

Abstract
Although essential for the development of the nervous system, Wnt1 also has been associated with neurodegenerative disease and cognitive loss during periods of oxidative stress. Here we show that endogenous expression of Wnt1 is suppressed during oxidative stress in both in vitro and in vivo experimental models. Loss of endogenous Wnt1 signaling directly correlates with neuronal demise and increased functional deficit, illustrating that endogenous neuronal Wnt1 offers a vital level of intrinsic cellular protection against oxidative stress. Furthermore, transient overexpression of Wnt1 or application of exogenous Wnt1 recombinant protein is necessary to preserve neurological function and rescue neurons from apoptotic membrane phosphatidylserine externalization and genomic DNA degradation, since blockade of Wnt1 signaling with a Wnt1 antibody or dickkopf related protein 1 abrogates neuronal protection by Wnt1. Wnt1 ultimately relies upon the activation of Akt1, the modulation of mitochondrial membrane permeability, and the release of cytochrome c to control the apoptotic cascade, since inhibition of Wnt1 signaling, the phosphatidylinositol 3-kinase pathway, or Akt1 activity abrogates the ability of Wnt1 to block these apoptotic components. Our work identifies Wnt1 and its downstream signaling as cellular targets with high clinical potential for novel treatment strategies for multiple disorders precipitated by oxidative stress.
Key words: Akt, apoptosis, dickkopf, oxidative stress, phosphatidylserine, protein kinase B, stroke, wingless
Introduction
Wnt proteins derived from the Drosophila Wingless (Wg) and the mouse Int-1 genes are secreted cysteine-rich glycosylated proteins that oversee multiple cellular functions such as stem cell development, vascular regeneration, and maturation of the nervous system.1–8 Wnt proteins also are intimately tied to disorders that involve metabolic homeostasis and cardiovascular disease.9–11 For example, abnormalities in Wnt pathways, such as with transcription factor 7-like 2 gene, can have an increased risk for type 2 diabetes12–14 and be associated with the development of obesity.15 In addition, patients with coronary artery disease and the combined metabolic syndrome with hypertension, hyperlipidemia, and diabetes have shown impaired Wnt signaling through a missense mutation in LRP-6.16 Wnt family members also may provide cellular protection during periods of elevated glucose. Wnt can promote increased insulin sensitivity17 and protect glomerular mesangial cells from elevated glucose induced apoptosis.18 For the Wnt1 family member, studies demonstrate that Wnt1 works in conjunction with the cytokine erythropoietin to impart cellular protection during elevated glucose exposure.19–21
The Wnt pathway also plays a role in nervous system disorders and can be upregulated in models of frontotemporal dementia.22 It is conceivable that the increased expression of Wnt may improve memory function since agents such as lithium chloride that enhance Wnt signaling activity have shown to be beneficial for cognition.23 Genetic analysis also suggests that LRP-6 variants of the Wnt pathway may be associated with late onset Alzheimer disease24 and that upregulation of the Wnt pathway, such as with agents as cannabidiol, may provide alternative treatments for Alzheimer disease.25 In cell culture models of Alzheimer disease, β-amyloid production and cellular injury is reduced through increased Wnt signaling and β-catenin activity.26 In particular, Wnt pathways controlled by Wnt1 are of considerable interest since Wnt1 appears critical for neuronal protection during β-amyloid exposure against apoptotic neuronal injury and microglial activation.27
Here we show that endogenous and exogenous Wnt1 is vital for the protection of primary neurons during oxidative stress in both in vitro and in vivo experimental models. In primary neurons with oxygen-glucose deprivation (OGD), suppression of endogenous Wnt1 correlates with neuronal cell demise. Transient overexpression of Wnt1 or application of exogenous recombinant Wnt1 is crucial for the prevention of early apoptotic membrane PS externalization and subsequent DNA degradation, since loss of Wnt1 signaling through blockade by Wnt1 antibody (Wnt1Ab) or the recombinant Wnt antagonist dickkopf related protein 1 (DKK-1) leads to the loss of neuronal protection. Furthermore, Wnt1 reduces cerebral infarction and markedly improves neurological recovery during reversible middle cerebral artery occlusion (MCAO) in rats. Yet, blockade of Wnt1 signaling during MCAO significantly enhances cerebral infarction and neurological deficit, illustrating that endogenous neuronal Wnt1 also provides an important intrinsic level of neuroprotection during oxidative stress. Control of neuronal apoptotic injury by Wnt1 ultimately relies upon the activation of Akt1, the modulation of mitochondrial membrane permeability, and the release of cytochrome c, since inhibition of Wnt1 signaling, the phosphatidylinositol 3-kinase (PI 3-K) pathway, or Akt1 activity abrogates the ability of Wnt1 to control these apoptotic pathways. Our work identifies Wnt1 and its downstream signaling as possible pathways with high clinical potential for the targeting of multiple disorders precipitated by oxidative stress.
