

Abstract
While mammals cannot survive oxygen deprivation for more than a few minutes without sustaining severe organ damage, some animals have mastered anaerobic life. Freshwater turtles belonging to the Trachemys and Chrysemys genera are the champion facultative anaerobes of the vertebrate world, often surviving without oxygen for many weeks at a time. The physiological and biochemical mechanisms that underlie anoxia tolerance in turtles include profound metabolic rate depression, post-translational modification of proteins, strong antioxidant defenses, activation of specific stress-responsive transcription factors, and enhanced expression of cyto-protective proteins. Turtles are also known for their incredible longevity and display characteristics of “negligible senescence.” We propose that the robust stress-tolerance mechanisms that permit long term anaerobiosis by turtles may also support the longevity of these animals. Many of the mechanisms involved in natural anoxia tolerance, such as hypometabolism or the induction of various protective proteins/pathways, have been shown to play important roles in mammalian oxygen-related diseases and improved understanding of how cells survive without oxygen could aid in the understanding and treatment of various pathological conditions that involve hypoxia or oxidative stress. In the present review we discuss the recent advances made in understanding the molecular nature of anoxia tolerance in turtles and the potential links between this tolerance and longevity.
Key words: Trachemys scripta elegans, longevity, anoxia tolerance, reoxygenation, reactive oxygen species, oxidative damage, antioxidant defenses, heat shock proteins, NFκB, anti-apoptotic proteins
Introduction
Some two billion years ago primitive life forms learned how to use oxygen as the final acceptor in their electron transport pathways. The high redox potential of oxygen allows it to easily accept electrons from reduced substrates, making it extremely useful for energy production. The ability to extract greater amounts of energy from organic molecules by using oxygen-linked catabolism has driven life to evolve into high complexities and has made oxygen vital to many life forms on Earth. Mammals are particularly sensitive to oxygen deprivation and situations of hypoxia or anoxia can lead rapidly to severe tissue damage or even death. In contrast, some ectothermic vertebrates are particularly well-adapted to surviving oxygen limitation. Various species of turtles have very well-developed capacities for living without oxygen (called anaerobiosis).1,2 For example, freshwater turtles living in northern regions of the US and Canada typically hibernate under water to elude freezing temperatures on land. However, bodies of water often become ice-locked, limiting the ability for animals to surface for lung breathing. Some turtle species compensate for this with a good capacity for extrapulmonary gas exchange across other epithelia whereas others have perfected strategies that allow survival without breathing oxygen for three months or more.3 Those species that tolerate extreme hypoxia or anoxia employ various biochemical and physiological mechanisms for survival, the major ones including prehibernation accumulation of huge glycogen stores to fuel anaerobic glycolysis, the use of the shell and skeletal systems for buffering lactate accumulation and minimizing acidosis, and the capacity for profound metabolic rate depression to only 10–20% of the corresponding aerobic rate. Metabolic depression allows tissues to minimize their energy needs, and therefore survive for extended periods of time using the ATP generated by anaerobic glycolysis alone. In addition to the challenges presented to tissues by severe oxygen limitation, turtles must also deal with the potential for oxidative stress occurring with the re-introduction of oxygen. The mitochondrial electron transport chain becomes reduced during hypoxia/anoxia because there is little or no oxygen to accept electrons. This makes it particularly susceptible to generating reactive oxygen species (ROS) upon reoxygenation when the oxygen tension is high. This can be extremely damaging to cells and indeed, in situations of ischemia-reperfusion, mammalian tissues show profound damage caused by ROS during the recovery phase.4,5 Situations of environmental oxygen variability, such as those experienced by diving or hibernating turtles, could present many opportunities for oxidative stress and oxidative damage, and yet, turtles emerge unharmed from repeated cycles of anoxia and reoxygenation.
Turtles are also known for their extraordinary longevity. Lifespans of over a hundred and fifty years have been documented for some species of the Chelonia, and more importantly, these animals do not appear to display the senescent phenotypes characteristic of many other species. Humans have long sought a cure for aging, and turtles present a unique model of “negligible senescence.”6,7 Criteria for “negligible senescence” include: (1) mortality rate should not increase with age and (2) reproduction should not decrease with age.7 Studies on painted turtles (Chrysemys picta marginata) showed that survivorship of old turtles does not differ from that of their younger counterparts8 and no decline in the reproductive output is apparent with age.8 Turtles, therefore, appear to meet the criteria necessary for “negligible senescence”. Other research has shown that telomere length, which generally decreases with age, does not appear to change between embryos and adults of European freshwater turtles (Emys orbicularis).9 These characteristics could make turtles a valuable model for studies that attempt to understand how cells can avoid the wear and tear of daily life.
A potential link between anoxia tolerance in turtles and their extraordinary longevity has been previously suggested.10 The mechanisms that turtles employ to survive anoxia might be both directly and indirectly linked to longevity. For example, in order to deal with a variable oxygen environment, turtles must have extraordinary stress-resistance mechanisms. Resistance to stress is generally correlated with longevity11 and organisms that are tolerant of one kind of stress are typically more likely to be tolerant to others, since many cytoprotective mechanisms operate over a wide variety of stresses.11 Whereas aging is often characterized by decreased stress-resistance and subsequent cellular degeneration, an animal that has robust stress-resistance mechanisms is more likely to deal better with aging. In addition, an understanding of the molecular nature of anoxia tolerance in turtles can lead to improved understanding of many age-related pathological conditions that involve oxygen restriction/deprivation (e.g., stroke or myocardial ischemia) or oxidative stress (e.g., Parkinson, Alzheimer disease). Finally, such studies might suggest ways of making cells and tissues of oxygen-sensitive animals, such as humans, more tolerant of oxygen limitation. This could lead to improved techniques for handling ischemic insults to organs, to aid in preserving organ viability during surgery, and even to enhanced technology for use in hypothermic- or cryopreservation of tissues and organs.
In the present review we discuss recent advances in the studies of the molecular nature of anoxia tolerance in turtles, with emphasis on the potential links between these mechanisms and the negligible senescence shown by these animals.
Oxidative Stress and Aging
Whereas oxygen-based metabolism has allowed life to evolve to ever new complexities, this did not come without a price. The catch is oxidative stress, the damage done by ROS generated from single electron reductions of oxygen.12 When ROS generation exceeds the capacity of antioxidant defenses, oxidative stress ensues and has been implicated in cellular degradation during aging as well as in a variety of disease states.
Formation of most ROS occurs at the site where the vast majority of intracellular oxygen is consumed, the mitochondria.13 It is here that reduced substrates donate their electrons to form an electrochemical gradient, resulting ultimately in the production of ATP, the energy currency of the cell. This occurs through a series of oxidative reactions conducted by protein complexes on the inner membrane of the mitochondria. Leakage of electrons from these complexes results in the one-electron reduction of oxygen to form superoxide, the precursor to most ROS. Dismutation of superoxide results in the production of hydrogen peroxide, whereas the subsequent interaction between superoxide and hydrogen peroxide in the Haber-Weiss reaction or cleavage of hydrogen peroxide in the Fenton reaction results in the formation of hydroxyl radicals.14 It is estimated that approximately 1–2% of the oxygen consumed during respiration is reduced to superoxide.15
The chemistry of superoxide differs dramatically based on its environment. In aqueous solutions, superoxide is a weak oxidizing agent and a much stronger reducing agent.16 Superoxide rapidly disappears in aqueous solutions due to its dismutation reaction, in which hydrogen peroxide and oxygen are formed:
Hydrogen peroxide is a weak oxidant and a weak reducing agent and is relatively stable in the absence of transition metals.16 However, whereas both superoxide and hydrogen peroxide have low oxidative toxicity themselves, toxicity themselves, they are both components of the net Haber-Weiss reaction that readily produces hydroxyl radicals, catalyzed by copper or iron ions:
The hydroxyl radical is highly reactive and is responsible for most of the damage incurred to biological macromolecules by ROS. The main source of biological hydroxyl radicals is the metal-catalyzed breakdown of hydrogen peroxide by the Fenton reaction, one-half of the net Haber-Weiss reaction:
Because of their short half life (less than 1 ns), hydroxyl radicals attack molecules very close to their site of formation17 and their very high reactivity makes them virtually impossible to scavenge.18 The reaction of hydroxyl radicals with biomacromolecules produces new radicals, usually with lower reactivity. Once unleashed upon the intracellular milieu, ROS react with many cellular macromolecules, including lipids, proteins and DNA, inflicting damaging effects on multiple subcellular structures.
ROS damage to macromolecules.
Lipid peroxidation is major source of damage causing cellular injury and death (reviewed in refs. 19 and 20). It is caused by the interaction of ROS (chiefly hydroxyl radical) with unsaturated bonds in membrane lipids. Polyunsaturated fatty acids in membrane phospholipids are particularly susceptible to peroxidation, which can result in the rearrangement or loss of double bonds or degradation of lipid side chains.21 This leads to disturbance of membrane structure, causing changes in membrane fluidity,22 permeability,23 alternations in ion transport24 and inhibition of metabolic processes.25 Lipid peroxidation in the mitochondria can suppress mitochondrial metabolism, as well as affect vital mitochondrial functions such as respiration, inner membrane barrier properties, maintenance of mitochondrial membrane potential and mitochondrial Ca2+ buffering capacity.26,27
Almost all kinds of amino acid residues in proteins are potential targets for oxidation by ROS (reviewed in ref. 28). Oxidation of some amino acid residues (lysine, arginine, proline) can also lead to the formation of carbonyl derivatives, that are considered to be the most relevant products of free radical attack on proteins.29 The presence of carbonyl proteins in cell and tissue samples has become widely accepted as a marker for oxidative stress. Copper or iron binding sites on proteins are other important targets of ROS, since these sites can serve as centers for additional production of hydroxyl radicals via the Fenton reaction.30 These radicals can then attack side chains of the amino acid residues at the metal-binding site.
ROS appear to produce several forms of DNA damage, including modification of all bases, production of base-free sites, deletions, frame shifts, strand breaks, DNA-protein cross-links and chromosomal rearrangements (reviewed in ref. 17). Modifications of DNA can be mutagenic and might contribute to cancer, neurodegenerative diseases and aging.31 The hydroxyl radical is known to react with all components of the DNA mole-cule—the purine and pyrimidine bases as well as the deoxyribose backbone.32,33 In addition to base and sugar modifications, covalent DNA-protein cross-links can also form by exposure to free radicals.34 The mechanism for the formation of these cross-links involves the addition of a DNA base radical to an aromatic amino acid of a protein or the combination of a DNA base radical with an amino acid radical.35
Free radical damage and aging.
Aging is characterized by an increase in many detrimental changes at different levels of biological organization. The idea that ROS are among the main perpetrators of cellular damage has gained much support over the past few decades (reviewed in ref. 36 and 37). Lipid damage has been shown to increase with age,38 one result of this being a buildup of lipofuscin, a pigment that accumulates with age and correlates with animal senescence. Tissue damage in the form of oxidized and cross-linked proteins also increases as a function of age.28,39 For example, 30–50% of the total cellular proteins in an old animal might be oxidized, possibly accounting for the observation that the catalytic activity of many enzymes decreases with age.40 Evidence also exists that the rate of degradation of oxidized proteins decreases with age.39 Oxidative DNA damage also increases with age.41 Since such damage can lead to mutations and the subsequent synthesis of dysfunctional proteins, it is likely to be a major contributor to many age-associated detrimental changes.
A wide range of evidence shows mitochondrial decline as a function of aging.42 Since mitochondria consume >90% of the oxygen utilized in cells, and are the major site of ROS production, they appear to bear the brunt of the free radical damage observed in cells during aging. Many different studies have shown that mtDNA mutations increase with age in mammals, especially in post-mitotic highly aerobic tissues.42 One consequence of this damage is decreased ATP production, reducing the energy to drive many cellular processes. In addition, it has been shown that mitochondria of older animals produce significantly more ROS than mitochondria of younger animals.43,44 This can be a result of an escalating cycle, whereby damaged mitochondria leak more free radicals to inflict additional self-damage, as well as damage to the rest of the cell.
