides at the given genomic positions (i.e. P = p(A)2+p(T)2+p(C)2+p(G)2 and p(A,C,G,T) =(0.295, 0.205, 0.205, 0.295) were estimated from the local background). A one-sided Kolmogorov-Smirnov test (KS-test) was performed to test the enrichment of microhomologies in NHR compared to the local background. The size of the effect was calculated as the fold enrichment of microhomology stretches between NHR and the background.
Mapping SVs with a Junction Library
The breakpoint junction mapping approach that we developed works as follows. The junction library for SV mapping is created by joining 30bp flanking sequences on each side of a breakpoint. A deletion event is represented with a single junction sequence in the library, while an insertion has both a left and right junction sequence corresponding to each of its breakpoints. DNA reads from personal genomes are aligned against the junction library. Reads are required to overlap a breakpoint by at least 10bp on each side. All successfully mapped reads are then aligned against the reference genome. Only those reads that do not map onto the reference genome are labeled as “unique” in the personal genome; the other reads are labeled as “non-unique”. A short-read aligner, Bowtie49, is used to perform all the alignments (allowing for two mismatches). To score the SV candidates on the basis of supportive read-matches (hits), the following formula is used:
where Si is the score representing the effective number of hits (supportive hits) in log2 scale for SV i, with unique and non-unique hits denoted as Ti and Ri respectively. If Ti or Ri is 0, the log term is replaced by 0. A score of 1 thus indicates 2 supportive hits, whereas scores > 2 (high-support) indicate the presence of >4 supportive hits.
The mapping process showed a linear time complexity in practice. On average, it required 8 hours to run our junction-mapping program (open-sourced and available for download at http://sv.gersteinlab.org/breakseq) against a sequenced genome at 40X physical coverage on a 3GHz quad-core computer node with 16GB physical memory. All identified SVs for the YRI and HCH genomes are listed in Supplementary Table 2; for NA12891, in accordance with pre-publication agreements for 1000 Genomes Project data, we only provide the coordinates of SVs identified on a single chromosome (i.e., chromosome 6).
Intersection of the breakpoint junction library with RefSeq genes
RefSeq gene annotations were downloaded from the UCSC Genome Browser. Intersection of the SVs in our breakpoint junction library and RefSeq genes were found by comparing the start- and end-coordinates of the two datasets. For insertion events whose inserted sequences could be traced, the positions from which the insertions were derived were compared to the RefSeq gene annotations. In particular, 60 out of 146 NAHR deletions and 193 out of 580 NHR deletions intersected with annotated exons from RefSeq genes. Insertions were also found to have an impact on coding regions, with 19 out of 51 NAHR insertions and 11 out of 30 NHR insertions intersecting with the exons. These included cases where exons at the insertion site were altered by the insertion event (19 NAHRs and 7 NHRs) and where the inserted sequence was itself derived from exonic DNA (3 NAHRs and 6 NHRs).
PCR Validation
We tested by PCR validation 24 insertion and 33 deletion calls predicted in NA12891 relative to the reference genome (Supplementary Table 3). Specifically, we designed PCR primers as previously described5 and amplified the predicted non-reference SV alleles. For the PCR, 10ng of genomic DNA (Coriell Institute, Camden, NJ, USA) were used with the SequalPrep Long PCR Kit (Invitrogen, Karlsruhe, Germany) in 20ul volumes using the following PCR conditions in a C1000 thermocycler (Biorad, Munich, Germany): 94°C for 3 minutes, followed by 10 cycles of 94°C for 10 seconds, 60°C for 30 seconds and 68°C for 10 minutes and 25 cycles of 94°C for 10 seconds, 56°C for 30 seconds and 68°C for 10 minutes (+10 sec/cycle), followed by a final cycle of 72°C for 10 minutes. Some of the reactions that failed with the SequalPrep enzyme were amplified with the LongAmp Taq DNA Polymerase (NEB, Frankfurt am Main, Germany) or the iProof High Fidelity DNA Polymerase (Biorad). PCR products were analyzed on a 1% agarose gel stained with Sybr Safe Dye (Invitrogen). Marker M1 was a 100bp ladder whereas M2 corresponded to a 1kb ladder (500, 1000, 1500, 2000, 3000, etc) (NEB). Primers and polymerases are listed in Supplementary Table 3.
Supplementary Material
Acknowledgments
We acknowledge support from the NIH, the AL Williams Professorship funds, and the European Molecular Biology Laboratory. We thank Roger Alexander and Ekta Khurana for proofreading the manuscript, and Alexej Abyzov, Zhengdong Zhang, Tobias Rausch, and Jiang Du for helpful discussions. Finally, we thank the 1000 Genomes Project for early data access.