Results
Oxygen-glucose deprivation (OGD) results in cellular injury and reduced Wnt1 expression in neurons
We initially investigated neuronal survival after exposure to OGD at various periods of exposure of 1 hour, 2 hours, 3 hours and 4 hours. Cell survival was assessed with trypan blue exclusion 24 hours after OGD exposure. In Figure 1A, neuronal survival was significantly reduced over progressive times following OGD application to 73 ± 3% (1 hour), 51 ± 4% (2 hours), 32 ± 3% (3 hours), and 20 ± 3% (4 hours) when compared with untreated control cultures (86 ± 3%, p < 0.01). Since OGD exposure for a period of 3 hours resulted in survival rate of approximately 30% (a 70% neuronal cell loss), this duration of OGD was used for the reminder of the experimental paradigms.
Figure 1.
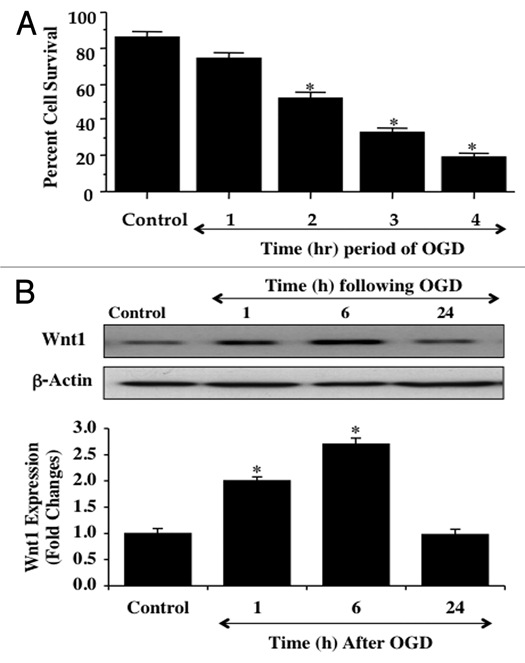
OGD leads to progressive injury in neurons and reduces endogenous Wnt1 expression over time. In A, primary hippocampal neurons were exposed to progressive durations of OGD at 1, 2, 3 and 4 hours and neuronal survival was determined 24 hours later by trypan blue dye exclusion assay. Neuronal survival was decreased to 73 ± 3% (1 hour), 51 ± 4% (2 hours), 32 ± 3% (3 hours), and 20 ± 3% (4 hours) following OGD exposure when compared with untreated control cultures (86 ± 3%, *p < 0.01 vs. Control). Each data point represents the mean and SEM from 6 experiments. In B, neuronal protein extracts (50 µg/lane) were immunoblotted with anti-Wnt1 (Wnt1) at 1, 6 and 24 hours following OGD exposure. Wnt1 expression is initially elevated at 1 hour and 6 hours, but then progressively and significantly is reduced at 24 hours following OGD exposure (*p < 0.01 vs. control)
We next examined whether OGD alters endogenous Wnt1 cellular expression. Western blot assay was performed for Wnt1 protein expression at 1, 6, and 24 hours following exposure to OGD that was applied for a 3 hour period. As shown in Figure 1B, OGD initially increased the expression of Wnt1 at 1 and 6 hours when compared to control, but a significant decrease in Wnt1 expression in neurons was observed at 24 hours following OGD exposure that was similar to control levels, suggesting that increases in Wnt1 expression are lost during OGD exposure. Quantitative results demonstrate that Wnt1 expression is significantly increased approximately two-fold at 1 hour and 2.5-fold at 6 hours following OGD, but is signifi- cantly decreased to approximately 1.0-fold at 24 hours after OGD exposure (Fig. 1B).