Antioxidant Defenses in Turtle Anoxia Tolerance
Anoxia tolerant turtles appear to possess excellent antioxidant defenses that aid in the damage-free survival of multiple episodes of anoxia-reoxygenation. Studies with red-eared sliders (Trachemys scripta elegans) show that these include both constitutive and inducible defenses.
Constitutive antioxidant defenses.
T.s. elegans maintain high constitutive activities of various antioxidant enzymes, including catalase, superoxide dismutase (SOD) and alkyl hydroperoxide reductase.45 These activities are much higher than those in other ectothermic vertebrates, and are actually comparable to mammalian activities even though the metabolic rate (and oxygen consumption) of turtles is much lower than mammalian values. Tissue pools of glutathione are also higher in turtle organs compared to other ectotherms,46 and ascorbic acid levels in turtle brain cortex are 2–3 times higher than in mammals.47 High activities of antioxidant defenses also appear to serve freezing-induced anoxia/ischemia. For example, freeze-tolerant wood frogs (Rana sylvatica) that undergo cycles of ischemia/reperfusion upon freezing and thawing also show high activities of antioxidant enzymes, significantly higher than those activities in freeze-intolerant frogs.48
The structure of turtle antioxidant enzymes might also be especially suitable for frequent anoxic excursions. For example, turtle glutathione reductase (GR), an enzyme that catalyzes the reduction of oxidized glutathione (GSSG), has a higher affinity for GSSG than the enzyme in most other vertebrates,49 allowing for better enzyme efficiency under different redox conditions. Turtle GR also showed high activity over a broad pH range (including at the low pH typical of anoxic conditions).49 In another recent study, two alpha classes of GSTs were found in T.s. elegans liver, a homodimer and a heterodimer.50 Both exhibited a major reduction in specific activity in response to anoxia and the heterodimer showed a major change in substrate preference, indicating an anoxia-responsive stable modification of the enzyme.50 Therefore, it appears that GST undergoes modifications in response to changing oxygen availability to optimize its function.
These mechanisms could help the turtle to avoid oxidative damage during situations of oxygen variability. Indeed, it was shown that the ratio of GSH/GSSG, which decreases under oxidative stress, actually increases during recovery from anoxia exposure in turtles (compared to control) suggesting that no oxidative stress occurs during reoxygenation.46 In addition, oxidative damage products were largely unaffected during anoxia/recovery in turtle organs, as evident from minimal changes in lipid peroxidation products.45 Similarly, organs of freeze-tolerant C. picta hatchlings also showed no increase in markers of oxidative damage after exposure to freezing, supercooling or hypoxia.51
The constitutive presence of such strong antioxidant defenses in turtle tissues could be directly linked to longevity. Maintenance of strong constitutive defenses throughout the turtle's lifetime would help it to avoid the day-to-day accumulation of oxidative damage to macromolecules that is observed in mammals and, hence, extend long-term organ viability.
Inducible antioxidant defenses.
In addition to the maintenance of constitutively-high antioxidant defenses, induction of proteins associated with antioxidant defense has also been observed in turtle organs in response to anoxia. Upregulation of the transcripts coding for the heavy chain of ferritin occurs in response anoxia in liver of adult T.s. elegans.52 In addition, the heavy and light chains of ferritin were upregulated in response to anoxia or freezing exposures in the heart and liver of hatchling C.p. marginata.53,54 Ferritin plays a key role in sequestering iron in cells. As previously mentioned, free ferrous iron (Fe2+) can participate in the Fenton reaction, catalyzing hydroxyl radical formation. Therefore, it is of benefit to increase the capacity to sequester iron during situations of oxygen variability, and thereby decrease the potential for ROS formation. The importance of ferritin upregulation under situations of anoxia/ischemia is observed across phylogeny; for example, ferritin heavy chain was upregulated in an anoxia-tolerant marine snail,55 whereas the light chain was freeze-responsive in wood frog heart.56
Heme oxygenase-1 (HO-1) is another enzyme that might have antioxidant functions during anoxia/reoxygenation episodes. It is involved in the degradation of heme, a major source of redox active iron in the cell, and its induction is also typically tied to the simultaneous upregulation of ferritin, which sequesters the iron released by the heme degradation reaction.57,58 HO-1 was upregulated in response to anoxia in turtle brain,59 which might be a further demonstration of the importance of minimizing redox-active iron during anoxia. In mammals, the expression of HO-1 is known to be induced in response to various forms of oxidative stress60–62 and its expression is believed to be protective in different disease states.63 The important antioxidant properties of HO-1 also stem from biliverdin, one of the products of heme catabolism by HO-1. Biliverdin and its catabolite, bilirubin, have antioxidant properties with demonstrated beneficial effects in a number of disease/injury states including ischemiareperfusion, organ transplantation, graft rejection and autoimmune inflammation.57,64,65
The transcript levels of several other antioxidant enzymes have been shown to increase in response to anoxia in the turtle. Both manganese and copper/zinc forms of SOD increase in turtle liver in response to anoxia.52 In addition, array screening showed that several antioxidant genes were upregulated by anoxia exposure in heart and liver of hatchling C. p. marginata: SOD-1, glutathione peroxidase (GPX) isozymes 1 and 4, glutathione-S-transferase (GST) isozymes M5 and A2 and peroxiredoxin 1.54 The activity of the antioxidant enzyme, γ-glutamyltranspeptidase, also increases significantly during freezing in livers of both R. sylvatica and C. picta,66,67 whereas catalase activity increased under anoxia exposure in livers of several hatchling turtle species.68 In addition, the protein levels of the transcription factor Nrf2 and several downstream antioxidant enzymes were significantly elevated in the brains of C.p. marginata hatchlings in response to freezing exposure.69
Protein levels of some members of the peroxiredoxin family were elevated in response to anoxia in several tissues of T.s. elegans (Krivoruchko and Storey, unpublished results). As in the case of ferritin and HO-1, upregulation of these enzymes in response to anoxia could serve as a preparatory step to deal with a burst of ROS production when tissue reoxygenation occurs, and might be responsible, at least in part, for the lack of oxidative damage observed in turtle tissues during aerobic recovery after anoxia exposure.
Besides their role in antioxidant defenses, the anoxia-responsive upregulation of peroxiredoxins might be of additional importance. Peroxiredoxins function in reducing H2O2, a molecule that has second messenger actions.70,71 Metabolic rate depression is a crucial factor in anoxia tolerance, and is expected to include reduced cell responsiveness to growth signals. This could potentially be achieved by reducing the levels of intracellular second messengers such as H2O2, which mediate growth factor and cytokine effects. Interestingly, upregulation of peroxiredoxins has also been shown to occur during mammalian hibernation, another situation of hypometabolism.72,73
Other Protective Mechanisms
While strong antioxidant defenses clearly play a key role in protecting turtle cells during anoxia and reoxygenation, other protective proteins also contribute to survival under these stressful conditions. These include various molecular chaperones, stress-responsive transcription factors and pro-survival proteins.
High constitutive HSPs.
Proteins that are particularly sensitive to changes in intracellular pH, redox state and ionic milieu, which occur in cells during anoxia and reoxygenation, can lose their native folded conformation to become misfolded. Proliferation of heat shock proteins (HSPs) is one of the best known cytoprotective mechanisms in response to stress.74 Figure 1 shows a generalized scheme for activation of the heat shock response in vertebrates. HSPs act as molecular chaperones, helping to fold nascent proteins, as well as aiding in the refolding of misfolded proteins under stress conditions.74,75 By their chaperone action, HSPs help to preserve cellular proteins and extend their functional life. Increased expression of HSPs has been shown to be a widespread response to many environmental stresses.74 HSPs also play a protective role in a number of pathological conditions associated with oxidative stress, including ischemia, cardiovascular disease and neurodegeneration.76
Figure 1.

Activation of the heat shock response. Various stresses can lead to unfolding of proteins, calling heat shock proteins (HSPs) into action to aid refolding. As a consequence, HSPs dissociate from association with the heat shock transcription factor (HSF). The freed HSF then becomes activated through trimerization and hyperphosphorylation, binds to the heat shock element (HSE) and mediates the upregulation of hsp genes. This results in the proliferation of cytoprotective HSPs in the cell.
Studies have found significantly higher levels of Hsp60 in the hearts of anoxia-tolerant painted turtles compared to anoxia intolerant softshell turtles, rabbits and rats.77 Hsp60 is a predominantly mitochondrial chaperone involved in the folding of proteins entering the mitochondria. Hsp60 also has protective effects against oxidative stress.78 Levels of this protein are reduced with aging,79 possibly accounting for some age-associate cell damage. High constitutive levels of Hsp60 are of particular interest, since oxidative damage to the mitochondria appears to be instrumental in the escalation of age-related damage. The presence of high levels of protective HSPs, such as Hsp60, in the mitochondria could protect mitochondrial proteins from unfolding and ensure long-term mitochondrial viability as well as potentially prevent age-associated increases in ROS-production by mitochondrial proteins involved in oxidative reactions. This is certainly an interesting possibility that warrants further investigation.
High constitutive levels of Hsp72 have also been reported in turtle brain.80 Hsp72 is known as the inducible HSP70 family member, and is generally found at very low levels under normal conditions. The constitutive expression of Hsp72 in turtle brain suggests that is has some importance in neuroprotection in this animal. Members of the HSP70 family and their co-chaperones play a crucial role in protein sorting and quality control by selecting and directing aberrant proteins to the proteasome for degradation.81 A decline in protein quality control is related to many neurodegenerative diseases involving aggregation-prone proteins, including Parkinson disease, amyotrophic lateral sclerosis, prion diseases, Alzheimer disease and CAG-repeat and polyQ expansion diseases, including Huntington disease.82 The constitutive presence of Hsp72 might be instrumental in preventing protein aggregation and age-associated neurodegenerative diseases in the turtle brain.
Anoxia-inducible molecular chaperones.
As in the case of antioxidant proteins, HSPs and other molecular chaperones also show inducibility in response to anoxia in turtle tissues. This included Hsp25/27, Hsp40, Hsp60, Hsp70, Hsc70 and Hsp90.59,80,83,84 Array screening of turtle brain comparing aerobic and anoxic animals also showed upregulation of transcripts coding for αB-crystallin, another known chaperone.2 In addition, the heat shock transcription factor (HSF1) that is responsible for the activation of the heat shock response was activated in response to anoxia in turtle tissues.84 Activation of the heat shock response in response to anoxia might help maintain protein stability under anoxic conditions as well as serve as a preparative mechanism for re-oxygenation, since increased HSP expression might also actively prevent damage following oxidative stress.
Small HSPs have been shown to have key functions in protection against protein unfolding, especially under conditions of oxidative stress. The αB-crystallin is known for its role as a molecular chaperone of cytoskeletal proteins that would otherwise aggregate and precipitate, something that has been strongly linked with neurodegenerative diseases.85 αB-Crystallin also responds to oxidative stress86 and its upregulation in turtle brain suggests an important role for this protein in neuronal protection against anoxia. Besides protecting against protein misfolding during stress, Hsp27 has been shown to possess antioxidant properties. It is able to hold glutathione in its reduced form, even under oxidative conditions, and by doing so restore redox homeostasis.87,88
Some HSPs, including Hsp27, Hsp70 and Hsp90 can also regulate apoptosis by binding and inhibiting members of the apoptotic cascade. For example, in stressed cells, Hsp70 and Hsp90 can bind Apaf-1 (apoptosis protease activating factor-1) and by doing so inhibit events downstream of Apaf-1 activation, such as activation of caspases.89,90 Hsp70 can also sequester released AIF from the mitochondria and prevent caspase-independent cell death.91 Hsp27 can inhibit apoptosis by either inhibiting the release of mitochondrial cytochrome c,92 or by binding to cytochdorme c directly.93
In mammals, the HSP response declines with aging. Age-dependent waning of the HSP response has been reported in neuronal tissues,94 skeletal and cardiac muscle95 and liver.96 The loss of HSP inducibility with age could contribute to age-related degenerative diseases in humans. On the other hand, since maintenance of HSP inducibility is crucial to anoxic survival by the turtle, this, in conjunction with high constitutive expression of HSPs, might contribute to their negligible senescence.
