

Abstract
About 246 million people worldwide had diabetes in 2007. The global figure of people with diabetes is projected to increase to 370 million in 2030. As the prevalence of diabetes has risen to epidemic proportions worldwide, diabetic nephropathy has become one of the most challenging health problems. Therapeutic options such as strict blood glucose and blood pressure controls are effective for preventing diabetic nephropathy, but are far from satisfactory, and the number of diabetic patients on end-stage renal disease is still increasing. Therefore, a novel therapeutic strategy that could halt the progression of diabetic nephropathy should be developed. There is accumulating evidence that advanced glycation end products (AGEs), senescent macroprotein derivatives formed at an accelerated rate under diabetes, play a role in diabetic nephropathy via oxidative stress generation. In this paper, we review the pathophysiological role of AGEs and their receptor (RAGE)-oxidative stress system in diabetic nephropathy.
Key words: diabetic nephropathy, AGEs, RAGE, oxidative stress, renin-angiotensin system
Introduction
Diabetic nephropathy is the most common cause of end-stage renal disease in the world, and could account for disability and high mortality rate in patients with diabetes. According to the World Health Organization, it is expected that the number of patients with diabetes will rise to 370 million by 2030 in the world.1 It has also been reported that about 25–40% of type 1 or type 2 diabetic patients develop diabetic nephropathy within 20–25 year of the onset of diabetes.2 Large clinical trials such as United Kingdom Prospective Diabetes Study (UKPDS) and Diabetes Control and Complications Trial (DCCT) revealed that strict control of blood glucose or blood pressure significantly reduced the development and progression of diabetic nephropathy in both type 1 and type 2 diabetes.3,4 However, current therapeutic options are far from satisfactory, and the effects of intensive therapy on diabetic nephropathy are insufficient. Indeed, intensive therapy of blood glucose and/or blood pressure is often difficult to maintain and may increase the risk of hypoglycemia and/or hypotension in diabetic patients, and number of diabetic patients with end-stage renal failure is still increasing in industrialized countries. Therefore, development of novel therapeutic strategies that could specially target diabetic nephropathy is urgently needed.
Various hyperglycemia-induced metabolic and hemodynamic derangements, including increased formation of advanced glycation end products (AGEs), enhanced reactive oxygen species (ROS) generation and activation of protein kinase C (PKC), polyol pathway and renin-angiotensin system (RAS), are considered to contribute to the development and progression of diabetic nephropathy.5 However, a follow-up study of DCCT called DCCT-Epidemiology of Diabetes Interventions and Complications (DCCT-EDIC) study, has shown that the reduction in the risk of progressive retinopathy and nephropathy resulting from intensive therapy in patients with type 1 diabetes persisted for at least several years, despite increasing hyperglycemia.6,7 Intensive therapy during the DCCT resulted in decreased progression of intimamedia thickness and subsequently reduced the risk of nonfatal myocardial infarction, stroke, or death from cardiovascular disease by 57 percent 11 years after the end of the trials.8,9 Further, a recent follow-up study of UKPDS called UKPDS80 also has shown that benefits of an intensive therapy in patients with type 2 diabetes are sustained after the cessation of the trial.10 In this study, despite an early loss of glycemic differences between intensive and conventional therapy, a continued reduction in microvascular risk and emergent risk reductions for myocardial infarction and death from any cause were observed during 10 years of post-trial follow-up.10 These observations indicate that intensive therapy to control blood glucose has long-term beneficial effects (legacy effects) on the risk of diabetic retinopathy, nephropathy, cardiovascular disease (CVD) and death in patients with both type 1 and type 2 diabetes, strongly suggesting that so-called ‘metabolic memory’ could cause chronic damage in diabetic vessels that are not easily reversed, even by subsequent, relatively good control of blood glucose.
Reducing sugars can react non-enzymatically with the amino groups of proteins to form reversible Schiff bases, and then Amadori products.5,11–14 These early glycation products undergo further complex reactions such as rearrangement, dehydration and condensation to become irreversibly cross-linked, heterogeneous fluorescent derivatives termed AGEs.5,11–14 The formation and accumulation of AGEs have been reported to progress at an accelerated rate under diabetes. The pathological role of the nonenzymatic glycation of proteins has become increasingly evident in various disorders including diabetic vascular complications.5,15–19 Furthermore, there is a growing body of evidence that AGEs and their signal-transducing receptor interaction evokes oxidative stress and subsequently elicits vascular inflammation and thrombosis, thereby playing a central role in the pathogenesis of vascular complications in diabetes.5,20–24
Although the exact pathophysiological mechanisms responsible for a legacy effect of the intensive glycemic control or metabolic memory are unclear, biochemical nature of AGEs, that is, gradual accumulation of AGEs under hyperglycemic conditions that are subsequently slowly degraded with intensive glycemic control, is compatible with the concept ‘metabolic memory’.5,11–14 Further, AGEs and/or diabetic conditions are shown to upregulate receptor for AGEs (RAGE) expression in various cell types and induce sustained activation of transcriptional factor nuclear factor-κB (NFκB).5,25–27 Therefore, it is conceivable that the AGE-RAGE-induced oxidative stress generation further potentiates the formation and accumulation of AGEs under diabetic conditions, 28 and these positive feedback loops between AGEs and RAGE-downstream pathways could make a vicious cycle, thus providing a mechanistic basis for understanding the concept of ‘metabolic memory’ in vascular complications in diabetes. Since the above-mentioned observations suggest that the AGE-RAGE-induced ROS generation is a novel molecular target for vascular complications in diabetes, we review here the pathophysiological role of AGE-RAGE-oxidative stress system and its therapeutic interventions in diabetic nephropathy.
Role of AGEs in Diabetic Nephropathy
In type 1 and type 2 diabetic patients, serum and tissue AGEs levels were significantly increased compared with non-diabetic control subjects.29,30 Further, diabetic patients with end-stage renal disease had almost twice as much AGEs in tissue as diabetic patients without renal disease.29 Both enhanced formation and decreased clearance are responsible for the accumulation of AGEs in patients with diabetic nephropathy.31,32 Indeed, serum levels of low molecular weight-AGEs, which are degradated products of tissue AGEs by macrophages, are reported to increase in diabetic and non-diabetic subjects with renal dysfunction.33–35 Since low molecular weight-AGEs are filtered by the kidney and then reabsorbed by proximal tubular cells,36 decreased glomerular filtration rate and tubulointerstitial cell damage could also be involved in AGEs accumulation in patients with diabetic nephropathy. Further, we have previously found that non-Ne-(carboxymethyl)lysine (CML) AGEs are increased in parallel to the severity of diabetic nephropathy in type 1 diabetic patients.37 AGEs-modified collage levels in the skin are reported to be one of the predictors for diabetic nephropathy and correlated with the severity of nephropathy in patients with long-standing type 1 diabetes as well.31,32
Accumulation of AGEs in the kidney may contribute to the progressive alteration in renal architecture and loss of renal function in patients and rodents via various mechanisms, including their cross-linking (β-sheets or cross-β structure) properties of matrix proteins and activation of the downstream signalings.38–41 AGE formation on extracellular matrix proteins alters both matrix-matrix and cell-matrix interactions, being involved in diabetic glomerulosclerosis. For example, non-enzymatic glycations of type IV collagen and laminin reduce their ability to interact with negatively charged proteoglycans, increasing vascular permeability to albumin.42 Further, AGE formation on various types of matrix proteins impairs their degradation by matrix metalloproteinases, contributing to basement membrane thickening and mesangial expansion, hallmarks of diabetic nephropathy.43,44 AGEs formed on the matrix components can trap and covalently cross-link with the extravasated plasma proteins such as lipoproteins, thereby exacerbating diabetic glomerulosclerosis.44 AGEs including glycoxidation or lipoxidation products such as CML, pentosidine, malondialdehyde-lysine accumulate in the expanded mesangial matrix and thickened glomerular basement membranes of early diabetic nephropathy, and in nodular lesions of advanced disease, further suggesting the active role of AGEs for diabetic nephropathy.45
AGEs induce apoptotic cell death and vscular endothelial growth factor (VEGF) expression in human cultured mesangial cells, as the case in pericytes, a counterpart of mesangial cells in retinas.46 Mesangial cells occupy a central anatomical position in the glomerulus, playing crucial roles in maintaining structure and function of glomerular capillary tufts.47 They actually provide structural support for capillary loops and modulate glomerular filtration by its smooth muscle activity.47–49 Therefore, the AGE-induced mesangial apoptosis and dysfuction may contribute in part to glomerular hyperfiltration, an early renal dysfunction in diabetes. Recently, antibodies against VEGF have been found to improve hyperfiltration and albuminuria in experimental diabetes, supproting our speculation.50 Further, we have recently found that AGEs stimulate monocyte chemoattractant protein-1 (MCP-1) expression in mesangial cells.46 Increased MCP-1 expression associated with monocyte infiltration in mesangium has been observed in the early phase of diabetic nephropathy.51 Urinary MCP-1/creatinine ratios in type 2 diabetic patients with microalbuminuria were much higher than those in normal controls, and intensive insulin treatment decreased significantly the urinary MCP-1/creatinine ratios.52 Therefore, AGE accumulation in glomerulus could also be implicated in the initiation of diabetic nephropathy by promoting the secretion of MCP-1 in mesangial cells.