Wnt1 protects against neuronal injury during OGD
In Figure 2A, representative figures show significant trypan blue staining in wildtype neurons 24 hours after OGD or with vector expression during OGD. In contrast, trypan blue uptake is significantly reduced in neurons during Wnt1 overexpression in the presence of OGD. On further analysis in Figure 2A, neuronal survival was significantly increased to approximately 70% during OGD with Wnt1 overexpression but not in wildtype cells or during vector expression with OGD.
Figure 2.

Transient transfection of Wnt1 increases neuronal survival and prevents genomic DNA degradation and membrane ps externalization. In A, overexpression of Wnt1 was performed under the control of a CMV promoter with Wnt1 cDNA and cell survival was determined 24 hours after OGD exposure through the trypan blue dye exclusion method. Representative images illustrate decreased trypan blue staining during Wnt1 transient transfection. Significant cell injury and trypan blue staining occurs during OGD alone in wildtype cells and during vector transfection. Quantification of data demonstrates that OGD significantly decreased percent cell survival when compared to the control cells. In contrast, Wnt1 significantly increased cell survival (*p < 0.01 vs. OGD). Each data point represents the mean and SEM from six experiments. Control = untreated neurons. In B, overexpression of Wnt1 was performed under the control of a CMV promoter with Wnt1 cDNA and genomic DNA degradation was determined 24 hours after OGD exposure through TUNEL. Representative images illustrate decreased TUNEL staining during Wnt1 transient transfection. Significant DNA fragmentation occurs during OGD alone in wildtype cells and during vector transfection. Quantification of data demonstrates that OGD results in marked DNA fragmentation when compared to the control cells. In contrast, Wnt1 significantly prevents DNA fragmentation (*p < 0.01 vs. OGD). Each data point represents the mean and SEM from six experiments. Control = untreated neurons. In C, overexpression of Wnt1 was performed under the control of a CMV promoter with Wnt1 cDNA and membrane PS externalization was determined 24 hours after OGD exposure through annexin V phycoerythrin (green fluorescence). Representative images illustrate decreased PS staining during Wnt1 transient transfection. Significant membrane PS externalization occurs during OGD alone in wildtype cells and during vector transfection. Quantification of data demonstrates that OGD results in marked PS exposure when compared to the control cells. In contrast, Wnt1 significantly prevents PS externalization (*p < 0.01 vs. OGD). Each data point represents the mean and SEM from six experiments. Control, untreated neurons.
Wnt1 blocks apoptotic early phosphatidylserine (PS) exposure and subsequent nuclear DNA degradation during OGD
Apoptotic neuronal cell injury consists of both later genomic DNA degradation as well as early membrane PS exposure.28,29 In addition, the externalization of membrane PS residues in neurons undergoing apoptosis can result in inflammatory cell activation and identify these cells for phagocytosis.30,31 We therefore examined the ability of Wnt1 to alter genomic DNA degradation and membrane PS exposure in neurons during OGD exposure. In representative Figures 2B and 2C, untreated control wildtype neurons were without DNA fragmentation assessed by TUNEL or PS externalization assessed by annexin V. In wildtype neurons exposed to OGD or in neurons with vector expression during OGD, significant DNA fragmentation (TUNEL) and membrane PS exposure (annexin V) occurs 24 hours after exposure. In contrast, Wnt1 overexpression in the presence of OGD significantly prevented DNA fragmentation and membrane PS exposure, demonstrating that Wnt1 blocks apoptotic programs during OGD.
Wnt1 signaling provides necessary protection against cell injury in neurons
In Figure 3A, application of recombinant human Wnt1 protein (5–100 ng/ml) in neuronal cultures 1 hour prior to OGD exposure significantly increased neuronal cell survival. Neuronal survival was significantly reduced to 40 ± 3% following exposure to OGD when compared with untreated control cultures (92 ± 2%, p < 0.01). Yet, application of Wnt1 prior to OGD significantly increases neuronal cell survival to a maximum of 69 ± 3% with a Wnt1 concentration of 100 ng/ml. As a result, for subsequent experimental protocols, we used recombinant Wnt1 protein of 100 ng/ml.
Figure 3.