References
- 1.Sebat J, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 2.Iafrate AJ, et al. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 3.Tuzun E, et al. Fine-scale structural variation of the human genome. Nat Genet. 2005;37:727–732. doi: 10.1038/ng1562. [DOI] [PubMed] [Google Scholar]
- 4.Redon R, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Korbel JO, et al. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kidd JM, et al. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turner DJ, et al. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat Genet. 2008;40:90–95. doi: 10.1038/ng.2007.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Ommen GJ. Frequency of new copy number variation in humans. Nat Genet. 2005;37:333–334. doi: 10.1038/ng0405-333. [DOI] [PubMed] [Google Scholar]
- 9.Korbel JO, et al. The genetic architecture of Down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proc Natl Acad Sci U S A. 2009;106:12031–12036. doi: 10.1073/pnas.0813248106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharp AJ, et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet. 2006;38:1038–1042. doi: 10.1038/ng1862. [DOI] [PubMed] [Google Scholar]
- 11.McCarroll SA, et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nat Genet. 2008;40:1107–1112. doi: 10.1038/ng.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Cid R, et al. Deletion of the late cornified envelope LCE3B and LCE3C genes as a susceptibility factor for psoriasis. Nat Genet. 2009;41:211–215. doi: 10.1038/ng.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gonzalez E, et al. The Influence of CCL3L1 Gene-Containing Segmental Duplications on HIV-1/AIDS Susceptibility. Science. 2005;307:1434–1440. doi: 10.1126/science.1101160. [DOI] [PubMed] [Google Scholar]
- 14.Aitman TJ, et al. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439:851–855. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- 15.Hastings PJ, Lupski JR, Rosenberg SM, Ira G. Mechanisms of change in gene copy number. Nat Rev Genet. 2009;10:551–564. doi: 10.1038/nrg2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim PM, et al. Analysis of copy number variants and segmental duplications in the human genome: Evidence for a change in the process of formation in recent evolutionary history. Genome Res. 2008;18:1865–1874. doi: 10.1101/gr.081422.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 18.Lee JA, Carvalho CM, Lupski JR. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 19.Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. 2006;7:85–97. doi: 10.1038/nrg1767. [DOI] [PubMed] [Google Scholar]
- 20.Korbel JO, et al. PEMer: a computational framework with simulation-based error models for inferring genomic structural variants from massive paired-end sequencing data. Genome Biol. 2009;10:R23. doi: 10.1186/gb-2009-10-2-r23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wheeler DA, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- 22.Bentley DR, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang J, et al. The diploid genome sequence of an Asian individual. Nature. 2008;456:60–65. doi: 10.1038/nature07484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perry GH, et al. The fine-scale and complex architecture of human copy-number variation. Am J Hum Genet. 2008;82:685–695. doi: 10.1016/j.ajhg.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mills RE, Bennett EA, Iskow RC, Devine SE. Which transposable elements are active in the human genome? Trends Genet. 2007;23:183–191. doi: 10.1016/j.tig.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 27.Xing J, et al. Mobile elements create structural variation: analysis of a complete human genome. Genome Res. 2009;19:1516–1526. doi: 10.1101/gr.091827.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Myers S, Bottolo L, Freeman C, McVean G, Donnelly P. A fine-scale map of recombination rates and hotspots across the human genome. Science. 2005;310:321–324. doi: 10.1126/science.1117196. [DOI] [PubMed] [Google Scholar]
- 29.Sharp AJ, et al. Segmental Duplications and Copy-Number Variation in the Human Genome. The American Journal of Human Genetics. 2005;77:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meunier J, Duret L. Recombination drives the evolution of GC-content in the human genome. Mol Biol Evol. 2004;21:984–990. doi: 10.1093/molbev/msh070. [DOI] [PubMed] [Google Scholar]
- 31.Breslauer KJ, Frank R, Blocker H, Marky LA. Predicting DNA duplex stability from the base sequence. Proc Natl Acad Sci U S A. 1986;83:3746–3750. doi: 10.1073/pnas.83.11.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sarai A, Mazur J, Nussinov R, Jernigan RL. Sequence dependence of DNA conformational flexibility. Biochemistry. 1989;28:7842–7849. doi: 10.1021/bi00445a046. [DOI] [PubMed] [Google Scholar]
- 33.Bailey JA, Eichler EE. Primate segmental duplications: crucibles of evolution, diversity and disease. Nat Rev Genet. 2006;7:552–564. doi: 10.1038/nrg1895. [DOI] [PubMed] [Google Scholar]
- 34.Bailey TL, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Myers S, Freeman C, Auton A, Donnelly P, McVean G. A common sequence motif associated with recombination hot spots and genome instability in humans. Nat Genet. 2008;40:1124–1129. doi: 10.1038/ng.213. [DOI] [PubMed] [Google Scholar]
- 36.Linardopoulou EV, et al. Human subtelomeres are hot spots of interchromosomal recombination and segmental duplication. Nature. 2005;437:94–100. doi: 10.1038/nature04029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee S, Cheran E, Brudno M. A robust framework for detecting structural variations in a genome. Bioinformatics. 2008;24:i59–67. doi: 10.1093/bioinformatics/btn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Campbell PJ, et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat Genet. 2008;40:722–729. doi: 10.1038/ng.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chiang DY, et al. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat Methods. 2009;6:99–103. doi: 10.1038/nmeth.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang LY, Abyzov A, Korbel JO, Snyder M, Gerstein M. MSB: a mean-shift-based approach for the analysis of structural variation in the genome. Genome Res. 2009;19:106–117. doi: 10.1101/gr.080069.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009 doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paten B, et al. Genome-wide nucleotide-level mammalian ancestor reconstruction. Genome Res. 2008;18:1829–1843. doi: 10.1101/gr.076521.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spencer CC, et al. The influence of recombination on human genetic diversity. PLoS Genet. 2006;2:e148. doi: 10.1371/journal.pgen.0020148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Levy S, et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mills RE, et al. An initial map of insertion and deletion (INDEL) variation in the human genome. Genome Res. 2006;16:1182–1190. doi: 10.1101/gr.4565806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kent WJ, Baertsch R, Hinrichs A, Miller W, Haussler D. Evolution’s cauldron: Duplication, deletion, and rearrangement in the mouse and human genomes. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:11484–11489. doi: 10.1073/pnas.1932072100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schwartz S, et al. Human-mouse alignments with BLASTZ. Genome Res. 2003;13:103–107. doi: 10.1101/gr.809403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Z, et al. PseudoPipe: an automated pseudogene identification pipeline. Bioinformatics. 2006;22:1437–1439. doi: 10.1093/bioinformatics/btl116. [DOI] [PubMed] [Google Scholar]
- 49.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.