AGEs stimulate insulin-like growth factor-I, -II, platelet-derived growth factor and transforming growth factor-β (TGFβ) in mesangial cells, which in turn mediate production of type IV collagen, laminin and fibronectin.53,54 AGEs induce TGFβ overexpression in both podocytes and proximal tubular cells as well.55,56 Recently, Ziyadeh et al. reported that long-term treatment of type 2 diabetic model mice with blocking antibodies against TGFβ suppressed excess matrix gene expression, glomerulosclerosis, and prevented the development of renal insufficiency.57 These observations suggest that AGE-induced TGFβ expression plays an important role in the pathogenesis of glomerulosclerosis and tubulointerstitial fibrosis in diabetic nephropathy.58,59
In vivo, administration of AGE-albumin to normal healthy mice for 4 weeks has been found to induce glomerular hypertrophy with overexpression of type IV collagen, laminin B1 and TGFβ genes.60 Furthermore, chronic infusion of AGE-albumin to otherwise healthy rats leads to focal glomerulosclerosis, mesangial expansion, and albuminuria.61
Role of RAGE and Oxidative Stress in Diabetic Nephropathy
There is accumulating evidence that AGE-RAGE axis is involved in the pathogenesis of diabetic nephropathy.55,62–64 Among various types of AGE receptors, RAGE is a signal transducing receptor for AGEs that could mediate the inflammatory reactions evoked by AGEs.5,20–24 In humans, RAGE expression is enhanced in podocytes and mesangial cells in diabetic patients with nephropathy.62,63 Several animal studies have also supported the crucial role for RAGE in the development and progression of diabetic nephropathy. Indeed, RAGE-overexpressing diabetic mice have been found to show progressive glomerulosclerosis with renal dysfunction, compared with diabetic littermates lacking the RAGE transgene.64 Further, Wendt et al. reported that diabetic homozygous RAGE null mice failed to develop mesangial matrix expansion or thickening of the glomerular basement membrane.55 They also claimed in their report that activation of RAGE in podocytes could contribute to expression of VEGF and enhanced attraction/activation of inflammatory cells in the diabetic glomeruli, causing albuminuria and glomerulosclerosis in diabetes.55 In addition, it has been reported that db/db or streptozotocin-induced diabetic mice develop renal changes seen in human diabetic nephropathy such as glomerular hypertrophy, glomerular basement membrane thickening, mesangial matrix expansion, connective tissue growth factor (CTGF) overexpression, and NFκB activation, all of which are blocked by the administration of neutralizing antibody raised against RAGE.65,66 The AGE-RAGE interaction can also induce sustained activation of NFκB as a result of increased levels of de novo synthesized NFκBp65 overriding endogenous negative feedback mechanisms and thus might contribute to the persistent damage to diabetic kidney.27
Engagement of RAGE with AGEs elicits oxidative stress generation, thus participating in diabetic nephropathy (Table 1).5,20–24 Indeed, ROS are cytotoxic to renal cells and promote inflammatory and fibrogenic reactions in diabetic kidney.46,56,67–69 The AGE-RAGE-mediated ROS generation stimulates production of pro-sclerotic growth factors such as TGFβ and CTGF via mitogen-activated protein kinase (MAPK), NFκB and/or PKC pathways in both mesangial and renal tubulointerstitial cells.46,56,67–69 Moreover, Tallas-Bonke et al. have recently reported that inhibition of NADPH oxidase by apocynin prevents the AGE-elicited renal damage in experimental diabetic nephropathy through a PKC-α dependent pathway.70 Therefore, the inhibition of NADPH oxidase-derived ROS generation elicited by AGE-RAGE system may be a novel therapeutic target for the treatment of diabetic patients with nephropathy.
Table 1.
Downstream pathways of the AGE-RAGE axis in diabetic nephropathy
| Intracellular signals | Target genes | Pathology |
| ROS, NADPH oxidase activation, NFκB, PKC, MAPK | TGFβ, CTGF, Ang II, ICAM-1, VCAM-1, VEGF, MCP-1 | inflammation, glomerulosclerosis, tubulointerstitial fibrosis, epithelial-to-mesenchymal transdifferentiation |
TGFβ is a well-known pro-fibrogenic factor.71 It not only stimulates matrix synthesis, but also inhibits matrix degradation, being involved in tubuloglomerular sclerosis in diabetes.71 TGFβ mRNA and protein levels are significantly increased in glimeruli and tubulointerstitium in type 1 and 2 diabetic animals and patients.69,72,73 AGE accumulation in diabetic kidney is shown to be closely linked to renal expression of TGFβ55–57,72,73 and administration of AGEs was reported to increase renal TGFβ levels in conjunction with increase in AGEs accumulation in diabetic rodents.74 In addition, we have previously found that AGEs activate TGFβ-Smad system though the interaction with RAGE in cultured mesangial cells.75 Moreover, Oldfield et al. have reported that AGEs cause TGFβ-induced epithelial-tomesenchymal transdifferentiation via interaction with RAGE in normal rat kidney epithelial cell line, NRK 52E cells as well.76 These observations suggest the pathological role for the AGE-RAGE axis in glomerular sclerosis and tubulointerstitial fibrosis, which is a molecular target for prevention of diabetic nephropathy (Fig. 1). In support of this speculation, inhibition of AGE formation by pylidoxamine was shown to reduce renal TGFβ mRNA levels in association with decrease in urinary albumin excretion rate in KK-A(y)/Ta mice, an animal model of type 2 diabetes.77 An AGEs-crosslink breaker, ALT-711, or OPB-9195, an inhibitor of AGE formation was reported to ameliorate renal injury in diabetic animals by suppressing TGFβ overexpression in diabetic animals as well.78,79
Figure 1.
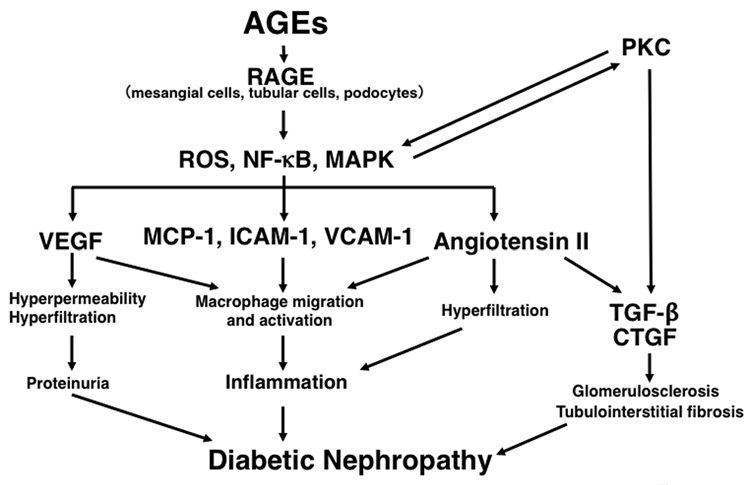
Pathophysiological role of the AGE-RAGE axis in diabetic nephropathy.