Wnt1 signaling is necessary for neuronal protection against OGD. In A, increasing concentrations of Wnt1 protein result in significantly increased neuronal survival assessed by trypan blue exclusion 24 hours after OGD (*p < 0.01 vs. OGD). In B, increasing concentrations of an antibody to Wnt1 (Wnt1Ab) during OGD do not alter neuronal survival assessed by trypan blue exclusion 24 hours after OGD when compared to neurons exposed to OGD alone. In C, increasing concentrations of an antibody to Wnt1 (Wnt1Ab) applied with Wnt1 (100 ng/ml) resulted in progressive loss of Wnt1 protection and increased neuronal cell injury assessed by trypan blue staining 24 hours after OGD (*p < 0.01 vs. OGD). In D, inhibition of Wnt1 signaling with DKK-1 (0.5 µg/ml), an antagonist of the Wnt/β-catenin pathway, administered with Wnt1 (100 ng/ml) significantly reduces protection by Wnt1 and neuronal cell survival assessed by trypan blue staining 24 hours after OGD (*p < 0.01 vs. OGD; †p < 0.01 vs. Wnt1/OGD). In all cases control = untreated neurons. Each data point represents the mean and SEM from six experiments.
We next examined whether specific antagonism against exogenous Wnt1 application with an antibody to Wnt1 (Wnt1Ab) could neutralize the protective capacity of Wnt1 during OGD. Initially, we examined application of the Wnt1Ab alone in Figure 3B. Administration of Wnt1Ab (0.01 – 1 µg/ml) alone did not significantly alter neuronal survival when compared to untreated control cultures (data not shown) or during exposure to OGD (Fig. 3B). In the presence of progressive concentrations of the Wnt1Ab (0.01 – 1 µg/ml) in combination with recombinant Wnt1 (100 ng/ml) during OGD (Fig. 3C), the protective capacity of Wnt1 was significantly reduced and reached a level that was similar to neurons exposed to OGD without Wnt1 at the Wnt1Ab concentration of 1 µg/ml. In addition, we examined the application of DKK-1 (0.5 µg/ml), an antagonist of the Wnt/β-catenin pathway that binds to LRP5/LRP6 and prevents formation of the Wnt/Frizzled/LRP complex,7,32 that has been shown to reduce the accumulation of free β-catenin.33 In Figure 3D, Wnt (100 ng/ml) significantly protected neurons form OGD exposure. In contrast, co-administration of Wnt1 (100 ng/ml) and DKK-1 (0.5 µg/ml) negated the protective effects of Wnt1, illustrating that Wnt1 and its signaling pathways are necessary for neuronal protection during OGD.
Wnt1 uses the PI 3-K pathway for neuronal protection during OGD
Wnt1 has been shown to employ protein kinase B (Akt1) for neuronal protection against amyloid toxicity.27 In addition, Akt alone is cytoprotective under a number of injury paradigms, such as hyperglycemia,34,35 hypoxia,36 β-amyloid (Aβ) toxicity,37 cellular aging,38 neurodegeneration39,40 and oxidative stress.41–43 We therefore examined the ability of Wnt1 to alter activity of Akt1 through phosphorylation during OGD (Fig. 4A). Western blot assay was performed for phosphorylated Akt1 (p-Akt1) and total Akt1 at 6 hours following OGD exposure. For Akt1 phosphorylation, Ser473 was examined since phosphorylation of Ser473 is considered to be critical for the complete activation of Akt1.44 In Figure 4A, application of Wnt1 (100 ng/ml) in untreated wildtype neurons or in the presence of OGD significantly elevated p-Akt1 expression to a greater extent than OGD alone. This increased expression of p-Akt1 was blocked by the PI 3-K inhibitor wortmannin (0.5 µM) that forms a covalent link with the lysine residue of PI 3-K45 (Fig. 4A) and by the specific Akt1 inhibitor SH-5 (20 µM).46 In Figure 4B, primary neurons treated with Wnt1 (100 ng/ml) following OGD exposure increased neuronal survival from 40 ± 3% to 67 ± 3%. However, application of wortmannin (0.5 µM) or SH-5 (20 µM) at concentrations that block activation of Akt1 activation (Fig. 4A) significantly reduced protection by Wnt1 during OGD, suggesting that Wnt1 requires activation of the PI 3-K and Akt for neuronal protection during OGD.
Figure 4.