NFκB.
The transcriptional responses involved in anoxia-tolerance and the transcription factors involved in facilitating these responses are now becoming an active field of research. NFκB is an oxygen-responsive transcription factor, known to be activated in response to a variety of stimuli and controlling the expression of hundreds of target genes, including genes involved in the immune response, stress response, antioxidant defenses, cell growth and differentiation and apoptosis (reviewed in ref. 97). NFκB is a dimeric transcription factor and is composed of proteins containing the Rel Homology domain. The Rel family is comprised of five members, p50 and its precursor p105 (also known as NFκB1), p52 and its precursor p100 (also known as NFκB2), p65 (also known as RelA), RelB and c-Rel. The best studied dimer, and the one frequently referred to as ‘NFκB’ occurs between p65 (RelA) and p50. Under normoxic conditions NFκB is retained in the cytoplasm through association with the inhibitor I.B, but in response to anoxia, it undergoes nuclear translocation, mediating the expression of target genes (Fig. 2). This control mechanism allows NFκB to respond rapidly to stress. In mammals, NFκB is an important mediator of transcriptional responses during ischemia/reperfusion (reviewed in ref. 98). In turtles, NFκB is activated in the turtle liver in response to anoxia.52 Some data on turtle liver NFκB are shown in Figure 3; protein levels of the two NFκB subunits increase in liver nuclei in response to anoxia (Fig. 3A), and the DNA-binding activity of NFκB also increases (Fig. 3B). NFκB was also shown to be anoxia-responsive in the turtle brain.99 These data suggest that NFκB has key functions in protecting these organs under anoxic conditions.
Figure 3.

Activation of NFκB in T. s. elegans liver. (A) Effect of 5 and 20 h of anoxic submergence on the nuclear content of the proteins making up NFκB dimer, p50 and p65 in T. s. elegans liver. Representative western blots bands of p50 and p65, as well as a histogram showing normalized protein levels are shown. (B) Effect of 5 and 20 h of anoxic submergence on the DNA-binding activity of NFκB in T. s. elegans liver, as assessed by a transcription factor ELISA. (a) Significantly different from the corresponding control (p < 0.05); (b) significantly different from the 5 h anoxic value (p < 0.05). Data are taken from ref. 52.
While many of the genes under NFκB control could play beneficial roles in anoxia tolerance, the vast array of genes that lie under NFκB control, as well as the lack of tools available to accurately analyze large-scale transcriptional responses in the turtle, makes it rather difficult to pinpoint all the genes that are activated by NFκB in the turtle in response to anoxia. Some potential candidates include the antioxidant genes, ferritin heavy chain and SOD, as well as the pro-survival genes Bcl-2 and Bcl-xL, which are indeed upregulated in response to anoxia in turtle liver.52
Anti-apoptotic proteins.
Studies in mammals have shown that apoptosis is oxygen-responsive, with exposure to hypoxia promoting cell-survival and exposure to anoxia promoting apoptosis.100,101 However, tissues of T.s. elegans can survive anoxia for many weeks, without any apparent damage, suggesting that inhibition of apoptosis must occur on a molecular level during anaerobiosis. The promotion or inhibition of apoptosis is controlled by two conserved pathways, the death receptor pathway and the mitochondrial pathway (shown in Fig. 4), and the decision to initiate apoptosis often depends on the levels of pro- and anti-apoptotic proteins.102 The transcript levels of two major anti-apoptotic proteins, Bcl-2 and Bcl-xL increase, possibly in an NFκB-dependent manner, in response to anoxia in turtle liver (Fig. 5).52 These proteins reside in the outer mitochondrial membrane and inhibit apoptosis by preventing the loss of outer mitochondrial membrane integrity, which leads to lethal downstream events such as activation of caspases.103,104 Studies in mammals have shown that enhanced expression of these proteins limits apoptosis in cells,105,106 and plays a protective role in mammalian ischemic preconditioning.107,108
Figure 4.
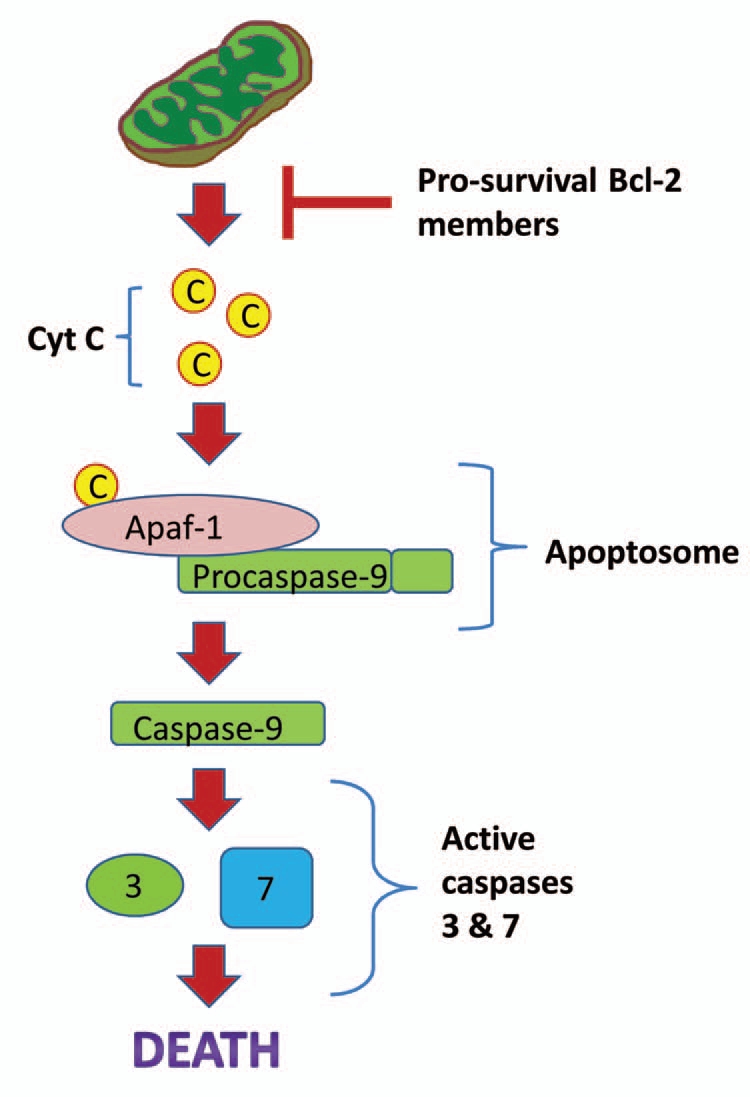
A generalized schematic for the mitochondrial apoptosis pathway. During apoptosis, the mitochondrial membrane swells and cytochrome c leaks out. This leads to binding with the pro-apoptotic protein Apaf-1, subsequent formation of the apoptosome and activation of caspase-9. Caspase-9 subsequently activates other caspases and results in cell death. Pro-survival Bcl-2 family members (such as Bcl-2 and Bcl-xL) can inhibit apoptosis by binding to the mitochondrial membrane and blocking its swelling.
Figure 5.

Effect of 5 and 20 h of anoxic submergence on transcript levels of prosurvival target genes of NFκB in T. s. elegans liver. Representative RT-PCR bands and a histogram showing normalized transcript levels under control and anoxic conditions are shown. Data were normalized against α-tubulin and are shown as means ± S.E.M., n = 3 independent trials. (a) Significantly different from the corresponding control (p < 0.05). Data are taken from ref. 52.
Like many of the proteins discussed in this review, these proteins potentially have dual roles, protecting against oxidative damage as well as anoxia. Bcl-2 has been previously reported to have antioxidant effects.109,110 Transfection of cells with Bcl-2 suppressed lipid peroxidation109 and Bcl-2 also reduced intracellular ROS levels.110 In addition, studies have shown that hyperglycemia-induced lipid peroxidation and advanced glycation end products (AGE) in endothelial cells are suppressed by Bcl-2.111
Brain-Specific Adaptations
The mammalian brain is especially prone to oxygen limitation and even short bouts of hypoxia can cause irreversible damage. The turtle brain, however, can survive many weeks of anoxia unharmed. Many of the mechanisms involved in brain anoxia-tolerance are also linked to age-related neurodegeneration, and as such may contribute to turtle longevity.10
Channels and neurotransmitters.
A robust reduction in ion channels occurs in response to anoxia in turtle brain. This includes reduction in voltage-gated Na+ channel density,112 decrease in mRNA of voltage gated K+ channel,113 decrease in K+ leak channel114 and silencing of the Ca2+-dependent NMDA receptor.115 Because damage to ion channels is involved in several age-related diseases in the brain,116 it has been suggested that the turtle's ability to both maintain channel integrity while downregulating activity in response to anoxia, as well as restore those channels upon reoxygenation, could be linked to longevity.10
A major characteristic of the response to oxygen limitation by mammalian brain is the uncontrolled and destructive release of excitatory neurotransmitters. The turtle brain, however, avoids such toxic release by maintaining a balance between dopamine and glutamate release and active uptake mechanisms.117,118 An increase in the inhibitory neurotransmitter GABA and GABA receptors in the brain is also observed,112 and likely contributes to strengthening of the inhibitory tone. Adenosine is also involved in the suppression of ion channels in response to anoxia119 and also plays a role in anoxia-mediated ROS suppression.120 An increased susceptibility to excitatory amino acids has been previously associated with age-related neurodegenerative diseases,121 and the turtle's ability to avoid this could be linked to its longevity.112
Inosine, the deamination product of adenosine, could also have a role to play in anoxia tolerance. Recent publications suggest a potentially important role for inosine in the activation of antioxidant defenses in response to hypoxia, hyperoxia or H2O2 insult.122–124 Interestingly, the enzyme that makes inosine, 5′ nucleotidase cytosolic II isozyme (NT5C2), is upregulated in turtle brain in response to anoxia.2 As previously discussed, enhancement of antioxidant defenses is an important part of anoxic survival and an increase in inosine could be a potential trigger for this response.
Neuroglobin.
Neuroglobin (Ngb) is another protein that has a potentially important protective role in turtle anoxia tolerance. Ngb is preferentially expressed in the nervous system, as well as some endocrine tissues125,126 and has been identified in the brains of rodents, fish, amphibians, birds and reptiles.125,127,128 Since globins generally function as either O2 storage or transport proteins, Ngb is suspected of playing a role in transferring oxygen to the mitochondria in the brain and retina.127 Other possible roles for Ngb are in the decomposition of reactive oxygen and nitrogen species, acting as a terminal oxidase that regenerates NAD+ under anaerobic conditions or as a detector of cellular oxygen concentrations.129–132 Recent studies have also shown that Ngb expression is reduced with increasing age133 and also plays a protective role in Alzheimer disease by attenuating amyloid beta neurotoxicity.134 In mammals, Ngb also plays an anti-apoptotic role by intervening in the activation of pro-caspase 9 through interaction with cytochrome c in the intrinsic mitochondrial pathway.135 A role for neuroglobin in the protection against oxidative damage in turtle brain has also been suggested and knockout of Ngb in neuronally enriched turtle cell cultures resulted in significant increases in H2O2 release.136
The levels of Nrb mRNA increased significantly in response to hypoxia, as well as post-anoxic reoxygenation, in T.s. elegans brain with a lesser degree of upregulation in response to 1 and 4 h of anoxia.128 These results suggest that Ngb may play an important role in turtle adaptation to hypoxia, possibly by ensuring adequate supply of O2 to the brain. The upregulation of Ngb during reoxygenation could also suggest a role in antioxidant defense of the brain.