CTGF has been considered to act as a downstream target of TGFβ in diabetic nephropathy.80 Several papers have suggested an active role for CTGF in diabetic nephropathy.80–82 CTGF levels in the glomeruli are increased in diabetic animals, and plasma levels of CTGF are reported to be elevated in patients with diabetic nephropathy.81,82 Further, Twigg et al. have recently found that an inhibitor of AGEs, aminoguanidine decreases renal CTGF and fibronectin levels in experimental diabetic nephropathy.82 They also showed that ALT-711 reduced renal CTGF levels in their models.82 Since CTGF also plays a role in the AGE-induced epithelial-to-mesenchymal transdifferentiation,83 suppression of CTGF expression may be a potential therapeutic target for tubuloglomerulosclerosis in diabetic nephropathy.
Therapeutic Interventions of the AGE-RAGE-Oxidative Stress System in Diabetic Nephropathy
Several large clinical studies have reported the potential utility of angiotensin-converting enzyme inhibitors (ACE-Is) or angiotensin II (Ang II) type 1 receptor blockers (ARBs) for the treatment of hypertensive diabetic patients with microalbuminuria or overt nephropathy (Table 2).84–88 Although blood pressure-lowering property could largely explain the beneficial effects of these agents on diabetic nephropathy, there is accumulating evidence to suggest that ACE-Is or ARBs may exert salutary effects on diabetic nephropathy, at least in part, by blocking the pathological crosstalk between the RAS and the metabolic pathways such as AGE-RAGE axis.89 Indeed, angiotensinogen production by cultured proximal tubular cells is increased in response to high glucose concentration, and the intrarenal Ang II level is significantly higher than that in serum in patients with diabetic nephropathy.90,91 Further, high glucose stimulates Ang II generation in association with increased TGFβ1 production by cultured mesangial cells.92
Table 2.
Clinical trials of the RAS inhibitors in diabetic nephropathy
| Agents | Subjects | Clinical outcomes |
| Captopril vs. placebo (ref. 84) | Type 1 diabetic patients with proteinuria more than 500 mg/day | Captopril treatment was associated with a 50 percent reduction in the risk of the combined end points of death, dialysis, and transplantation. |
| Enalapril vs. placebo (ref. 85) | Normotensive, type II diabetic patients with microalbuminuria and normal renal function | Enalapril treatment resulted in long-term stabilization of plasma creatinine levels and of the degree of urinary loss of albumin. |
| Irbesartan vs. placebo (ref. 86) | Hypertensive patients with type 2 diabetes and microalbuminuria | 5.2% in the 300-mg group and 9.7% in the 150-mg group reached the primary end point (the time to the onset of diabetic nephropathy), as compared with 14.9% in the placebo group; hazard ratios were 0.30 and 0.61 for the two irbesartan groups, respectively. |
| Irbesartan vs. amlodipine vs. placebo (ref. 87) | Hypertensive patients with nephropathy due to type 2 diabetes | Irbesartan treatment was associated with a risk of the primary composite end point (a doubling of the base-line serum creatinine concentration, the development of end-stage renal disease, or death from any cause) that was 20% lower than that in the placebo group and 23% lower than that in the amlodipine group. |
| Losartan vs. placebo (ref. 88) | Type 2 diabetic patients with nephropathy | Losartan treatment significantly reduced the risk of the primary outcome (the composite of a doubling of the base-line serum creatinine concentration, end-stage renal disease, or death). |
Since Ang II increases intracellular ROS generation in renal cells, it may stimulate the production of AGEs and further augment the AGE-RAGE system in diabetic kidney.93–98 There is accumulating in vitro- and in vivo-evidence to suggest the pathophysiological crosstalk between the RAS and AGE-RAGE axis in diabetic nephropathy. Indeed, we have previously found that AGEs activate mesangial TGFβ-Smad system through the autocrine production of Ang II via oxidative stress generation.75 Miyata et al. have recently reported that an ACE-I temocapril and an ARB olmesartan significantly inhibit in vitro-formation of pentosidine and CML.95 In animal models, Forbes et al. have reported that ACE-I decreases circulating and renal tissue levels of AGEs in experimental diabetic nephropathy.93 An ARB reduced renal levels of AGEs in AGE-injected animals as well, while Ang II infusion accelerated the formation and accumulation of AGEs in both glomeruli and renal tubules in their models.94 In addition, administration of olmesartan medoxomil was found to inhibit the increase of systolic and diastolic blood pressure levels and urinary N-acetyl-beta-D-glucosaminidase activity and prevent glomerulosclerosis in exogenously AGE-injected rats.96 In humans, an ACE-I, ramipril treatment has been shown to result in a mild decline of fluorescent non-CML-AGE and malondialdehyde concentrations in non-diabetic nephropathy patients.97 In type 2 diabetic subjects, a low-dose of valsartan treatment was reported to decrease serum AGE levels in a blood pressureindependent manner.98 These observations suggest that beneficial effects of the blockade of the RAS on diabetic nephropathy could be ascribed at least in part to its inhibitory effects of the AGE-RAGE-oxidative stress system.
Several papers have shown that peroxisome proliferatoractivated receptor-γ (PPARγ) agonists block the deleterious effects of AGEs and exert beneficial actions on diabetic nephropathy.99–110 Indeed, activation of PPARγ by rosiglitazone inhibited AGE-induced inducible nitric oxide (NO) synthase expression, nitrite release, fibronectin and type IV collagen production by mesangial cells.99,100 Rosiglitazone attenuated the AGE-induced interleukin-8 and soluble intercellular adhesion molecule-1 (ICAM-1) generation by proximal tubular epithelial cells as well through the suppression of signal transducer and activator of transcription (STAT).101 Further, rosiglitazone was reported to inhibit renal extracellular matrix accumulation, fibronectin, type IV collagen and plasminogen activator inhibitor-1 (PAI-1) production and subsequently reduce proteinuria in AGE-injected rats.101
Suppression of RAGE expression may be a molecular target of PPARγ agonists.102–111 Marx et al. reported that stimulation of human endothelial cells with PPARγ agonists such as rosiglitazone and pioglitazone decreased basal as well as tumor necrosis factor-alpha-induced RAGE expression via suppression of NFκB activation. They also showed that PPARγ agonists decreased AGE-induced MCP-1 expression in endothelial cells.102 Further, we have found that telmisartan, an ARB, downregulates RAGE expression and suppresses its downstream signalings in various cell types through its unique PPARγ-modulating ability.104–109 Indeed, telmisartan was found to reduce RAGE mRNA levels and subsequently inhibit superoxide generation as well as MCP-1 expression in mesangial cells, all of which were prevented by GW9662, an inhibitor of PPARγ.106 In addition, we have recently found that nifedipine, but not amlodipine, a control calcium channel blocker, decreased RAGE mRNA levels and subsequently reduced ROS generation, and vascular cell adhesion molecule- 1 (VCAM-1) and MCP-1 expression in AGE-exposed mesangial cells, all of which were blocked by the simultaneous treatment of GW9662.111 Although nifedipine did not affect expression levels of PPARγ, it increased the PPARγ transcriptional activity in mesangial cells. Taken together, these observations provide unique beneficial aspect of telmisartsan and nifedipine on diabetic nephropathy; it could work as an anti-inflammatory agent against AGEs by suppressing RAGE expression in cultured mesangial cells via PPARγ activation.
Pigment epithelium-derived factor (PEDF) is a glycoprotein that belongs to the superfamily of serine protease inhibitors with complex neurotrophic, neuroprotective, anti-angiogenic, anti-oxidative and anti-inflammatory properties, any of which could potentially be exploited as a therapeutic option for the treatment of vascular complications in diabetes.112–119 There are a couple of papers to suggest the protective effects of PEDF against diabetic nephropathy.120,121 PEDF was decreased at both the mRNA and protein levels in the kidney of diabetic rats, whereas TGFβ and fibronectin levels were increased in the same diabetic kidneys.120 In vitro-studies showed that high concentrations of glucose significantly decreased PEDF secretion in primary human glomerular mesangial cells, thus suggesting that hyperglycemia is a direct cause of the PEDF decrease in the kidney. Further, PEDF blocked the high-glucose-induced overexpression of TGFβ, a major pathogenic factor in diabetic nephropathy, and fibronectin in primary cultured mesangial cells.120 Therefore, decreased expression of PEDF in diabetic kidneys could contribute to extracellular matrix overproduction and the development of diabetic nephropathy. In vivo, overexpression of PEDF was found to alleviate microalbuminuria, to prevent the expression of two major fibrogenic factors, TGFβ and CTGF, and to significantly reduce the production of an extracellular matrix protein in the diabetic kidney.121 Moreover, PEDF upregulated metalloproteinase-2 expression in diabetic kidney, which is responsible for extracellular matrix degradation. Taken together, these findings suggest that PEDF functions as an endogenous anti-TGFβ and anti-fibrogenic factor in the kidney. A therapeutic potential of PEDF in diabetic nephropathy is supported by its downregulation in diabetes, its prevention of the overexpression of TGFβ, CTGF and extracellular matrix proteins accumulation in diabetic kidney and its amelioration of albuminuria. Since we have recently found that PEDF inhibits the AGE-induced vascular permeability by blocking the ROS-mediated VEGF expression,114,115 the salutary effects of PEDF on proteinuria may be ascribed partly to its inhibitory actions on the AGE-signaling to VEGF expression in diabetic nephropathy.