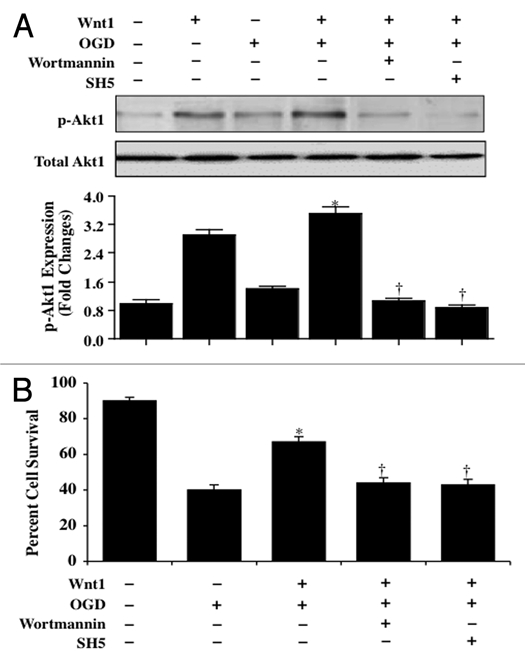
Wnt1 relies upon the PI 3-K pathway and Akt1 to provide neuronal protection. In A, primary neuronal protein extracts (50 µ/lane) were immunoblotted with anti-phosphorylated-Akt1 (p-Akt1, Ser473) or anti-total Akt1 at 6 hours following OGD. Application of Wnt1 (100 ng/ml) in untreated wildtype neurons or in the presence of OGD significantly elevated p-Akt1 expression to a greater extent than OGD alone. This increased expression of p-Akt1 by Wnt1 was blocked by the PI 3-K inhibitor wortmannin (0.5 µM) and by the specific Akt1 inhibitor SH-5 (20 µM). Total Akt1 is not altered (*p < 0.01, vs. OGD; †p < 0.01 vs. Wnt1/OGD). Each data point represents the mean and SEM from six experiments. In B, primary neurons treated with Wnt1 (100 ng/ml) increased neuronal survival assessed by trypan blue staining 24 hours after OGD. Yet, application of wortmannin (0.5 µM) or SH-5 (20 µM) at concentrations that block activation of Akt1 activation significantly reduced protection by Wnt1 24 hours after OGD (*p < 0.01, vs. OGD; †p < 0.01 vs. Wnt1/OGD). Each data point represents the mean and SEM from six experiments.
Wnt1 through Akt1 is required for neuronal survival during apoptotic membrane PS exposure and genomic DNA degradation
In Figure 5A, representative images illustrate that OGD alone leads to significant TUNEL staining and membrane PS exposure in neurons. In contrast and similar to our studies with Wnt1 overexpression in Figure 2, Wnt1 (100 ng/ml) administration significantly prevented DNA degradation assessed by TUNEL labeling and membrane PS externalization assessed by annexin V at 24 hours after OGD exposure (Fig. 5A and B). Furthermore, during inhibition of Wnt1 activity with Wnt1Ab (1 µg/ml) or with application of DKK-1 (0.5 µg/ml), neuronal protection was lost by Wnt1 with increased TUNEL labeling and membrane PS externalization in neurons during OGD exposure (Fig. 5A and B). In addition, inhibition of Akt1 activity by SH-5 (20 µM) during Wnt1 application also result in the loss of Wnt1 cytoprotection, demonstrating that the activation of Akt1 is an essential component for Wnt1 to prevent neuronal genomic DNA degradation and early membrane PS exposure during OGD.
Figure 5.