Interestingly, whereas a strong increase in Ngb levels is not generally observed in response to hypoxia in rodents,132 another species that lives in environments of changing oxygen availability, the zebrafish (Danio rerio), shows strong upregulation of Ngb in response to hypoxia,137 as in turtles. These findings suggest that inducibility of this protein in response to hypoxia may be an important adaptation to low oxygen environments. Other animals that live in hypoxic environments or deal with oxygen variability show constitutively high levels of neuroglobin. This includes the subterranean blind mole rat Spalax ehrenbergi and the goldfish.132
The Role of Hypometabolism
A key element to turtle anoxia tolerance is a profound metabolic rate depression to 10–20% of the corresponding aerobic resting rate at the same temperature. This suppresses, rebalances and reprioritizes the rates of ATP-utilizing and ATP-generating processes so that turtles can sustain long term viability without oxygen. Entrance into hypometabolism is not just an anoxia-specific phenomenon, but is a common survival strategy across phylogeny and is observed in phenomena including hibernation, torpor, aestivation, anaerobiosis, diapause and anhydrobiosis. Understanding the mechanisms of hypometabolism could be of potential use to medical sciences. For example, decreasing the metabolic rate of human organs meant for transplantation could lengthen the time organ explants remain viable. The topic of hypometabolism in turtles and other systems has been thoroughly reviewed elsewhere.138,139 Therefore, in the present review we will only touch on some of the key strategies associated with metabolic rate depression in the anoxic turtle.
Entrance into hypometabolism in response to anoxia in turtles is characterized by a reorganization of energy-consuming processes. For example, studies with turtle hepatocytes showed a 94% decrease in overall ATP turnover under anoxia, but found that five main ATP-consuming processes were suppressed to different degrees.140 Under anoxia, the Na+K+-ATPase became the dominant energy consumer of the cell, consuming 62% of total ATP turnover as compared with 28% in normoxia. Meanwhile, protein synthesis and degradation were strongly suppressed by >90%, whereas gluconeogenesis and urea synthesis were virtually halted. Similar reductions in protein synthesis were also observed in other organs.141 This reorganization of metabolic processes allows turtle cells to direct energy into the most vital processes for cellular survival under anoxia and away from less vital processes, and results in the most efficient ATP utilization under this energy-limited condition.
Reversible protein phosphorylation is considered to be the most widespread and powerful mechanism utilized by cells to make stable changes to enzyme activities in response to various stimuli. This mechanism also plays an important role in the regulation of metabolic depression in other hypometabolic systems.138,139 Reversible protein phosphorylation regulates the activities and kinetic properties of many enzymes involved in diverse cellular processes. These include membrane receptors and transporters, ion channels and ATPases, proteins involved in gene transcription, protein synthesis and degradation, cell cycle control and signal transduction cascades. For example, it was shown that reversible phosphorylation is involved in the regulation of voltage-gated ion channels (Na+, Ca2+, K+) and membrane receptors (e.g., N-methyl-D-aspartate-type glutamate receptor) in anoxia-tolerant turtles.142–144 In addition, changes in the phosphorylation status of various ribosomal translation factors were observed in turtle tissues in response to anoxia,145 suggesting that such changes are involved in anoxia-induced suppression of protein synthesis in turtles.
It is estimated that 1–10% of cellular energy is devoted to gene transcription, depending on the tissue;146 this is also a significant metabolic cost that must be reduced in situations of hypometabolism. Suppressed rates of gene transcription have been observed in a number of hypometabolic systems in response to anoxia including brine shrimp (Artemia franciscana) and intertidal snails (Littorina littorea),147,148 as well as in mammalian hibernation.149,150 A prominent mechanism of global or localized transcriptional control is the post-translational modification of histones that are subject to a number of modifications that affect the state of transcriptional activity in cells.151,152 For example, his-tone acetylation is associated with higher transcriptional activity because this modification makes chromatin more accessible to proteins of the transcriptional apparatus.153 Histone deacetylases (HDACs) are a group of enzymes that catalyze the removal of acetyl groups from histones, and as such are associated with transcriptional repression. Increased HDAC expression and activity, as well as decreased histone acetylation was observed in skeletal muscle of hibernating ground squirrels.154 Such changes also occurred in turtle muscle in response to anoxia, with HDAC protein levels increasing by 1.7–3.5 fold, and HDAC activity increasing by 1.5-fold in response to anoxia.155 This suggests that histone deacetylation is an important general mode of global transcriptional suppression in hypometabolic systems.
Other Models for Longevity
Other models for longevity also show remarkable stress-resistance, and from a comparative perspective, it would be interesting to investigate whether their stress-resistance mechanisms are similar to those observed in turtles. Among mammals, bats and naked mole rats are particularly known for extreme longevity. Bats are the longest-lived mammals relative to their body size.156,157 Their longevity often surpasses 30 years, and the longest-lived bat species on record survived for at least 41 years in the wild.158 Naked mole rats have a recorded lifespan that exceeds 28 years, approximately nine times greater than predicated for their body size. They display no age-related changes to body mass, basal metabolic rate, percent body fat, fat-free mass or bone density,159 an indication of “negligible senescence.” Both animals show remarkable tolerance to stresses. For example, primary fibroblasts isolated from the little brown bat were reported to be much more resistant to H2O2, cadmium, UV light and heat, as compared to cells from mice.160 Cultured cells from this species also showed enhanced repair of γ-irradiation-induced DNA lesions.158 Hippocampal slices of mole rats are also significantly more tolerant of oxygen and nutrient deprivation than laboratory white rats161 and mole rat neurons maintain synaptic transmission much longer than mouse neurons and can recover from periods of anoxia exceeding 30 min.162 Furthermore, cultured cells from naked mole rats, including primary fibroblasts and endothelial cells, tend to be resistant to several forms of stress.163,164 Interestingly, no tumors have ever been found in a mole rat and they might possess extraordinary cancer-resistance mechanisms.165
Bats have lower H2O2 production166,167 and protein oxidation168 compared to other mammals, which might directly contribute to their longevity. Bats also show resistance to protein oxidation both in vivo and in vitro under conditions of acute oxidative stress.168 In addition, bats showed lower levels of protein ubiquitination, along with reduced proteasome activity and increased resistance to urea-induced protein unfolding compared to mice.168 Naked mole rats also display no increase in cysteine oxidation and ubiquitination over two decades of life.169 These results suggest that the longevity in these animals may be regulated by very efficient maintenance of protein homeostasis. A potential mechanism for the long-term maintenance of protein homeostasis could be via the increased expression of molecular chaperones and it would be interesting to investigate whether, as in turtles, this occurs in bats and naked mole rats. Interestingly, several species of bats are also known to hibernate, a condition characterized by strongly reduced metabolic rate, decreased body temperature and hypoxia/ischemia, as well as increased activation of various stress-tolerance mechanisms (reviewed in ref. 170). It is possible that some of the stress-tolerance mechanisms that support bat cells during long term torpor might also contribute to their enhanced longevity.
Conclusions
Many different molecular mechanisms appear to play protective roles in anoxia-tolerance (see Table 1 for a summary of the major pathways discussed) and a variety of these might have beneficial anti-aging properties. For example, the maintenance of constitutive protective mechanisms, while energy-expensive, would provide immediate protection against any assault on metabolic integrity as well as minimize damage accumulated throughout an animal's lifetime. On the other hand, while the inducibility of key protective mechanisms is known to decrease with age in normal-lived animals, the turtle's exposure to oxygen variability throughout its lifetime might put an emphasis on maintaining the inducibility of these mechanisms, making it less prone to aging-associated damage.
Table 1.
Major proteins/pathways discussed in this review and their roles
| Pathway/proteins discussed | Role |
| Antioxidant enzymes/proteins | Destroy intracellular rOS and provide relief against oxidative stress (reviewed in refs. 14, 16, 181). |
| Heat shock response | Enhance intracellular protein folding capacity (reviewed in refs. 74, 75). |
| NFκB | Enhanced expression of genes involved in the immune response, stress response, antioxidant defenses, cell growth and differentiation, and apoptosis (reviewed in ref. 97). |
| Anti-apoptotic proteins | Inhibition of apoptosis (reviewed in ref. 102). |
| Neuroglobin | Enhanced oxygen delivery to the brain; Possible antioxidant effects (reviewed in ref. 127, 179). |
Understanding the molecular adaptations associated with anoxia tolerance is of key importance not only from a comparative point of view, but also from a medical one, as many of these mechanisms are involved in human disease. For example, induction of a hypometabolic state in human organs could potentially prolong organ survival for transplantation,171 as well as prolong organ survival following trauma. Indeed, some research demonstrates that induced hypometabolism reduces the detrimental effects of hypoxic/ischemic organ injury.172–174 The natural mechanisms of anoxia tolerance are also of interest because the molecular nature of ischemic injury in mammals is not yet completely understood and the study of these responses in anoxia-tolerant animals could highlight those mechanisms that are beneficial for survival. For example, the induction of the transcription factor NFκB in turtle organs in response to anoxia suggests a beneficial role during oxygen deprivation. This transcription factor appears to play a dual role in mammalian ischemia/reperfusion injury, promoting inflammation and death, but also playing a crucial role in ischemic preconditioning.175–178 Hence, a better understanding of the role of this transcription factor and its target genes could lead to better treatment for a variety of pathogenic conditions involving ischemia/reperfusion injury, including heart disease and stroke. Similarly, other molecular mechanisms that appear to play roles in turtle hypoxia/anoxia tolerance are relevant to human disease. Neuroglobin overexpression confers neuroprotection against neuronal hypoxia/ischemia-induced damage,179 whereas enhanced expression of protective proteins such as heat shock proteins, antioxidant proteins and anti-apoptotic proteins has been tied with the enhanced organ protection observed during ischemic preconditioning.180
Despite the potential key importance of natural anoxia tolerance to medical sciences, there is still much to be learned about the mechanisms underlying this ability. It is expected that as we learn more about these processes, we will be able to better understand the molecular nature of many oxygen-associated pathological conditions in mammals and identify new treatments for these conditions, effectively improving human health and extending lifespan.
Figure 2.

Activation of the NFκB pathway. Under normal conditions, the NFκB dimer (composed of the proteins p50 and p65) is retained in the cytoplasm through interaction with its inhibitor protein, IκB. In response to various stimuli, the IκB kinase becomes activated and phosphorylates IκB, thereby targeting it for ubiquitination and subsequent degradation by the proteasome. This leaves NFκB free to move to the nucleus and activate transcription of various target genes.
Acknowledgements
We are grateful to J.M. Storey for editorial review of this manuscript. Research in our lab is supported by a discovery grant from the Natural Sciences and Engineering Research Council of Canada and the Canada Research Chairs program.