Conclusion
In this paper, we review the pathophysiological role of AGE-RAGE-oxidative stress in diabetic nephropathy. Inhibition of the AGE formation and/or the blockade of RAGE-downstream pathways will be a promising therapeutic strategy for the treatment of diabetic patients with nephropathy.
Acknowledgements
This work was supported in part by Grants of Collaboration with Venture Companies Project from the Ministry of Education, Culture, Sports, Science and Technology, Japan. There is no conflict of the interest in this paper.
Abbreviations
- UKPDS
United Kingdom prospective diabetes study
- DCCT
diabetes control and complication trial
- AGEs
advanced glycation end products
- ROS
reactive oxygen species
- PKC
protein kinase C
- RAS
renin-angiotensin system
- DCCT-EDIC
DCCT-epidemiology of diabetes interventions and complications
- CVD
cardiovascular disease
- RAGE
receptor for AGEs
- NFκB
nuclear factor-κB
- CML
Nϵ-carboxymethyllysine
- VEGF
vascular endothelial growth factor
- MCP-1
monocyte chemoattractant protein-1
- TGFβ
transforming growth factor-β
- CTGF
connective tissue growth factor
- MAPK
mitogen-activated protein kinase
- ACE-Is
angiotensin-converting enzyme inhibitors
- Ang II
angiotensin II
- ARBs
Ang II type 1 receptor blockers
- PPARγ
peroxisome proliferator-activated receptor-γ
- NO
nitric oxide
- ICAM-1
intercellular adhesion molecule-1
- STAT
signal transducer and activator of transcription
- PAI-1
plasminogen activator inhibitor-1
- VCAM-1
vascular cell adhesion molecule-1
- PEDF
pigment epithelium-derived factor
Footnotes
Previously published online: www.landesbioscience.com/journals/oximed/article/11148
References
- 1.Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 2.Remuzzi G, Schieppati A, Ruggenenti P. Clinical practice. Nephropathy in patients with type 2 diabetes. N Engl J Med. 2002;346:1145–1151. doi: 10.1056/NEJMcp011773. [DOI] [PubMed] [Google Scholar]
- 3.Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352:837–853. [PubMed] [Google Scholar]
- 4.The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med. 1993;329:977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 5.Yamagishi S, Imaizumi T. Diabetic vascular complications: pathophysiology, biochemical basis and potential therapeutic strategy. Curr Pharm Des. 2005;11:2279–2299. doi: 10.2174/1381612054367300. [DOI] [PubMed] [Google Scholar]
- 6.Retinopathy and nephropathy in patients with type 1 diabetes four years after a trial of intensive therapy. The Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. N Engl J Med. 2000;342:381–389. doi: 10.1056/NEJM200002103420603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Writing Team For The Diabetes Control And Complications Trial/Epidemiology Of Diabetes Interventions And Complications Research Group, author. Sustained effect of intensive treatment of type 1 diabetes mellitus on development and progression of diabetic nephropathy: the Epidemiology of Diabetes Interventions and Complications (EDIC) study. JAMA. 2003;290:2159–2167. doi: 10.1001/jama.290.16.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nathan DM, Lachin J, Cleary P, Orchard T, Brillon DJ, Backlund JY, et al. Intensive diabetes therapy and carotid intima-media thickness in type 1 diabetes mellitus. N Engl J Med. 2003;348:2294–2303. doi: 10.1056/NEJMoa022314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nathan DM, Cleary PA, Backlund JY, Genuth SM, Lachin JM, Orchard TJ, et al. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med. 2005;353:2643–2653. doi: 10.1056/NEJMoa052187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med. 2008;359:1577–1589. doi: 10.1056/NEJMoa0806470. [DOI] [PubMed] [Google Scholar]
- 11.Brownlee M, Cerami A, Vlassara H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N Engl J Med. 1988;318:1315–1321. doi: 10.1056/NEJM198805193182007. [DOI] [PubMed] [Google Scholar]
- 12.Dyer DG, Blackledge JA, Thorpe SR, Baynes JW. Formation of pentosidine during nonenzymatic browning of proteins by glucose. Identification of glucose and other carbohydrates as possible precursors of pentosidine in vivo. J Biol Chem. 1991;266:11654–11660. [PubMed] [Google Scholar]
- 13.Grandhee SK, Monnier VM. Mechanism of formation of the Maillard protein cross-link pentosidine. Glucose, fructose and ascorbate as pentosidine precursors. J Biol Chem. 1991;266:11649–11653. [PubMed] [Google Scholar]
- 14.Rahbar S, Figarola JL. Novel inhibitors of advanced glycation endproducts. Arch Biochem Biophys. 2003;419:63–79. doi: 10.1016/j.abb.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 15.Vlassara H, Palace MR. Diabetes and advanced glycation endproducts. J Intern Med. 2002;251:87–101. doi: 10.1046/j.1365-2796.2002.00932.x. [DOI] [PubMed] [Google Scholar]
- 16.Yamagishi S, Fukami K, Ueda S, Okuda S. Molecular mechanisms of diabetic nephropathy and its therapeutic intervention. Curr Drug Targets. 2007;8:952–959. doi: 10.2174/138945007781386884. [DOI] [PubMed] [Google Scholar]
- 17.Bucala, Cerami A. Advanced glycosylation: chemistry, biology and implications for diabetes and aging. Adv Pharmacol. 1992;23:1–34. doi: 10.1016/s1054-3589(08)60961-8. [DOI] [PubMed] [Google Scholar]
- 18.Yamagishi S, Nakamura K, Matsui T, Ueda S, Fukami K, Okuda S. Agents that block advanced glycation end products (AGE)-RAGE (receptor for AGEs)-oxidative stress system: a novel therapeutic strategy for diabetic vascular complications. Expert Opin Investig Drugs. 2008;17:983–996. doi: 10.1517/13543784.17.7.983. [DOI] [PubMed] [Google Scholar]
- 19.Yamagishi S, Ueda S, Okuda S. Food-derived advanced glycation end products (AGEs): a novel therapeutic target for various disorders. Curr Pharm Des. 2007;13:2832–2836. doi: 10.2174/138161207781757051. [DOI] [PubMed] [Google Scholar]
- 20.Bierhaus A, Hofmann MA, Ziegler R, Nawroth PP. AGEs and their interaction with AGE-receptors in vascular disease and diabetes mellitus I. The AGE concept. Cardiovasc Res. 1998;37:586–600. doi: 10.1016/s0008-6363(97)00233-2. [DOI] [PubMed] [Google Scholar]
- 21.Wendt T, Bucciarelli L, Qu W, Lu Y, Yan SF, Stern DM, et al. Receptor for advanced glycation endproducts (RAGE) and vascular inflammation: insights into the pathogenesis of macrovascular complications in diabetes. Curr Atheroscler Rep. 2002;4:228–237. doi: 10.1007/s11883-002-0024-4. [DOI] [PubMed] [Google Scholar]
- 22.Schmidt AM, Stern D. Atherosclerosis and diabetes: the RAGE connection. Curr Atheroscler Rep. 2000;2:430–436. doi: 10.1007/s11883-000-0082-4. [DOI] [PubMed] [Google Scholar]
- 23.Yamagishi S, Nakamura K, Matsui T. regulation of advanced glycation end product (AGE)-receptor (RAGE) system by PPARgamma agonists and its implication in cardiovascular disease. Pharmacol Res. 2009;60:174–178. doi: 10.1016/j.phrs.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 24.Yamagishi S, Matsui T, Nakamura K. Kinetics, role and therapeutic implications of endogenous soluble form of receptor for advanced glycation end products (sRAGE) in diabetes. Curr Drug Targets. 2007;8:1138–1143. doi: 10.2174/138945007782151298. [DOI] [PubMed] [Google Scholar]