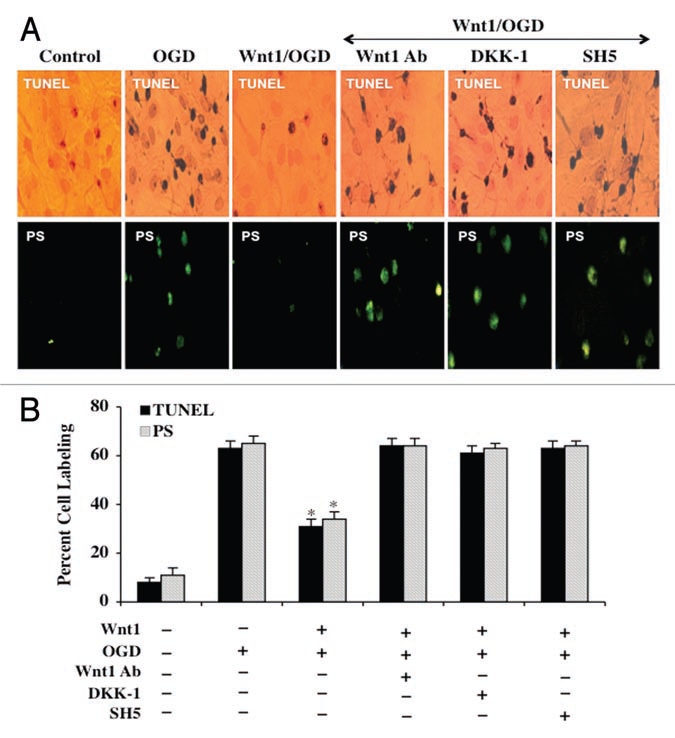
Wnt1 uses Akt1 to block apoptotic membrane PS exposure and genomic DNA degradation during OGD. In A, representative images illustrate that recombinant human Wnt1 protein (100 ng/ml) significantly blocks neuronal genomic DNA degradation assessed by TUNEL and membrane PS externalization assessed by annexin V phycoerythrin (green fluorescence) 24 hours following OGD. In contrast, inhibition of Wnt1 (100 ng/ml) signaling with Wnt1Ab (1 µg/ml) or with application of DKK-1 (0.5 µg/ml) or inhibition of Akt1 with SH-5 (20 µM) during Wnt1 application leads to the loss of Wnt1 protection. In B, quantification of data illustrates that DNA fragmentation and membrane PS externalization were significantly increased 24 hours following OGD when compared to untreated neuronal control cultures during Wnt1 (100 ng/ml) application with Wnt1Ab (1 µg/ml), DKK-1 (0.5 µg/ml), or SH-5 (20 µM). Each data point represents the mean and SEM from six experiments.
Wnt1 through Akt1 prevents mitochondrial depolarization and cytochrome c release during OGD
Mitochondrial depolarization in neurons was assessed during OGD by the cationic membrane potential indicator JC-1. In Figures 6A and 6B, OGD resulted in a significant decrease in the EC mitochondrial red/green fluorescence intensity ratio within 3 hours (42 ± 10%) when compared to untreated control mitochondria (100 ± 16%), demonstrating that OGD results in mitochondrial membrane depolarization. Administration of Wnt1 (100 ng/ml) in the presence of OGD significantly increased the red/green fluorescence intensity of the mitochondria to 86 ± 13%, illustrating that Wnt1 can significantly improve mitochondrial permeability transition pore membrane potential (Figs. 6A and B). Yet, inhibition of Wnt1 activity with Wnt1Ab (1 µg/ml) or with application of DKK-1 (0.5 µg/ml) during combined Wnt1 (100 ng/ml) application lead to significant mitochondrial depolarization during OGD, suggesting that the presence of Wnt1 activity is important for the control of mitochondrial permeability during OGD. Furthermore, inhibition of Akt1 activity by SH-5 (20 µM) during Wnt1 (100 ng/ml) application also resulted in the loss of mitochondrial permeability during OGD, demonstrating that Wnt1 uses Akt1 activation for the preservation of mitochondrial membrane function (Fig. 6A and B).
Figure 6.