Footnotes
Previously published online: www.landesbioscience.com/journals/oximed/article/12356
References
- 1.Storey KB. Metabolic adaptations supporting anoxia tolerance in reptiles: recent advances. Comp Biochem Physiol B. 1996;113:23–35. doi: 10.1016/0305-0491(95)02043-8. [DOI] [PubMed] [Google Scholar]
- 2.Storey KB. Anoxia tolerance in turtles: metabolic regulation and gene expression. Comp Biochem Physiol A. 2007;147:263–276. doi: 10.1016/j.cbpa.2006.03.019. [DOI] [PubMed] [Google Scholar]
- 3.Ultsch GR. The ecology of overwintering among turtles: where turtles overwinter and its consequences. Biol Rev Camb Philos Soc. 2006;81:339–367. doi: 10.1017/S1464793106007032. [DOI] [PubMed] [Google Scholar]
- 4.Churchill TA. Organ preservation for transplantation. In: Storey KB, editor. Functional Metabolism: Regulation and Adaptation. Hoboken, NJ: Wiley-Liss; 2004. pp. 383–413. [Google Scholar]
- 5.Ma A, Qi S, Chen H. Antioxidant therapy for prevention of inflammation, ischemic reperfusion injuries and allograft rejection. Cardiovasc Hematol Agents Med Chem. 2008;6:20–43. doi: 10.2174/187152508783329966. [DOI] [PubMed] [Google Scholar]
- 6.Finch CE. Longevity, Senescence and the Genome. Chicago: University of Chicago Press; 1990. [Google Scholar]
- 7.Finch CE. Update on slow aging and negligible senescence—a mini-review. Gerontology. 2009;55:307–313. doi: 10.1159/000215589. [DOI] [PubMed] [Google Scholar]
- 8.Congdon JD, Nagle RD, Kinney OM, van Loben Sels RC, Quinter T, Tinkle DW. Testing hypotheses of aging in long-lived painted turtles (Chrysemys picta) Exp Gerontol. 2003;38:765–772. doi: 10.1016/s0531-5565(03)00106-2. [DOI] [PubMed] [Google Scholar]
- 9.Girondot M, Garcia J. Senescence and longevity in turtles. What telomeres tell us. In: Miaud DC, Guyetant R, editors. Current Studies in Herpetology. France: Societa Europaea Herpetologica, Le Bouget du Lac; 1998. pp. 133–137. [Google Scholar]
- 10.Lutz PL, Prentice HM, Milton SL. Is turtle longevity linked to enhanced mechanisms for surviving brain anoxia and reoxygenation? Exp Gerontol. 2003;38:797–800. doi: 10.1016/s0531-5565(03)00111-6. [DOI] [PubMed] [Google Scholar]
- 11.Söti C, Csermely P. Protein stress and stress proteins: implications in aging and disease. J Biosci. 2007;32:511–515. doi: 10.1007/s12038-007-0050-z. [DOI] [PubMed] [Google Scholar]
- 12.Frank L. Oxygen toxicity in eukaryotes. In: Oberly LW, editor. Superoxide Dismutase, Pathological States. Vol. 3. Boca Raton: CRC Press; 1985. pp. 1–43. [Google Scholar]
- 13.Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. Biochem J. 1973;134:707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hermes-Lima M. Oxygen in biology and biochemistry: role of free radicals. In: Storey KB, editor. Functional Metabolism: Regulation and Adaptation. Hoboken, NJ.: Wiley-Liss; 2004. pp. 319–368. [Google Scholar]
- 15.Cadenas E, Boveris A, Ragan CI, Stoppani AO. Production of superoxide radicals and hydrogen peroxide by NADH-ubiquinone reductase and ubiquinolcytochrome c reductase from beef-heart mitochondria. Arch Biochem Biophys. 1977;180:248–257. doi: 10.1016/0003-9861(77)90035-2. [DOI] [PubMed] [Google Scholar]
- 16.Gutteridge JM. Biological origin of free radicals and mechanisms of antioxidant protection. Chem Biol Interact. 1994;91:133–140. doi: 10.1016/0009-2797(94)90033-7. [DOI] [PubMed] [Google Scholar]
- 17.Valko M, Izakovic M, Mazur M, Rhodes CJ, Telser J. Role of oxygen radicals in DNA damage and cancer incidence. Mol Cell Biochem. 2004;266:37–56. doi: 10.1023/b:mcbi.0000049134.69131.89. [DOI] [PubMed] [Google Scholar]
- 18.Halliwell B. Biochemistry of oxidative stress. Biochem Soc Trans. 2007;35:1147–1150. doi: 10.1042/BST0351147. [DOI] [PubMed] [Google Scholar]
- 19.Stark G. Functional consequences of oxidative membrane damage. J Membr Biol. 2005;205:1–16. doi: 10.1007/s00232-005-0753-8. [DOI] [PubMed] [Google Scholar]
- 20.Catalá A. Lipid peroxidation of membrane phospholipids generates hydroxy-alkenals and oxidized phospholipids active in physiological and/or pathological conditions. Chem Phys Lipids. 2009;157:1–11. doi: 10.1016/j.chemphyslip.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 21.Buege JA, Aust SD. Microsomal lipid peroxidation. Methods Enzymol. 1978;52:302–310. doi: 10.1016/s0076-6879(78)52032-6. [DOI] [PubMed] [Google Scholar]
- 22.Borchman D, Lamba OP, Salmassi S, Lou M, Yappert MC. The dual effect of oxidation on lipid bilayer structure. Lipids. 1992;27:261–265. doi: 10.1007/BF02536472. [DOI] [PubMed] [Google Scholar]
- 23.Goldstein RM, Weissmann G. Effects of the generation of superoxide anion on permeability of liposomes. Biochem Biophys Res Commun. 1977;75:604–609. doi: 10.1016/0006-291x(77)91515-7. [DOI] [PubMed] [Google Scholar]
- 24.Kourie JI. Interaction of reactive oxygen species with ion transport mechanisms. Am J Physiol. 1988;275:1–24. doi: 10.1152/ajpcell.1998.275.1.C1. [DOI] [PubMed] [Google Scholar]
- 25.Mattson MP, Pedersen WA, Duan W, Culmsee C, Camandola S. Cellular and molecular mechanisms underlying perturbed energy metabolism and neuronal degeneration in Alzheimer's and Parkinson's diseases. Ann NY Acad Sci. 1999;893:154–175. doi: 10.1111/j.1749-6632.1999.tb07824.x. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y, Marcillat O, Giulivi C, Ernster L, Davies KJ. The oxidative inactivation of mitochondrial electron transport chain components and ATPase. J Biol Chem. 1990;265:16330–16336. [PubMed] [Google Scholar]
- 27.Albano E, Bellomo G, Parola M, Carini R, Dianzani MU. Stimulation of lipid peroxidation increases the intracellular calcium content of isolated hepatocytes. Biochim Biophys Acta. 1991;1091:310–316. doi: 10.1016/0167-4889(91)90194-3. [DOI] [PubMed] [Google Scholar]
- 28.Stadtman ER. Protein oxidation and aging. Free Radic Res. 2006;40:1250–1258. doi: 10.1080/10715760600918142. [DOI] [PubMed] [Google Scholar]
- 29.Amici AR, Levine RL, Stadtman ER. Conversion of amino acids residues in proteins and amino acid homopolymers to carbonyl derivatives by metal-catalyzed reactions. J Biol Chem. 1989;264:3341–3346. [PubMed] [Google Scholar]
- 30.Chevion M. A site-specific mechanism for free radical induced biological damage: the essential role of redoxactive transition metals. Free Rad Biol Med. 1988;5:27–37. doi: 10.1016/0891-5849(88)90059-7. [DOI] [PubMed] [Google Scholar]
- 31.Bohr VA. Repair of oxidative DNA damage in nuclear and mitochondrial DNA and some changes with aging in mammalian cells. Free Radic Biol Med. 2002;32:804–812. doi: 10.1016/s0891-5849(02)00787-6. [DOI] [PubMed] [Google Scholar]
- 32.Lombardi V, Valko L, Stolc S, Valko M, Ondrejickova O, Horakova L, et al. Free radicals in rabbit spinal cord ischemia: Electron spin resonance spectroscopy and correlation with SOD activity. Cell Mol Neurobiol. 1998;18:399–412. doi: 10.1023/A:1022597431593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stolc S, Valko L, Valko M, Lombardi V. A technique for the fast sampling of biological tissues for electron paramagnetic resonance spectroscopy. Free Rad Biol Med. 1996;20:89–91. doi: 10.1016/0891-5849(95)02002-0. [DOI] [PubMed] [Google Scholar]
- 34.Oleinick NL, Chiu SM, Ramakrishnan N, Xue LY. The formation, identification and significance of DNAprotein cross-links in mammalian cells. Br J Cancer Suppl. 1987;8:135–140. [PMC free article] [PubMed] [Google Scholar]
- 35.Evans MD, Dizdaroglu M, Cooke MS. Oxidative DNA damage and disease: induction, repair and significance. Mutat Res. 2004;567:1–61. doi: 10.1016/j.mrrev.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 36.Wickens AP. Ageing and the free radical theory. Respir Physiol. 2001;128:379–391. doi: 10.1016/s0034-5687(01)00313-9. [DOI] [PubMed] [Google Scholar]
- 37.Gilca M, Stoian I, Atanasiu V, Virgolici B. The oxidative hypothesis of senescence. J Postgrad Med. 2007;53:207–213. doi: 10.4103/0022-3859.33869. [DOI] [PubMed] [Google Scholar]
- 38.Praticò D. Lipid peroxidation and the aging process. Sci Aging Knowledge Environ. 2002;2002:5. doi: 10.1126/sageke.2002.50.re5. [DOI] [PubMed] [Google Scholar]
- 39.Shringarpure R, Davies KJ. Protein turnover by the proteasome in aging and disease. Free Radic Biol Med. 2002;32:1084–1089. doi: 10.1016/s0891-5849(02)00824-9. [DOI] [PubMed] [Google Scholar]
- 40.Stadtman ER. The status of oxidatively modified proteins as a marker of aging. In: Esser K, Martin GM, editors. Molecular Aspects of Aging. Chichester: Wiley; 1995. pp. 129–144. [Google Scholar]
- 41.Chen JH, Hales CN, Ozanne SE. DNA damage, cellular senescence and organismal ageing: causal or correlative? Nucleic Acids Res. 2007;35:7417–7428. doi: 10.1093/nar/gkm681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Terzioglu M, Larsson NG. Mitochondrial dysfunction in mammalian ageing. Novartis Found Symp. 2007;287:197–208. doi: 10.1002/9780470725207.ch14. [DOI] [PubMed] [Google Scholar]
- 43.Weindruch RH, Cheung MK, Verity MA, Walford RL. Modification of mitochondrial respiration by aging and dietary restriction. Mech Ageing Dev. 1980;12:375–392. doi: 10.1016/0047-6374(80)90070-6. [DOI] [PubMed] [Google Scholar]
- 44.Nohl H. Oxygen radical release in mitochondria: influence of age. In: Johnson JE, Walford R Jr, Harman D, Miquel J, editors. Free Radicals, Aging and Degenerative Diseases. New York Liss; 1986. pp. 77–97. [Google Scholar]
- 45.Willmore WG, Storey KB. Antioxidant systems and anoxia tolerance in a freshwater turtle Trachemys scripta elegans. Mol Cell Biochem. 1997;170:177–185. doi: 10.1023/a:1006817806010. [DOI] [PubMed] [Google Scholar]
- 46.Willmore WG, Storey KB. Glutathione systems and anoxia tolerance in turtles. Am J Physiol. 1997;273:219–225. doi: 10.1152/ajpregu.1997.273.1.R219. [DOI] [PubMed] [Google Scholar]
- 47.Rice ME, Lee EJ, Choy Y. High levels of ascorbic acid, not glutathione, in the CNS of anoxia-tolerant reptiles contrasted with levels in anoxia-intolerant species. J Neurochem. 1995;64:1790–1799. doi: 10.1046/j.1471-4159.1995.64041790.x. [DOI] [PubMed] [Google Scholar]
- 48.Joanisse DR, Storey KB. Oxidative damage and antioxidants in Rana sylvatica, the freeze-tolerant wood frog. Am J Physiol. 1996;271:545–553. doi: 10.1152/ajpregu.1996.271.3.R545. [DOI] [PubMed] [Google Scholar]