- 25.Tanaka N, Yonekura H, Yamagishi S, Fujimori H, Yamamoto Y, Yamamoto H. The receptor for advanced glycation end products is induced by the glycation products themselves and tumor necrosis factor-alpha through nuclear factor-kappaB, and by 17beta-estradiol through Sp-1 in human vascular endothelial cells. J Biol Chem. 2000;275:25781–25790. doi: 10.1074/jbc.M001235200. [DOI] [PubMed] [Google Scholar]
- 26.Yamagishi S, Amano S, Inagaki Y, Okamoto T, Takeuchi M, Makita Z. Beraprost sodium, a prostaglandin I2 analogue, protects against advanced gycation end products-induced injury in cultured retinal pericytes. Mol Med. 2002;8:546–550. [PMC free article] [PubMed] [Google Scholar]
- 27.Bierhaus A, Schiekofer S, Schwaninger M, Andrassy M, Humpert PM, Chen J, et al. Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes. 2001;50:2792–2808. doi: 10.2337/diabetes.50.12.2792. [DOI] [PubMed] [Google Scholar]
- 28.Park L, Raman KG, Lee KJ, Lu Y, Ferran LJ, Jr, Chow WS, et al. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat Med. 1998;4:1025–1031. doi: 10.1038/2012. [DOI] [PubMed] [Google Scholar]
- 29.Makita Z, Radoff S, Rayfield EJ, Yang Z, Skolnik E, Delaney V, et al. Advanced glycosylation end products in patients with diabetic nephropathy. N Engl J Med. 1991;325:836–842. doi: 10.1056/NEJM199109193251202. [DOI] [PubMed] [Google Scholar]
- 30.Galler A, Muller G, Schinzel R, Kratzsch J, Kiess W, Munch G. Impact of metabolic control and serum lipids on the concentration of advanced glycation end products in the serum of children and adolescents with type 1 diabetes, as determined by fluorescence spectroscopy and nepsilon-(carboxymethyl)lysine ELISA. Diabetes Care. 2003;26:2609–2615. doi: 10.2337/diacare.26.9.2609. [DOI] [PubMed] [Google Scholar]
- 31.Monnier VM, Bautista O, Kenny D, Sell DR, Fogarty J, Dahms W, et al. Skin collagen glycation, glycoxidation and crosslinking are lower in subjects with long-term intensive versus conventional therapy of type 1 diabetes: relevance of glycated collagen products versus HbA1c as markers of diabetic complications. DCCT Skin Collagen Ancillary Study Group. Diabetes Control and Complications Trial. Diabetes. 1999;48:870–880. doi: 10.2337/diabetes.48.4.870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Genuth S, Sun W, Cleary P, Sell DR, Dahms W, Malone J, et al. Glycation and carboxymethyllysine levels in skin collagen predict the risk of future 10-year progression of diabetic retinopathy and nephropathy in the diabetes control and complications trial and epidemiology of diabetes interventions and complications participants with type 1 diabetes. Diabetes. 2005;54:3103–3111. doi: 10.2337/diabetes.54.11.3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deuther-Conrad W, Franke S, Sommer M, Henle T, Stein G. Differences in the modulating potential of advanced glycation end product (AGE) peptides versus AGE proteins. Kidney Int Suppl. 2001;78:63–66. doi: 10.1046/j.1523-1755.2001.59780063.x. [DOI] [PubMed] [Google Scholar]
- 34.Sharp PS, Rainbow S, Mukherjee S. Serum levels of low molecular weight advanced glycation end products in diabetic subjects. Diabet Med. 2003;20:575–579. doi: 10.1046/j.1464-5491.2003.00973.x. [DOI] [PubMed] [Google Scholar]
- 35.Thomas MC, Tsalamandris C, MacIsaac R, Medley T, Kingwell B, Cooper ME, et al. Low-molecular-weight AGEs are associated with GFR and anemia in patients with type 2 diabetes. Kidney Int. 2004;66:1167–1172. doi: 10.1111/j.1523-1755.2004.00868.x. [DOI] [PubMed] [Google Scholar]
- 36.Miyata T, Ueda Y, Horie K, Nangaku M, Tanaka S, van Ypersele de Strihou C, et al. Renal catabolism of advanced glycation end products: the fate of pentosidine. Kidney Int. 1998;53:416–422. doi: 10.1046/j.1523-1755.1998.00756.x. [DOI] [PubMed] [Google Scholar]
- 37.Miura J, Yamagishi S, Uchigata Y, Takeuchi M, Yamamoto H, Makita Z, et al. Serum levels of non-carboxymethyllysine advanced glycation endproducts are correlated to severity of microvascular complications in patients with Type 1 diabetes. J Diabetes Complications. 2003;17:16–21. doi: 10.1016/s1056-8727(02)00183-6. [DOI] [PubMed] [Google Scholar]
- 38.Bohlender JM, Franke S, Stein G, Wolf G. Advanced glycation end products and the kidney. Am J Physiol Renal Physiol. 2005;289:645–659. doi: 10.1152/ajprenal.00398.2004. [DOI] [PubMed] [Google Scholar]
- 39.Beisswenger PJ, Makita Z, Curphey TJ, Moore LL, Jean S, Brinck-Johnsen T, et al. Formation of immunochemical advanced glycosylation end products precedes and correlates with early manifestations of renal and retinal disease in diabetes. Diabetes. 1995;44:824–829. doi: 10.2337/diab.44.7.824. [DOI] [PubMed] [Google Scholar]
- 40.Soulis-Liparota T, Cooper M, Papazoglou D, Clarke B, Jerums G. Retardation by aminoguanidine of development of albuminuria, mesangial expansion and tissue fluorescence in streptozocin-induced diabetic rat. Diabetes. 1991;40:1328–1334. doi: 10.2337/diab.40.10.1328. [DOI] [PubMed] [Google Scholar]
- 41.Bouma B, Kroon-Batenburg LM, Wu YP, Brunjes B, Posthuma G, Kranenburg O, et al. Glycation induces formation of amyloid cross-beta structure in albumin. J Biol Chem. 2003;278:41810–41819. doi: 10.1074/jbc.M303925200. [DOI] [PubMed] [Google Scholar]
- 42.Silbiger S, Crowley S, Shan Z, Brownlee M, Satriano J, Schlondorff D. Nonenzymatic elevated glucose reduces collagen synthesis and proteoglycan charge. Kidney Int. 1993;43:853–864. doi: 10.1038/ki.1993.120. [DOI] [PubMed] [Google Scholar]
- 43.Mott JD, Khalifah RG, Nagase H, Shield CF, 3rd, Hudson JK, Hudson BG. Nonenzymatic glycation of type IV collagen and matrix metalloproteinase susceptibility. Kidney Int. 1997;52:1302–12. doi: 10.1038/ki.1997.455. [DOI] [PubMed] [Google Scholar]
- 44.Brownlee M. Lilly Lecture 1993. Glycation and diabetic complications. Diabetes. 1994;43:836–841. doi: 10.2337/diab.43.6.836. [DOI] [PubMed] [Google Scholar]
- 45.Suzuki D, Miyata T, Saotome N, Horie K, Inagi R, Yasuda Y, et al. Immunohistochemical evidence for an increased oxidative stress and carbonyl modification of proteins in diabetic glomerular lesions. J Am Soc Nephrol. 1999;10:822–832. doi: 10.1681/ASN.V104822. [DOI] [PubMed] [Google Scholar]
- 46.Yamagishi S, Inagaki Y, Okamoto T, Amano S, Koga K, Takeuchi M, et al. Advanced glycation end product-induced apoptosis and overexpression of vascular endothelial growth factor and monocyte chemoattractant protein-1 in human-cultured mesangial cells. J Biol Chem. 2002;277:20309–20315. doi: 10.1074/jbc.M202634200. [DOI] [PubMed] [Google Scholar]
- 47.Dworkin LD, Ichikawa I, Brenner BM. Hormonal modulation of glomerular function. Am J Physiol. 1983;244:95–104. doi: 10.1152/ajprenal.1983.244.2.F95. [DOI] [PubMed] [Google Scholar]