Wnt1 inhibits mitochondrial depolarization and prevents the release of cytochrome c during OGD. In A, OGD leads to a significant decrease in the red/green fluorescence intensity ratio of mitochondria using the cationic membrane potential indicator JC-1 within 3 hours when compared with untreated control neurons, demonstrating that OGD leads to mitochondrial membrane depolarization. Application of Wnt1 (100 ng/ml) during OGD significantly increased the red/green fluorescence intensity of mitochondria in neurons, illustrating that mitochondrial membrane potential was restored by Wnt1. In contrast, inhibition of Wnt1 (100 ng/ml) signaling with Wnt1Ab (1 µg/ml) or with application of DKK-1 (0.5 µg/ml) or inhibition of Akt1 with SH-5 (20 µM) during Wnt1 application and OGD resulted in mitochondrial depolarization similar to OGD exposure alone. In B, the relative ratio of red/green fluorescent intensity of mitochondrial staining in untreated control neurons, in neurons exposed to OGD, during Wnt1 (100 ng/ml)/OGD application alone or during Wnt1 (100 ng/ml)/OGD with Wnt1Ab (1 µg/ml), DKK-1 (0.5 µg/ml), or SH-5 (20 µM) was measured in six independent experiments with analysis performed using the public domain NIH Image program (http://rsb.info.nih.gov/nih-image) (Wnt1/OGD vs. OGD, *p < 0.01; Wnt1/OGD with Wnt1Ab, DKK-1, or SH-5 vs. Wnt1/OGD, †p < 0.01). In C and D, equal amounts of mitochondrial (mito) or cytosol (cyto) protein extracts (50 µg/lane) were immunoblotted demonstrating that Wnt1 (100 ng/ml) significantly prevented cytochrome c release from mitochondria within 3 hours after OGD but Wnt1Ab (1 µg/ml), DKK-1 (0.5 µg/ml), or SH-5 (20 µM) during Wnt1/OGD application prevented Wnt1 from maintaining cytochrome c in the mitochondria (Wnt1/OGD vs. OGD, *p < 0.01; Wnt1/OGD with Wnt1Ab, DKK-1, or SH-5 vs. Wnt1/OGD, †p < 0.01).
OGD within 3 hours also produced a significant release of cytochrome c from the mitochondria to a 2.1 ± 0.1-fold increase when compared to untreated control mitochondria using western analysis (Fig. 6C and D). Wnt1 (100 ng/ml) during OGD prevented cytochrome c release to a similar degree that occurs with untreated control neuronal mitochondria (Fig. 6C and D). However, inhibition of Wnt1 activity with Wnt1Ab (1 µg/ml) or with application of DKK-1 (0.5 µg/ml) as well as the inhibition of Akt1 activity by SH-5 (20 µM) during Wnt1 (100 ng/ml) administration prevent Wnt1 from controlling the release of cytochrome c during OGD, supporting the premise that Wnt1 signaling in conjunction with Akt1 is required for preventing the release of cytochrome c from mitochondria in neurons during OGD exposure (Fig. 6C and D).
Transient cerebral ischemia suppresses endogenous Wnt1 cortical expression but exogenous Wnt1 reduces cerebral infarction
Similar to our studies that investigated the effects of OGD on Wnt1 endogenous expression in primary neurons, we examined whether cerebral ischemia in a transient MCAO rat model alters Wnt1 expression the cerebral cortex. To investigate the expression of Wnt1 in the cerebral cortex, focal cerebral ischemia was induced by insertion of a monofilament thread (4-0) into internal carotid artery and blockade of the origin of MCA. Reperfusion was performed following 90 minutes ischemia by withdrawal of the thread. As shown in Figure 7A and similar to our in vitro studies, transient cerebral ischemia initially increased the expression of Wnt1 at 1 and 6 hours following MCAO when compared to control. However, a significant decrease in Wnt1 expression was lost in the cortex at 24 hours following focal MCAO. Quantitative results demonstrate that Wnt1 expression is significantly increased approximately 1.8-fold at 1 hour and 2.0-fold at 6 hours in the cortex following MCAO, but is significantly decreased to approximately 1.0-fold at 24 hours after MCAO (Fig. 7A). Wnt1 expression on the contralateral non-infarction hemisphere was not altered from control, illustrating that the generation of cerebral ischemia directly led to changes in endogenous Wnt1 expression (Fig. 7A).
Figure 7.