- 49.Willmore WG, Storey KB. Purification and properties of glutathione reductase from liver of the anoxia-tolerant turtle, Trachemys scripta elegans. Mol Cell Biochem. 2007;297:139–149. doi: 10.1007/s11010-006-9339-8. [DOI] [PubMed] [Google Scholar]
- 50.Willmore WG, Storey KB. Purification and properties of the glutathione S-transferases from the anoxia-tolerant turtle, Trachemys scripta elegans. FEBS J. 2005;272:3602–3614. doi: 10.1111/j.1742-4658.2005.04783.x. [DOI] [PubMed] [Google Scholar]
- 51.Baker PJ, Costanzo JP, Lee RE., Jr Oxidative stress and antioxidant capacity of a terrestrially hibernating hatchling turtle. J Comp Physiol B. 2007;177:875–883. doi: 10.1007/s00360-007-0185-0. [DOI] [PubMed] [Google Scholar]
- 52.Krivoruchko A, Storey KB. Molecular mechanisms of turtle anoxia tolerance: A role for NFκB. Gene. 2010;450:63–69. doi: 10.1016/j.gene.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 53.Storey KB. Reptile freeze tolerance: metabolism and gene expression. Cryobiology. 2006;52:1–16. doi: 10.1016/j.cryobiol.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 54.Storey KB. Gene hunting in hypoxia and exercise. Adv Exp Biol Med. 2006;588:293–309. doi: 10.1007/978-0-387-34817-9_24. [DOI] [PubMed] [Google Scholar]
- 55.Larade K, Storey KB. Accumulation and translation of ferritin heavy chain transcripts following anoxia exposure in a marine invertebrate. J Exp Biol. 2004;207:1353–1360. doi: 10.1242/jeb.00872. [DOI] [PubMed] [Google Scholar]
- 56.Storey KB. Strategies for exploration of freeze responsive gene expression: advances in vertebrate freeze tolerance. Cryobiology. 2004;48:134–145. doi: 10.1016/j.cryobiol.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 57.Soares MP, Bach FH. Heme oxygenase-1: from biology to therapeutic potential. Trends Mol Me. 2009;15:50–58. doi: 10.1016/j.molmed.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 58.Vile GF, Basu-Modak S, Waltner C, Tyrrell RM. Heme oxygenase 1 mediates an adaptive response to oxidative stress in human skin fibroblasts. Proc Natl Acad Sci USA. 1994;91:2607–2610. doi: 10.1073/pnas.91.7.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kesaraju S, Schmidt-Kastner R, Prentice HM, Milton SL. Modulation of stress proteins and apoptotic regulators in the anoxia tolerant turtle brain. J Neurochem. 2009;109:1413–1426. doi: 10.1111/j.1471-4159.2009.06068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Motterlini R, Foresti R, Intaglietta M, Winslow RM. NO-mediated activation of heme oxygenase: endogenous cytoprotection against oxidative stress to endothelium. Am J Physiol. 1996;270:107–114. doi: 10.1152/ajpheart.1996.270.1.H107. [DOI] [PubMed] [Google Scholar]
- 61.Doi K, Akaike T, Fujii S, Tanaka S, Ikebe N, Beppu T, et al. Induction of haem oxygenase-1 nitric oxide and ischaemia in experimental solid tumours and implications for tumour growth. Br J Cancer. 1999;80:1945–1954. doi: 10.1038/sj.bjc.6690624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Motterlini R, Foresti R, Bassi R, Calabrese V, Clark JE, Green CJ. Endothelial heme oxygenase-1 induction by hypoxia. Modulation by inducible nitric-oxide synthase and S-nitrosothiols. J Biol Chem. 2000;275:13613–13620. doi: 10.1074/jbc.275.18.13613. [DOI] [PubMed] [Google Scholar]
- 63.Abraham NG, Kappas A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol Rev. 2008;60:79–127. doi: 10.1124/pr.107.07104. [DOI] [PubMed] [Google Scholar]
- 64.Ollinger R, Wang H, Yamashita K, Wegiel B, Thomas M, Margreiter R, et al. Therapeutic applications of bilirubin and biliverdin in transplantation. Antioxid Redox Signal. 2007;9:2175–2185. doi: 10.1089/ars.2007.1807. [DOI] [PubMed] [Google Scholar]
- 65.Idriss NK, Blann AD, Lip GY. Hemoxygenase-1 in cardiovascular disease. J Am Coll Cardiol. 2008;52:971–978. doi: 10.1016/j.jacc.2008.06.019. [DOI] [PubMed] [Google Scholar]
- 66.Hemmings SJ, Storey KB. Characterization of gammaglutamyltranspeptidase in the liver of the frog: 3. Response to freezing and thawing in the freeze-tolerant wood frog Rana sylvatica. Cell Biochem Funct. 1996;14:139–148. doi: 10.1002/cbf.661. [DOI] [PubMed] [Google Scholar]
- 67.Hemmings SJ, Storey KB. Hepatic changes in the freeze-tolerant turtle Chrysemys picta marginata in response to freezing and thawing. Cell Biochem Funct. 2000;18:175–186. doi: 10.1002/1099-0844(200009)18:3<175::AID-CBF871>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 68.Dinkelacker SA, Costanzo JP, Lee RE., Jr Anoxia tolerance and freeze tolerance in hatchling turtles. J Comp Physiol B. 2005;175:209–217. doi: 10.1007/s00360-005-0478-0. [DOI] [PubMed] [Google Scholar]
- 69.Krivoruchko A, Storey KB. Activation of antioxidant defenses in response to freezing in freeze tolerant painted turtle hatchlings. Biochim Biophys Acta. 2010;1800:662–668. doi: 10.1016/j.bbagen.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 70.Rhee SG, Chae H, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med. 2005;38:1543–1552. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 71.Rhee SG, Kang SW, Jeong W, Chang TS, Yang KS, Woo HA. Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr Opin Cell Biol. 2005;17:183–189. doi: 10.1016/j.ceb.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 72.Eddy F, McNally JD, Storey KB. Upregulation of a thioredoxin peroxidase-like protein, proliferation associated gene, in hibernating bats. Arch Biochem Biophys. 2005;435:101–111. doi: 10.1016/j.abb.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 73.Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008;22:1427–1438. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lindquist S, Craig EA. The heat-shock proteins. Annu Rev Genet. 1988;22:631–677. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- 75.Winter J, Jakob U. Beyond transcription—new mechanisms for the regulation of molecular chaperones. Crit Rev Biochem Mol Biol. 2004;39:297–317. doi: 10.1080/10409230490900658. [DOI] [PubMed] [Google Scholar]
- 76.Kalmar B, Greensmith L. Induction of heat shock proteins for protection against oxidative stress. Adv Drug Deliv Rev. 2009;61:310–318. doi: 10.1016/j.addr.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 77.Chang J, Knowlton AA, Wasser JS. Expression of heat shock proteins in turtle and mammal hearts: relationship to anoxia tolerance. Am J Physiol. 2000;278:209–214. doi: 10.1152/ajpregu.2000.278.1.R209. [DOI] [PubMed] [Google Scholar]
- 78.Hollander JM, Lin KM, Scott BT, Dillmann WH. Overexpression of PHGPx and HSP60/10 protects against ischemia/reoxygenation injury. Free Radic Biol Med. 2003;35:742–751. doi: 10.1016/s0891-5849(03)00400-3. [DOI] [PubMed] [Google Scholar]
- 79.Colotti C, Cavallini G, Vitale RL, Donati A, Maltinti M, Del Ry S, et al. Effects of aging and anti-aging caloric restrictions on carbonyl and heat shock protein levels and expression. Biogerontology. 2005;6:397–406. doi: 10.1007/s10522-005-4906-z. [DOI] [PubMed] [Google Scholar]
- 80.Prentice HM, Milton SL, Scheurle D, Lutz PL. The upregulation of cognate and inducible heat shock proteins in the anoxic turtle brain. J Cereb Blood Flow Metab. 2004;24:826–828. doi: 10.1097/01.WCB.0000126565.27130.79. [DOI] [PubMed] [Google Scholar]
- 81.Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62:670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008;22:1427–1438. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ramaglia V, Buck LT. Time-dependent expression of heat shock proteins 70 and 90 in tissues of the anoxic western painted turtle. J Exp Biol. 2004;207:3775–3784. doi: 10.1242/jeb.01211. [DOI] [PubMed] [Google Scholar]
- 84.Krivoruchko A, Storey KB. Regulation of the heat shock response under anoxia in the turtle, Trachemys scripta elegans. J Comp Physiol B. 2010;180:403–414. doi: 10.1007/s00360-009-0414-9. [DOI] [PubMed] [Google Scholar]
- 85.Head MW, Goldman JE. Small heat shock proteins, the cytoskeleton and inclusion body formation. Neuropathol Appl Neurobiol. 2000;26:304–312. doi: 10.1046/j.1365-2990.2000.00269.x. [DOI] [PubMed] [Google Scholar]
- 86.Goldbaum O, Richter-Landsberg C. Stress proteins in oligodendrocytes: differential effects of heat shock and oxidative stress. J Neurochem. 2001;78:1233–1242. doi: 10.1046/j.1471-4159.2001.00507.x. [DOI] [PubMed] [Google Scholar]
- 87.Arrigo AP, Virot S, Chaufour S, Firdaus W, Kretz-Remy C, Diaz-Latoud C. Hsp27 consolidates intracellular redox homeostasis by upholding glutathione in its reduced form and by decreasing iron intracellular levels. Antioxid Redox Signal. 2005;7:414–422. doi: 10.1089/ars.2005.7.414. [DOI] [PubMed] [Google Scholar]
- 88.Arrigo AP. The cellular “networking” of mammalian Hsp27 and its functions in the control of protein folding, redox state and apoptosis. Adv Exp Med Biol. 2007;594:14–26. doi: 10.1007/978-0-387-39975-1_2. [DOI] [PubMed] [Google Scholar]
- 89.Pandey P, Saleh A, Nakazawa A, Kumar S, Srinivasula SM, Kumar V, et al. Negative regulation of cytochrome c-mediated oligomerization of Apaf-1 and activation of procaspase-9 by heat shock protein 90. EMBO J. 2000;19:4310–4322. doi: 10.1093/emboj/19.16.4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, et al. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol. 2000;2:469–475. doi: 10.1038/35019501. [DOI] [PubMed] [Google Scholar]
- 91.Matsumori Y, Hong SM, Aoyama K, Fan Y, Kayama T, Sheldon RA, et al. Hsp70 overexpression sequesters AIF and reduces neonatal hypoxic/ischemic brain injury. J Cereb Blood Flow Metab. 2005;25:899–910. doi: 10.1038/sj.jcbfm.9600080. [DOI] [PubMed] [Google Scholar]
- 92.Gorman AM, Szegezdi E, Quigney DJ, Samali A. Hsp27 inhibits 6-hydroxydopamineinduced cytochrome c release and apoptosis in PC12 cells. Biochem Biophys Res Commun. 2005;327:801–810. doi: 10.1016/j.bbrc.2004.12.066. [DOI] [PubMed] [Google Scholar]
- 93.Bruey JM, Ducasse C, Bonniaud P, Ravagnan L, Susin SA, Diaz-Latoud C, et al. Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat Cell Biol. 2000;2:645–652. doi: 10.1038/35023595. [DOI] [PubMed] [Google Scholar]
- 94.Winklhofer KF, Tatzelt J, Haass C. The two faces of protein misfolding: gain-and loss-of-function in neurodegenerative diseases. EMBO J. 2008;27:336–349. doi: 10.1038/sj.emboj.7601930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kayani AC, Morton JP, McArdle A. The exercise-induced stress response in skeletal muscle: failure during aging. Appl Physiol Nutr Metab. 2008;33:1033–1041. doi: 10.1139/H08-089. [DOI] [PubMed] [Google Scholar]