- 48.Kreisberg JI, Venkatachalam M, Troyer D. Contractile properties of cultured glomerular mesangial cells. Am J Physiol. 1985;249:457–463. doi: 10.1152/ajprenal.1985.249.4.F457. [DOI] [PubMed] [Google Scholar]
- 49.Schlondorff D. The glomerular mesangial cell: an expanding role for a specialized pericyte. FASEB J. 1987;1:272–281. doi: 10.1096/fasebj.1.4.3308611. [DOI] [PubMed] [Google Scholar]
- 50.De Vriese AS, Tilton RG, Elger M, Stephan CC, Kriz W, Lameire NH. Antibodies against vascular endothelial growth factor improve early renal dysfunction in experimental diabetes. J Am Soc Nephrol. 2001;12:993–1000. doi: 10.1681/ASN.V125993. [DOI] [PubMed] [Google Scholar]
- 51.Banba N, Nakamura T, Matsumura M, Kuroda H, Hattori Y, Kasai K. Possible relationship of monocyte chemoattractant protein-1 with diabetic nephropathy. Kidney Int. 2000;58:684–690. doi: 10.1046/j.1523-1755.2000.00214.x. [DOI] [PubMed] [Google Scholar]
- 52.Ye SD, Zheng M, Zhao LL, Qian Y, Yao XM, Ren A, et al. Intensive insulin therapy decreases urinary MCP-1 and ICAN-1 excretions in incipient diabetic nephropathy. Eur J Clin Invest. 2009;39:980–985. doi: 10.1111/j.1365-2362.2009.02203.x. [DOI] [PubMed] [Google Scholar]
- 53.Matsumura T, Yamagishi S, Brownlee M. Advanced glycation end products and the pathogenesis of diabetic complications. In: Leroith D, Taylor SI, Olefsky JM, editors. Diabetes Mellitus: A Fundamental and Clinical Text. New York: Lippincott-Raven Publishers; 2000. pp. 983–991. [Google Scholar]
- 54.Yamagishi S, Takeuchi M, Makita Z. In: Advanced glycation end products and the pathogenesis of diabetic nephropathy. Type-2 diabetic nephropathy in Japan, from bench to bedside. Tomino Y, editor. Basel: Karger; 2001. pp. 30–35. [DOI] [PubMed] [Google Scholar]
- 55.Wendt TM, Tanji N, Guo J, Kislinger TR, Qu W, Lu Y, et al. RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am J Pathol. 2003;162:123–137. doi: 10.1016/S0002-9440(10)63909-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yamagishi S, Inagaki Y, Okamoto T, Amano S, Koga K, Takeuchi M. Advanced glycation end products inhibit de novo protein synthesis and induce TGFbeta overexpression in proximal tubular cells. Kidney Int. 2003;63:464–473. doi: 10.1046/j.1523-1755.2003.00752.x. [DOI] [PubMed] [Google Scholar]
- 57.Ziyadeh FN, Hoffman BB, Han DC, Iglesias-De La Cruz MC, Hong SW, et al. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc Natl Acad Sci USA. 2000;97:8015–8020. doi: 10.1073/pnas.120055097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yamagishi S, Koga K, Inagaki Y, Amano S, Okamoto T, Takeuchi M. Dilazep hydrochloride, an antiplatelet drug, prevents progression of diabetic nephropathy in Otsuka Long-Evans Tokushima fatty rats. Drugs Exp Clin Res. 2002;28:221–227. [PubMed] [Google Scholar]
- 59.Raj DS, Choudhury D, Welbourne TC, Levi M. Advanced glycation end products: a Nephrologist's perspective. Am J Kidney Dis. 2000;35:365–380. doi: 10.1016/s0272-6386(00)70189-2. [DOI] [PubMed] [Google Scholar]
- 60.Yang CW, Vlassara H, Peten EP, He CJ, Striker GE, Striker LJ. Advanced glycation end products upregulate gene expression found in diabetic glomerular disease. Proc Natl Acad Sci USA. 1994;91:9436–9440. doi: 10.1073/pnas.91.20.9436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vlassara H, Striker LJ, Teichberg S, Fuh H, Li YM, Steffes M. Advanced glycation end products induce glomerular sclerosis and albuminuria in normal rats. Proc Natl Acad Sci USA. 1994;91:11704–11708. doi: 10.1073/pnas.91.24.11704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tanji N, Markowitz GS, Fu C, Kislinger T, Taguchi A, Pischetsrieder M, et al. Expression of advanced glycation end products and their cellular receptor RAGE in diabetic nephropathy and nondiabetic renal disease. J Am Soc Nephrol. 2000;11:1656–1666. doi: 10.1681/ASN.V1191656. [DOI] [PubMed] [Google Scholar]
- 63.Suzuki D, Toyoda M, Yamamoto N, Miyauchi M, Katoh M, Kimura M, et al. Relationship between the expression of advanced glycation end-products (AGE) and the receptor for AGE (RAGE) mRNA in diabetic nephropathy. Intern Med. 2006;45:435–441. doi: 10.2169/internalmedicine.45.1557. [DOI] [PubMed] [Google Scholar]
- 64.Yamamoto Y, Kato I, Doi T, Yonekura H, Ohashi S, Takeuchi M, et al. Development and prevention of advanced diabetic nephropathy in RAGE-overexpressing mice. J Clin Invest. 2001;108:261–268. doi: 10.1172/JCI11771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Flyvbjerg A, Denner L, Schrijvers BF, Tilton RG, Mogensen TH, Paludan SR, et al. Long-term renal effects of a neutralizing RAGE antibody in obese type 2 diabetic mice. Diabetes. 2004;53:166–172. doi: 10.2337/diabetes.53.1.166. [DOI] [PubMed] [Google Scholar]
- 66.Jensen LJ, Denner L, Schrijvers BF, Tilton RG, Rasch R, Flyvbjerg A. Renal effects of a neutralising RAGE-antibody in long-term streptozotocin-diabetic mice. J Endocrinol. 2006;188:493–501. doi: 10.1677/joe.1.06524. [DOI] [PubMed] [Google Scholar]
- 67.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 68.Baynes JW, Thorpe SR. Role of oxidative stress in diabetic complications: a new perspective on an old paradigm. Diabetes. 1999;48:1–9. doi: 10.2337/diabetes.48.1.1. [DOI] [PubMed] [Google Scholar]
- 69.Ha H, Hwang IA, Park H, Lee HB. Role of reactive oxygen species in the pathogenesis of diabetic nephropathy. Diabetes Res Clin Pract. 2008;82:42–45. doi: 10.1016/j.diabres.2008.09.017. [DOI] [PubMed] [Google Scholar]
- 70.Thallas-Bonke V, Thorpe SR, Coughlan MT, Fukami K, Yap FY, Sourris K, et al. Inhibition of NADPH oxidase prevents AGE mediated damage in diabetic nephropathy through a protein kinase C-{alpha} dependent pathway. Diabetes. 2008;57:460–469. doi: 10.2337/db07-1119. [DOI] [PubMed] [Google Scholar]
- 71.Border WA, Noble NA. Transforming growth factor beta in tissue fibrosis. N Engl J Med. 1994;331:1286–1292. doi: 10.1056/NEJM199411103311907. [DOI] [PubMed] [Google Scholar]
- 72.Yamamoto T, Nakamura T, Noble NA, Ruoslahti E, Border WA. Expression of transforming growth factor beta is elevated in human and experimental diabetic nephropathy. Proc Natl Acad Sci USA. 1993;90:1814–1818. doi: 10.1073/pnas.90.5.1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Davis BJ, Forbes JM, Thomas MC, Jerums G, Burns WC, Kawachi H, et al. Superior renoprotective effects of combination therapy with ACE and AGE inhibition in the diabetic spontaneously hypertensive rat. Diabetologia. 2004;47:89–97. doi: 10.1007/s00125-003-1256-8. [DOI] [PubMed] [Google Scholar]
- 74.Zimanyi MA, Denton KM, Forbes JM, Thallas-Bonke V, Thomas MC, Poon F, et al. A developmental nephron deficit in rats is associated with increased susceptibility to a secondary renal injury due to advanced glycation end-products. Diabetologia. 2006;49:801–810. doi: 10.1007/s00125-006-0175-x. [DOI] [PubMed] [Google Scholar]