- 96.Gagliano N, Grizzi F, Annoni G. Mechanisms of aging and liver functions. Digest Dis. 2007;25:118–123. doi: 10.1159/000099475. [DOI] [PubMed] [Google Scholar]
- 97.Pahl HL. Activators and target genes of Rel/NFkappaB transcription factors. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 98.Latanich CA, Toledo-Pereyra LH. Searching for NFkappaB-based treatments of ischemia reperfusion injury. J Invest Surg. 2009;22:301–315. doi: 10.1080/08941930903040155. [DOI] [PubMed] [Google Scholar]
- 99.Lutz PL, Prentice HM. Sensing and responding to hypoxia, molecular and physiological mechanisms. Integr Comp Biol. 2002;42:436–468. doi: 10.1093/icb/42.3.463. [DOI] [PubMed] [Google Scholar]
- 100.Santore MT, McClintock DS, Lee VY, Budinger GR, Chandel NS. Anoxia-induced apoptosis occurs through a mitochondria-dependent pathway in lung epithelial cells. Am J Physiol. 2002;282:727–734. doi: 10.1152/ajplung.00281.2001. [DOI] [PubMed] [Google Scholar]
- 101.Papandreou I, Krishna C, Kaper F, Cai D, Giaccia AJ, Denko NC. Anoxia is necessary for tumor cell toxicity caused by a low-oxygen environment. Cancer Res. 2005;65:3171–3178. doi: 10.1158/0008-5472.CAN-04-3395. [DOI] [PubMed] [Google Scholar]
- 102.Gupta S. Molecular steps of death receptor and mitochondrial pathways of apoptosis. Life Sci. 2001;69:2954–2957. doi: 10.1016/s0024-3205(01)01404-7. [DOI] [PubMed] [Google Scholar]
- 103.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 104.Janumyan YM, Sansam CG, Chattopadhyay A, Cheng N, Soucie EL, Penn LZ, et al. Bcl-XL/Bcl-2 coordinately regulates apoptosis, cell cycle arrest and cell cycle entry. EMBO J. 2003;22:5459–5470. doi: 10.1093/emboj/cdg533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.McClintock DS, Santore MT, Lee VY, Brunelle J, Budinger GR, Zong WX, et al. Bcl-2 family members and functional electron transport chain regulate oxygen deprivation-induced cell death. Mol Cell Biol. 2002;22:94–104. doi: 10.1128/MCB.22.1.94-104.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Piret JP, Minet E, Cosse JP, Ninane N, Debacq C, Raes M, et al. Hypoxia-inducible factor-1-dependent overexpression of myeloid cell factor-1 protects hypoxic cells against tert-butyl hydroperoxide-induced apoptosis. J Biol Chem. 2005;280:9336–9344. doi: 10.1074/jbc.M411858200. [DOI] [PubMed] [Google Scholar]
- 107.Peralta C, Perales JC, Bartrons R, Mitchell C, Gilgenkrantz H, Xaus C, et al. The combination of ischemic preconditioning and liver Bcl-2 overexpression is a suitable strategy to prevent liver and lung damage after hepatic ischemia-reperfusion. Am J Pathol. 2002;160:2111–2122. doi: 10.1016/S0002-9440(10)61160-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hu GH, Lu XS. Effect of normothermic liver ischemic preconditioning on the expression of apoptosis-regulating genes C-jun and Bcl-XL in rats. World J Gastroenterol. 2005;11:2579–2582. doi: 10.3748/wjg.v11.i17.2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hockenbery DM, Oltvai ZN, Yin XM, Milliman C, Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75:241–251. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- 110.Kane DJ, Sarafian TA, Anton R, Hahn H, Gralla EB, Valentine JS, et al. Bcl-2 inhibition of neural death: decreased generation of reactive oxygen species. Science. 1993;262:1274–1277. doi: 10.1126/science.8235659. [DOI] [PubMed] [Google Scholar]
- 111.Giardino I, Edelstein D, Brownlee M. BCL-2 expression or antioxidants prevent hyperglycemia-induced formation of intracellular advanced glycation endproducts in bovine endothelial cells. J Clin Invest. 1996;97:1422–1428. doi: 10.1172/JCI118563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lutz PL, Nilsson GE, Prentice HM. The Brain without Oxygen: Causes of Failure Molecular and Physiological Mechanisms for Survival. Third ed. Dordrecht: Kluwer; 2003. [Google Scholar]
- 113.Prentice HM, Milton SL, Scheurle D, Lutz PL. Gene transcription of brain voltage-gated potassium channels is reversibly regulated by oxygen supply. Am J Physiol. 2003;285:1317–1321. doi: 10.1152/ajpregu.00261.2003. [DOI] [PubMed] [Google Scholar]
- 114.Pek M, Lutz PL. ATP-sensitive K+ channel activation provides transient protection to the anoxic turtle brain. Am J Physiol. 1998;44:2023–2027. doi: 10.1152/ajpregu.1998.275.6.R2023. [DOI] [PubMed] [Google Scholar]
- 115.Bickler PE, Donohoe PH, Buck LT. Hypoxia-induced silencing of NMDA receptors in turtle neurons. J Neurosci. 2000;20:3522–3528. doi: 10.1523/JNEUROSCI.20-10-03522.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Annunziato L, Pannaccione A, Cataldi M, Secondo A, Castaldo P, Di Renzo G, et al. Modulation of ion channels by reactive oxygen and nitrogen species: a pathophysiological role in brain aging? Neurobiol Aging. 2002;23:819–834. doi: 10.1016/s0197-4580(02)00069-6. [DOI] [PubMed] [Google Scholar]
- 117.Milton SL, Lutz PL. Low extracellular dopamine levels are maintained in the anoxic turtle brain. J Cereb Blood Flow Metab. 1998;18:803–807. doi: 10.1097/00004647-199807000-00010. [DOI] [PubMed] [Google Scholar]
- 118.Milton SL, Thompson JW, Lutz PL. Mechanisms for maintaining extracellular glutamate in the anoxic turtle striatum. Am J Physiol. 2002;282:1317–1323. doi: 10.1152/ajpregu.00484.2001. [DOI] [PubMed] [Google Scholar]
- 119.Buck LT. Adenosine as a signal for ion channel arrest in anoxia-tolerant organisms. Comp Biochem Physiol B. 2004;139:401–414. doi: 10.1016/j.cbpc.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 120.Milton SL, Nayak G, Kesaraju S, Kara L, Prentice HM. Suppression of reactive oxygen species production enhances neuronal survival in vitro and in vivo in the anoxia-tolerant turtle Trachemys scripta. J Neurochem. 2007;101:993–1001. doi: 10.1111/j.1471-4159.2007.04466.x. [DOI] [PubMed] [Google Scholar]
- 121.Brewer G. Age-related toxicity to lactate, glutamate and β-amyloid in cultured adult neurons. Neurobiol Aging. 1998;19:561–568. doi: 10.1016/s0197-4580(98)00091-8. [DOI] [PubMed] [Google Scholar]
- 122.Gelain DP, De Souza LF, Ribeiro GR, Zim M, Jardim FR, Moreira JC, et al. Extracellular inosine is modulated by H2O2 and protects sertoli cells against lipoperoxidation and cellular injury. Free Radic Res. 2004;38:37–47. doi: 10.1080/10715760310001629056. [DOI] [PubMed] [Google Scholar]
- 123.Buckley S, Barsky L, Weinberg K, Warburton D. In vivo inosine protects alveolar epithelial type 2 cells against hyperoxia-induced DNA damage through MAP kinase signaling. Am J Physiol. 2005;288:569–575. doi: 10.1152/ajplung.00278.2004. [DOI] [PubMed] [Google Scholar]
- 124.Tomaselli B, Podhraski V, Heftberger V, Böck G, Baier-Bitterlich G. Purine nucleoside-mediated protection of chemical hypoxia-induced neuronal injuries involves p42/44 MAPK activation. Neurochem Int. 2005;46:513–521. doi: 10.1016/j.neuint.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 125.Burmester T, Weich B, Reinhardt S, Hankeln T. A vertebrate globin expressed in the brain. Nature. 2000;407:520–523. doi: 10.1038/35035093. [DOI] [PubMed] [Google Scholar]
- 126.Reuss S, Saaler-Reinhardt S, Weich B, Wystub S, Reuss M, Burmester T, et al. Expression analysis of neuroglobin mRNA in rodent tissues. Neuroscience. 2002;115:645–656. doi: 10.1016/s0306-4522(02)00536-5. [DOI] [PubMed] [Google Scholar]
- 127.Burmester T, Hankeln T. Neuroglobin: A respiratory protein of the nervous system. News Physiol Sci. 2004;19:110–113. doi: 10.1152/nips.01513.2003. [DOI] [PubMed] [Google Scholar]
- 128.Milton SL, Nayak G, Lutz PL, Prentice HM. Gene transcription of neuroglobin is upregulated by hypoxia and anoxia in the brain of the anoxia-tolerant turtle Trachemys scripta. J Biomed Sci. 2006;13:509–514. doi: 10.1007/s11373-006-9084-8. [DOI] [PubMed] [Google Scholar]
- 129.Weber RE, Vinogradov SN. Nonvertebrate hemoglobins: functions and molecular adaptations. Physiol Rev. 2001;81:569–628. doi: 10.1152/physrev.2001.81.2.569. [DOI] [PubMed] [Google Scholar]
- 130.Kriegl JM, Bhattacharyya AJ, Nienhaus K, Deng P, Minkow O, Nienhaus GU. Ligand binding and protein dynamics in neuroglobin. Proc Natl Acad Sci USA. 2002;99:7992–7997. doi: 10.1073/pnas.082244399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Herold S, Fago A, Weber RE, Dewilde S, Moens L. Reactivity studies of the Fe(III) and Fe(II)NO forms of human neuroglobin reveal a potential role against oxidative stress. J Biol Chem. 2004;279:22841–22847. doi: 10.1074/jbc.M313732200. [DOI] [PubMed] [Google Scholar]
- 132.Burmester T, Gerlach F, Hankeln T. Regulation and role of neuroglobin and cytoglobin under hypoxia. Adv Exp Med Biol. 2007;618:169–180. doi: 10.1007/978-0-387-75434-5_13. [DOI] [PubMed] [Google Scholar]
- 133.Szymanski M, Wang R, Fallin MD, Bassett SS, Avramopoulos D. Neuroglobin and Alzheimer's dementia: Genetic association and gene expression changes. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.10.003. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Khan AA, Mao XO, Banwait S, Jin K, Greenberg DA. Neuroglobin attenuates beta-amyloid neurotoxicity in vitro and transgenic Alzheimer phenotype in vivo. Proc Natl Acad Sci USA. 2007;104:19114–1949. doi: 10.1073/pnas.0706167104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Raychaudhuri S, Skommer J, Henty K, Birch N, Brittain T. Neuroglobin protects nerve cells from apoptosis by inhibiting the intrinsic pathway of cell death. Apoptosis. 2010;15:401–411. doi: 10.1007/s10495-009-0436-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Nayak G, Prentice HM, Milton SL. Role of neuroglobin in regulating reactive oxygen species in the brain of the anoxia-tolerant turtle Trachemys scripta. J Neurochem. 2009;110:603–162. doi: 10.1111/j.1471-4159.2009.06157.x. [DOI] [PubMed] [Google Scholar]
- 137.Roesner A, Hankeln T, Burmester T. Hypoxia induces a complex response of globin expression in zebrafish (Danio rerio) J Exp Biol. 2006;209:2129–2137. doi: 10.1242/jeb.02243. [DOI] [PubMed] [Google Scholar]
- 138.Storey KB, Storey JM. Putting life on ‘pause’—molecular regulation of hypometabolism. J Exp Biol. 2007;210:1700–1714. doi: 10.1242/jeb.02716. [DOI] [PubMed] [Google Scholar]