- 75.Fukami K, Ueda S, Yamagishi S, Kato S, Inagaki Y, Takeuchi M, et al. AGEs activate mesangial TGFbeta-Smad signaling via an angiotensin II type I receptor interaction. Kidney Int. 2004;66:2137–2147. doi: 10.1111/j.1523-1755.2004.66004.x. [DOI] [PubMed] [Google Scholar]
- 76.Oldfield MD, Bach LA, Forbes JM, Nikolic-Paterson D, McRobert A, Thallas V, et al. Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE) J Clin Invest. 2001;108:1853–1863. doi: 10.1172/JCI11951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tanimoto M, Gohda T, Kaneko S, Hagiwara S, Murakoshi M, Aoki T, et al. Effect of pyridoxamine (K-163), an inhibitor of advanced glycation end products, on type 2 diabetic nephropathy in KK-A(y)/Ta mice. Metabolism. 2007;56:160–167. doi: 10.1016/j.metabol.2006.08.026. [DOI] [PubMed] [Google Scholar]
- 78.Forbes JM, Thallas V, Thomas MC, Founds HW, Burns WC, Jerums G, et al. The breakdown of preexisting advanced glycation end products is associated with reduced renal fibrosis in experimental diabetes. FASEB J. 2003;17:1762–1764. doi: 10.1096/fj.02-1102fje. [DOI] [PubMed] [Google Scholar]
- 79.Tsuchida K, Makita Z, Yamagishi S, Atsumi T, Miyoshi H, Obara S, et al. Suppression of transforming growth factor beta and vascular endothelial growth factor in diabetic nephropathy in rats by a novel advanced glycation end product inhibitor, OPB-9195. Diabetologia. 1999;42:579–588. doi: 10.1007/s001250051198. [DOI] [PubMed] [Google Scholar]
- 80.Riser BL, Denichilo M, Cortes P, Baker C, Grondin JM, Yee J, et al. Regulation of connective tissue growth factor activity in cultured rat mesangial cells and its expression in experimental diabetic glomerulosclerosis. J Am Soc Nephrol. 2000;11:25–38. doi: 10.1681/ASN.V11125. [DOI] [PubMed] [Google Scholar]
- 81.Twigg SM, Cao Z, McLennan SV, Burns WC, Brammar G, Forbes JM, et al. Renal connective tissue growth factor induction in experimental diabetes is prevented by aminoguanidine. Endocrinology. 2002;143:4907–4915. doi: 10.1210/en.2002-220619. [DOI] [PubMed] [Google Scholar]
- 82.Roestenberg P, van Nieuwenhoven FA, Wieten L, Boer P, Diekman T, Tiller AM, et al. Connective tissue growth factor is increased in plasma of type 1 diabetic patients with nephropathy. Diabetes Care. 2004;27:1164–1170. doi: 10.2337/diacare.27.5.1164. [DOI] [PubMed] [Google Scholar]
- 83.Burns WC, Twigg SM, Forbes JM, Pete J, Tikellis C, Thallas-Bonke V, et al. Connective tissue growth factor plays an important role in advanced glycation end product-induced tubular epithelial-to-mesenchymal transition: implications for diabetic renal disease. J Am Soc Nephrol. 2006;17:2484–2494. doi: 10.1681/ASN.2006050525. [DOI] [PubMed] [Google Scholar]
- 84.Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med. 1993;329:1456–1462. doi: 10.1056/NEJM199311113292004. [DOI] [PubMed] [Google Scholar]
- 85.Ravid M, Savin H, Jutrin I, Bental T, Katz B, Lishner M. Long-term stabilizing effect of angiotensin-converting enzyme inhibition on plasma creatinine and on proteinuria in normotensive type II diabetic patients. Ann Intern Med. 1993;118:577–581. doi: 10.7326/0003-4819-118-8-199304150-00001. [DOI] [PubMed] [Google Scholar]
- 86.Parving HH, Lehnert H, Brochner-Mortensen J, Gomis R, Andersen S, Arner P. The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. N Engl J Med. 2001;345:870–878. doi: 10.1056/NEJMoa011489. [DOI] [PubMed] [Google Scholar]
- 87.Lewis EJ, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med. 2001;345:851–860. doi: 10.1056/NEJMoa011303. [DOI] [PubMed] [Google Scholar]
- 88.Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345:861–869. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- 89.Fukami K, Yamagishi S, Ueda S, Okuda S. Novel therapeutic targets for diabetic nephropathy. Endocr Metab Immune Disord Drug Targets. 2007;7:83–92. doi: 10.2174/187153007780832118. [DOI] [PubMed] [Google Scholar]
- 90.Leehey DJ, Singh AK, Alavi N, Singh R. Role of angiotensin II in diabetic nephropathy. Kidney Int Suppl. 2000;77:93–98. doi: 10.1046/j.1523-1755.2000.07715.x. [DOI] [PubMed] [Google Scholar]
- 91.Zhang SL, Filep JG, Hohman TC, Tang SS, Ingelfinger JR, Chan JS. Molecular mechanisms of glucose action on angiotensinogen gene expression in rat proximal tubular cells. Kidney Int. 1999;55:454–464. doi: 10.1046/j.1523-1755.1999.00271.x. [DOI] [PubMed] [Google Scholar]
- 92.Singh R, Alavi N, Singh AK, Leehey DJ. Role of angiotensin II in glucose-induced inhibition of mesangial matrix degradation. Diabetes. 1999;48:2066–2073. doi: 10.2337/diabetes.48.10.2066. [DOI] [PubMed] [Google Scholar]
- 93.Forbes JM, Cooper ME, Thallas V, Burns WC, Thomas MC, Brammar GC, et al. Reduction of the accumulation of advanced glycation end products by ACE inhibition in experimental diabetic nephropathy. Diabetes. 2002;51:3274–3282. doi: 10.2337/diabetes.51.11.3274. [DOI] [PubMed] [Google Scholar]
- 94.Thomas MC, Tikellis C, Burns WM, Bialkowski K, Cao Z, Coughlan MT, et al. Interactions between renin angiotensin system and advanced glycation in the kidney. J Am Soc Nephrol. 2005;16:2976–2984. doi: 10.1681/ASN.2005010013. [DOI] [PubMed] [Google Scholar]
- 95.Miyata T, van Ypersele de Strihou C, Ueda Y, Ichimori K, Inagi R, Onogi H, et al. Angiotensin II receptor antagonists and angiotensin-converting enzyme inhibitors lower in vitro the formation of advanced glycation end products: biochemical mechanisms. J Am Soc Nephrol. 2002;13:2478–2487. doi: 10.1097/01.asn.0000032418.67267.f2. [DOI] [PubMed] [Google Scholar]
- 96.Yamagishi S, Takeuchi M, Inoue H. Olmesartan medoxomil, a newly developed angiotensin II type 1 receptor antagonist, protects against renal damage in advanced glycation end product (age)-injected rats. Drugs Exp Clin Res. 2005;31:45–51. [PubMed] [Google Scholar]
- 97.Sebekova K, Gazdikova K, Syrova D, Blazicek P, Schinzel R, Heidland A, et al. Effects of ramipril in nondiabetic nephropathy: improved parameters of oxidatives stress and potential modulation of advanced glycation end products. J Hum Hypertens. 2003;17:265–270. doi: 10.1038/sj.jhh.1001541. [DOI] [PubMed] [Google Scholar]
- 98.Saisho Y, Komiya N, Hirose H. Effect of valsartan, an angiotensin II receptor blocker, on markers of oxidation and glycation in Japanese type 2 diabetic subjects: blood pressure-independent effect of valsartan. Diabetes Res Clin Prac. 2006;74:201–203. doi: 10.1016/j.diabres.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 99.Chang PC, Chen TH, Chang CJ, Hou CC, Chan P, Lee HM. Advanced glycosylation end products induce inducible nitric oxide synthase (iNOS) expression via a p38 MAPK-dependent pathway. Kidney Int. 2004;65:1664–1675. doi: 10.1111/j.1523-1755.2004.00602.x. [DOI] [PubMed] [Google Scholar]
- 100.Yu X, Li C, Li X, Cai L. Rosiglitazone prevents advanced glycation end products-induced renal toxicity likely through suppression of plasminogen activator inhibitor-1. Toxicol Sci. 2007;96:346–356. doi: 10.1093/toxsci/kfm010. [DOI] [PubMed] [Google Scholar]
- 101.Tang SC, Leung JC, Chan LY, Tsang AW, Lai KN. Activation of tubular epithelial cells in diabetic nephropathy and the role of the peroxisome proliferatoractivated receptor-gamma agonist. J Am Soc Nephrol. 2006;17:1633–1643. doi: 10.1681/ASN.2005101113. [DOI] [PubMed] [Google Scholar]
- 102.Marx N, Walcher D, Ivanova N, Rautzenberg K, Jung A, Friedl R. Thiazolidinediones reduce endothelial expression of receptors for advanced glycation end products. Diabetes. 2004;53:2662–2668. doi: 10.2337/diabetes.53.10.2662. [DOI] [PubMed] [Google Scholar]
- 103.Wang K, Zhou Z, Zhang M, Fan L, Forudi F, Zhou X, et al. Peroxisome proliferator-activated receptor gamma downregulates receptor for advanced glycation end products and inhibits smooth muscle cell proliferation in a diabetic and nondiabetic rat carotid artery injury model. J Pharmacol Exp Ther. 2006;31:37–43. doi: 10.1124/jpet.105.095125. [DOI] [PubMed] [Google Scholar]
- 104.Yamagishi S, Matsui T, Nakamura K, Takeuchi M, Inoue H. Telmisartan inhibits advanced glycation end products (AGEs)-elicited endothelial cell injury by suppressing AGE receptor (RAGE) expression via peroxisome proliferator-activated receptor-gamma activation. Protein Pept Lett. 2008;15:850–853. doi: 10.2174/092986608785203746. [DOI] [PubMed] [Google Scholar]
- 105.Matsui T, Nakamura K, Takeuchi M, Yamagishi S. Telmisartan blocks advanced glycation end-product (AGE)-induced plasminogen activator inhibitor-1 gene expression in endothelial cells through activation of peroxisome proliferator-activated receptor-γ. Lett Drug Des Dis. 2008;5:477–480. [Google Scholar]
- 106.Matsui T, Yamagishi S, Ueda S, Nakamura K, Imaizumi T, Takeuchi M, et al. Telmisartan, an angiotensin II type 1 receptor antagonist, inhibits advanced glycation end-product (AGE)-induced monocyte chemoattractant protein-1 expression in mesangial cells through downregulation of receptor for AGEs via peroxisome proliferator-activated receptor-γ activation. J Int Med Res. 2007;35:482–489. doi: 10.1177/147323000703500407. [DOI] [PubMed] [Google Scholar]
- 107.Yoshida T, Yamagishi S, Nakamura K, Matsui T, Imaizumi T, Takeuchi M, et al. Telmisartan inhibits AGE-induced C-reactive protein production through downregulation of the receptor for AGE via peroxisome proliferator-activated receptor-gamma activation. Diabetologia. 2006;49:3094–3099. doi: 10.1007/s00125-006-0437-7. [DOI] [PubMed] [Google Scholar]
- 108.Yoshida T, Yamagishi S, Matsui T, Nakamura K, Ueno T, Takeuchi M, et al. Telmisartan, an angiotensin II type 1 receptor blocker, inhibits advanced glycation end-product (AGE)-elicited hepatic insulin resistance via peroxisome proliferator-activated receptor-gamma activation. J Int Med Res. 2008;36:237–243. doi: 10.1177/147323000803600204. [DOI] [PubMed] [Google Scholar]
- 109.Yamagishi S, Nakamura K, Matsui T. Potential utility of telmisartan, an angiotensin II type 1 receptor blocker with peroxisome proliferator-activated receptor-gamma (PPARgamma)-modulating activity for the treatment of cardiometabolic disorders. Curr Mol Med. 2007;7:463–469. doi: 10.2174/156652407781387073. [DOI] [PubMed] [Google Scholar]
- 110.Ma L, Gao HQ, Li BY, Ma YB, You BA, Zhang FL. Grape seed proanthocyanidin extracts inhibit vascular cell adhesion molecule expression induced by advanced glycation end products through activation of peroxisome proliferators-activated receptor gamma. J Cardiovasc Pharmacol. 2007;49:293–298. doi: 10.1097/FJC.0b013e31803c5616. [DOI] [PubMed] [Google Scholar]
- 111.Matsui T, Yamagishi S, Tekuchi M, Ueda S, Fukami K, Okuda S. Nifedipine, a calcium channel blocker, inhibits advanced glycation end product (AGE)-elicited mesangial cell damage by suppressing AGE receptor (RAGE) expression via peroxisome proliferator-activated receptor-gamma activation. Biochem Biophys Res Commun. 2009;385:269–272. doi: 10.1016/j.bbrc.2009.05.061. [DOI] [PubMed] [Google Scholar]
- 112.Tombran-Tink J, Barnstable CJ. PEDF: a multifaceted neurotrophic factor. Nat Rev Neurosci. 2003;4:628–636. doi: 10.1038/nrn1176. [DOI] [PubMed] [Google Scholar]
- 113.Yamagishi S, Matsui T, Nakamura K, Ueda S, Noda Y, Imaizumi T. Pigment epithelium-derived factor (PEDF): Its potential therapeutic implication in diabetic vascular complications. Curr Drug Targets. 2008;9:1025–1029. doi: 10.2174/138945008786786154. [DOI] [PubMed] [Google Scholar]
- 114.Yoshida Y, Yamagishi S, Matsui T, Jinnouchi Y, Fukami K, Imaizumi T, et al. Protective role of pigment epithelium-derived factor (PEDF) in early phase of experimental diabetic retinopathy. Diabetes Metab Res Rev. 2009;25:678–686. doi: 10.1002/dmrr.1007. [DOI] [PubMed] [Google Scholar]
- 115.Yamagishi S, Nakamura K, Matsui T, Inagaki Y, Takenaka K, Jinnouchi Y, et al. Pigment epitheliumderived factor inhibits advanced glycation end product-induced retinal vascular hyperpermeability by blocking reactive oxygen species-mediated vascular endothelial growth factor expression. J Biol Chem. 2006;281:20213–20220. doi: 10.1074/jbc.M602110200. [DOI] [PubMed] [Google Scholar]
- 116.Yamagishi SI, Nakamura K, Matsui T, Yoshida T, Takeuchi M, Imaizumi T. Pigment epithelium-derived factor (PEDF) blocks advanced glycation end product (AGE)-induced angiogenesis in vitro. Horm Metab Res. 2007;39:233–235. doi: 10.1055/s-2007-970425. [DOI] [PubMed] [Google Scholar]
- 117.Yamagishi S, Matsui T, Nakamura K, Takeuchi M, Imaizumi T. Pigment epithelium-derived factor (PEDF) prevents diabetes- or advanced glycation end products (AGE)-elicited retinal leukostasis. Microvasc Res. 2006;72:86–90. doi: 10.1016/j.mvr.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 118.Yamagishi S, Matsui T, Nakamura K, Yoshida Y, Takeuchi M, Inoue H. Pigment-epithelium-derived factor suppresses expression of receptor for advanced glycation end products in the eye of diabetic rats. Ophthalmic Res. 2007;39:92–97. doi: 10.1159/000099244. [DOI] [PubMed] [Google Scholar]
- 119.Yamagishi S, Amano S, Inagaki Y, Okamoto T, Koga K, Sasaki N, et al. Advanced glycation end products-induced apoptosis and overexpression of vascular endothelial growth factor in bovine retinal pericytes. Biochem Biophys Res Commun. 2002;290:973–978. doi: 10.1006/bbrc.2001.6312. [DOI] [PubMed] [Google Scholar]
- 120.Wang JJ, Zhang SX, Lu K, Chen Y, Mott R, Sato S, et al. Decreased expression of pigment epithelium-derived factor is involved in the pathogenesis of diabetic nephropathy. Diabetes. 2005;54:243–250. doi: 10.2337/diabetes.54.1.243. [DOI] [PubMed] [Google Scholar]
- 121.Wang JJ, Zhang SX, Mott R, Knapp RR, Cao W, Lau K, et al. Salutary effect of pigment epithelium-derived factor in diabetic nephropathy: evidence for antifibrogenic activities. Diabetes. 2006;55:1678–1685. doi: 10.2337/db05-1448. [DOI] [PubMed] [Google Scholar]