- 139.Storey KB, Storey JM. Metabolic rate depression in animals: transcriptional and translational controls. Biol Rev Camb Philos Soc. 2004;79:207–233. doi: 10.1017/s1464793103006195. [DOI] [PubMed] [Google Scholar]
- 140.Hochachka PW, Buck LT, Doll CJ, Land SC. Unifying theory of hypoxia tolerance: molecular/metabolic defense and rescue mechanisms for surviving oxygen lack. Proc Natl Acad Sci USA. 1996;93:9493–9498. doi: 10.1073/pnas.93.18.9493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Fraser KP, Houlihan DF, Lutz PL, Leone-Kabler S, Manuel L, Brechin JG. Complete suppression of protein synthesis during anoxia with no post-anoxia protein synthesis debt in the red-eared slider turtle Trachemys scripta elegans. J Exp Biol. 2001;204:4353–4360. doi: 10.1242/jeb.204.24.4353. [DOI] [PubMed] [Google Scholar]
- 142.Hochachka PW, Lutz PL. Mechanism, origin and evolution of anoxia tolerance in animals. Comp Biochem Physiol. 2001;130:435–459. doi: 10.1016/s1096-4959(01)00408-0. [DOI] [PubMed] [Google Scholar]
- 143.Bickler PE, Donohoe PH, Buck LT. The hypoxic brain: suppressing energy-expensive membrane functions by regulation of receptors and ion channels. In: Storey KB, editor. Molecular Mechanisms of Metabolic Arrest. Oxford: BIOS Scientific Publishers; 2001. pp. 77–102. [Google Scholar]
- 144.Bickler PE, Buck LT. Hypoxia tolerance in reptiles, amphibians and fishes: life with variable oxygen availability. Annu Rev Physiol. 2007;69:145–170. doi: 10.1146/annurev.physiol.69.031905.162529. [DOI] [PubMed] [Google Scholar]
- 145.Rider MH, Hussain N, Dilworth SM, Storey KB. Phosphorylation of translation factors in response to anoxia in turtles, Trachemys scripta elegans: role of the AMP-activated protein kinase and target of rapamycin signalling pathways. Mol Cell Biochem. 2009;332:207–213. doi: 10.1007/s11010-009-0193-3. [DOI] [PubMed] [Google Scholar]
- 146.Rolfe DF, Brown GC. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev. 1997;77:731–758. doi: 10.1152/physrev.1997.77.3.731. [DOI] [PubMed] [Google Scholar]
- 147.Van Breukelen F, Maier R, Hand SC. Depression of nuclear transcription and extension of mRNA half-life under anoxia in Artemia franciscana embryos. J Exp Biol. 2000;203:1123–1130. doi: 10.1242/jeb.203.7.1123. [DOI] [PubMed] [Google Scholar]
- 148.Larade K, Storey KB. A profile of the metabolic responses to anoxia in marine invertebrates. In: Storey KB, Storey JM, editors. Sensing, Signaling and Cell Adaptation. Amsterdam: Elsevier Science; 2002. pp. 27–46. [Google Scholar]
- 149.Bocharova LS, Gordon RY, Arkhipov VI. Uridine uptake and RNA synthesis in the brain of torpid and awakened ground squirrels. Comp Biochem Physiol B. 1992;101:189–192. doi: 10.1016/0305-0491(92)90177-s. [DOI] [PubMed] [Google Scholar]
- 150.Van Breukelen F, Martin SL. Reversible depression of transcription during hibernation. J Comp Physiol B. 2002;172:355–361. doi: 10.1007/s00360-002-0256-1. [DOI] [PubMed] [Google Scholar]
- 151.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci USA. 1964;51:786–794. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Spotswood HT, Turner BM. An increasingly complex code. J Clin Invest. 2002;110:577–582. doi: 10.1172/JCI16547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hebbes TR, Thorne AW, Crane-Robinson C. A direct link between core histone acetylation and transcriptionally active chromatin. EMBO J. 1988;7:1395–1402. doi: 10.1002/j.1460-2075.1988.tb02956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Morin P, Storey KB. Evidence for a reduced transcriptional state during hibernation in ground squirrels. Cryobiology. 2006;53:310–318. doi: 10.1016/j.cryobiol.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 155.Krivoruchko A, Storey KB. Epigenetics in anoxia tolerance: a role for histone deacetylases. Mol Cell Biochem. 2010 doi: 10.1007/s11010-010-0479-5. In press. [DOI] [PubMed] [Google Scholar]
- 156.Austad SN, Fischer KE. Mammalian aging, metabolism and ecology: evidence from the bats and marsupials. J Gerontol. 1991;46:47–53. doi: 10.1093/geronj/46.2.b47. [DOI] [PubMed] [Google Scholar]
- 157.Wilkinson GS, South JM. Life history, ecology and longevity in bats. Aging Cell. 2002;1:124–131. doi: 10.1046/j.1474-9728.2002.00020.x. [DOI] [PubMed] [Google Scholar]
- 158.Podlutsky AJ, Khritankov AM, Ovodov ND, Austad SN. A new field record for bat longevity. J Gerontol A. 2005;60:1366–1368. doi: 10.1093/gerona/60.11.1366. [DOI] [PubMed] [Google Scholar]
- 159.O'Connor TP, Lee A, Jarvis JU, Buffenstein R. Prolonged longevity in naked mole-rats: age-related changes in metabolism, body composition and gastrointestinal function. Comp Biochem Physiol A. 2002;133:835–842. doi: 10.1016/s1095-6433(02)00198-8. [DOI] [PubMed] [Google Scholar]
- 160.Harper JM, Salmon AB, Leiser SF, Galecki AT, Miller RA. Skin-derived fibroblasts from long-lived species are resistant to some, but not all, lethal stresses and to the mitochondrial inhibitor rotenone. Aging Cell. 2007;6:1–13. doi: 10.1111/j.1474-9726.2006.00255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Nathaniel TI, Saras A, Umesiri FE, Olajuyigbe F. Tolerance to oxygen nutrient deprivation in the hippocampal slices of the naked mole rats. J Integr Neurosci. 2009;8:123–136. doi: 10.1142/s0219635209002149. [DOI] [PubMed] [Google Scholar]
- 162.Larson J, Park TJ. Extreme hypoxia tolerance of naked mole-rat brain. Neuroreport. 2009;20:1634–1637. doi: 10.1097/WNR.0b013e32833370cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Labinskyy N, Csiszar A, Orosz Z, Smith K, Rivera A, Buffenstein R, Ungvari Z. Comparison of endothelial function, O2-• and H2O2 production and vascular oxidative stress resistance between the longest-living rodent, the naked mole rat and mice. Am J Physiol. 2006;291:2698–2704. doi: 10.1152/ajpheart.00534.2006. [DOI] [PubMed] [Google Scholar]
- 164.Salmon AB, Sadighi Akha A, Buffenstein R, Miller RA. Fibroblasts from naked mole-rats are resistant to multiple forms of cell injury, but sensitive to peroxide, UV light and ER stress. J Gerontol A. 2008;63:232–241. doi: 10.1093/gerona/63.3.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Buffenstein R. Negligible senescence in the longest living rodent, the naked mole-rat: Insights from a successfully aging species. J Comp Physiol B. 2008;178:439–445. doi: 10.1007/s00360-007-0237-5. [DOI] [PubMed] [Google Scholar]
- 166.Brunet-Rossinni AK. Reduced free-radical production and extreme longevity in the little brown bat (Myotis lucifugus) versus two non-flying mammals. Mech Ageing Dev. 2004;125:11–20. doi: 10.1016/j.mad.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 167.Brown JC, McClelland GB, Faure PA, Klaiman JM, Staples JF. Examining the mechanisms responsible for lower ROS release rates in liver mitochondria from the long-lived house sparrow (Passer domesticus) and big brown bat (Eptesicus fuscus) compared to the short-lived mouse (Mus musculus) Mech Ageing Dev. 2009;130:467–476. doi: 10.1016/j.mad.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 168.Salmon AB, Leonard S, Masamsetti V, Pierce A, Podlutsky AJ, Podlutskaya N, et al. The long lifespan of two bat species is correlated with resistance to protein oxidation and enhanced protein homeostasis. FASEB J. 2009;23:2317–2326. doi: 10.1096/fj.08-122523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Pérez VI, Buffenstein R, Masamsetti V, Leonard S, Salmon AB, Mele J, et al. Protein stability and resistance to oxidative stress are determinants of longevity in the longest-living rodent, the naked mole-rat. Proc Natl Acad Sci USA. 2009;106:3059–3064. doi: 10.1073/pnas.0809620106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Storey KB. Out cold: biochemical regulation of mammalian hibernation—a mini-review. Gerontology. 2010;56:220–230. doi: 10.1159/000228829. [DOI] [PubMed] [Google Scholar]
- 171.Storey KB. Cold ischemic organ preservation: lessons from natural systems. J Invest Med. 2004;52:315–322. doi: 10.1136/jim-52-05-31. [DOI] [PubMed] [Google Scholar]
- 172.Nozari A, Safar P, Wu X, Stezoski WS, Henchir J, Kochanek P, et al. Suspended animation can allow survival without brain damage after traumatic exsanguination cardiac arrest of 60 minutes in dogs. J Trauma. 2004;57:1266–1275. doi: 10.1097/01.ta.0000124268.24159.8b. [DOI] [PubMed] [Google Scholar]
- 173.Bos EM, Leuvenink HG, Snijder PM, Kloosterhuis NJ, Hillebrands JL, Leemans JC, et al. Hydrogen sulfide-induced hypometabolism prevents renal ischemia/reperfusion injury. J Am Soc Nephrol. 2009;20:1901–1905. doi: 10.1681/ASN.2008121269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Blackstone E, Roth MB. Suspended animation-like state protects mice from lethal hypoxia. Shock. 2007;27:370–372. doi: 10.1097/SHK.0b013e31802e27a0. [DOI] [PubMed] [Google Scholar]
- 175.Ridder DA, Schwaninger M. NFkappaB signaling in cerebral ischemia. Neuroscience. 2009;158:995–1006. doi: 10.1016/j.neuroscience.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 176.Sarnico I, Lanzillotta A, Benarese M, Alghisi M, Baiguera C, Battistin L, et al. NFkappaB dimers in the regulation of neuronal survival. Int Rev Neurobiol. 2009;85:351–362. doi: 10.1016/S0074-7742(09)85024-1. [DOI] [PubMed] [Google Scholar]
- 177.Gutiérrez SH, Kuri MR, del Castillo ER. Cardiac role of the transcription factor NFkappaB. Cardiovasc Hematol Disord Drug Targets. 2008;8:153–160. doi: 10.2174/187152908784533702. [DOI] [PubMed] [Google Scholar]
- 178.Luedde T, Trautwein C. Intracellular survival pathways in the liver. Liver Int. 2006;26:1163–1174. doi: 10.1111/j.1478-3231.2006.01366.x. [DOI] [PubMed] [Google Scholar]
- 179.Yu Z, Fan X, Lo EH, Wang X. Neuroprotective roles and mechanisms of neuroglobin. Neurol Res. 2009;31:122–127. doi: 10.1179/174313209X389866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Obrenovitch TP. Molecular physiology of preconditioning-induced brain tolerance to ischemia. Physiol Rev. 2008;88:211–247. doi: 10.1152/physrev.00039.2006. [DOI] [PubMed] [Google Scholar]
- 181.Elahi MM, Kong YX, Matata BM. Oxidative stress as a mediator of cardiovascular disease. Oxid Med Cell Longev. 2009;2:259–269. doi: 10.4161/oxim.2.5.9441. [DOI] [PMC free article] [PubMed] [Google Scholar]