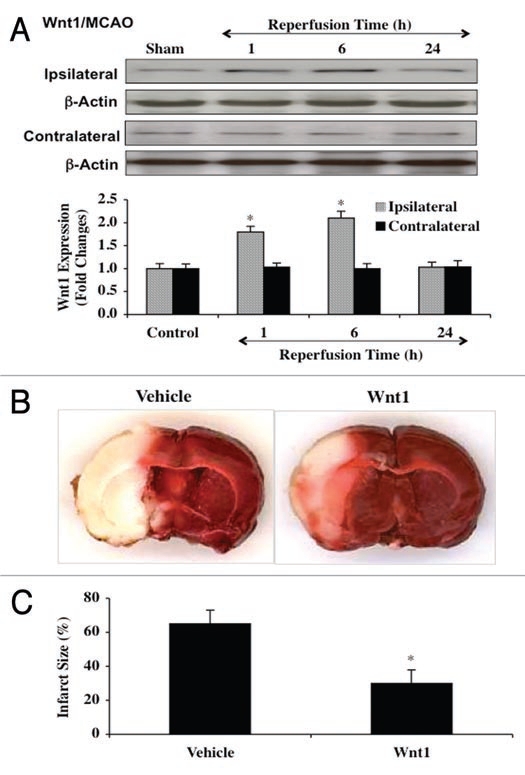
Transient cerebral ischemia blocks endogenous Wnt1 cortical expression but exogenous Wnt1 is protective against cerebral ischemia. In A and B, focal cerebral ischemia was induced by insertion of a monofilament thread (4-0) into the internal carotid artery and blockade of the origin of MCA. Reperfusion was performed following 90 minutes ischemia by withdrawal of the thread. For A, cortical protein extracts (50 µg/lane) were immunoblotted with anti-Wnt1 (Wnt1) at 1, 6 and 24 hours following OGD exposure. Similar to the effects upon Wnt1 expression with OGD in neuronal cultures, Wnt1 expression is initially elevated at 1 hour and 6 hours, but then progressively and significantly is reduced at 24 hours following OGD exposure (*p < 0.01 vs. control). Wnt1 expression on the contralateral non-infarction hemisphere was not altered from control, illustrating that the generation of cerebral ischemia directly led to changes in endogenous Wnt1 expression. In B, Wnt1 protein (24 µg/kg) was injected into the internal carotid artery through the external carotid artery at 30 minutes prior to the onset of MCAO and at the onset of reperfusion. Animals were euthanized 24 hours following ischemia and the infarct size was determined by 2,3,5-triphenytetrazolium (TTC) staining. Representative images show that the infarct size (white in color) was significantly reduced by treatment with Wnt1 protein. In C, quantitative results demonstrate that infarct size was significantly decreased by Wnt1 (24 µg/kg) treatment following reperfusion after cerebral ischemia. The total infarct size was expressed as a percentage of the contralateral hemisphere (*p < 0.05 vs. vehicle). In all cases, each data point represents the mean and SEM from six experiments.
Loss of Wnt1 expression following MCAO may be associated with hemisphere infarction. Following focal MCAO and reperfusion 90 minutes later, we assessed cerebral infarct size with 2,3,5-triphenytetrazolium (TTC) staining 24 hours following MCAO and reperfusion. In Figure 7B and 7C, MCAO resulted in an approximate 65% infarction of the hemisphere over the same period that endogenous Wnt1 expression is lost as shown in Fig. 7A. However, treatment with recombinant Wnt1 protein (24 µg/kg) administered into internal carotid artery through the external carotid artery prior to the onset of MCAO and once again during the onset of reperfusion markedly reduced cerebral infarction to a level of 30 ± 8% (Fig. 7B and C).
Wnt1 is necessary for reduction in cerebral infarction following transient cerebral MCAO and preserves neurological recovery
Given that exogenous Wnt1 application prior to cerebral MCAO infarction and reperfusion resulted in a significant reduction in cerebral infarct size per our studies in Figure 7B and C, we next evaluated whether blockade of Wnt1 and its signaling pathways could alter cerebral protection by Wnt1. In Figure 8A, representative serial cortical images reveal significant infarction in animals treated only with vehicle. Compilation of the total hemisphere infarction resulted in an approximate 50% infarction shown in Figure 8B. In contrast, treatment with recombinant Wnt1 protein (24 µg/kg) prior to the onset of MCAO and once again during the onset of reperfusion resulted in significant cerebral hemisphere protection with an infarct size of approximate 28% (Fig. 8A and B). Furthermore, treatment with Wnt1Ab (60 µg/kg) or DKK-1 (30 µg/kg) prior to the onset of MCAO and once again during the onset of reperfusion significantly increased infarct size to approximately 62% for each agent, illustrating that loss of Wnt1 activity or the induction of its downstream signal transduction pathways leads to marked cerebral injury following transient MCAO. In addition, cerebral infarction with these agents was worse than during vehicle administration alone, suggesting that an endogenous level of Wnt1 in the cerebral hemisphere may be required for endogenous protection against cerebral injury (Fig. 8A and B).
Figure 8.
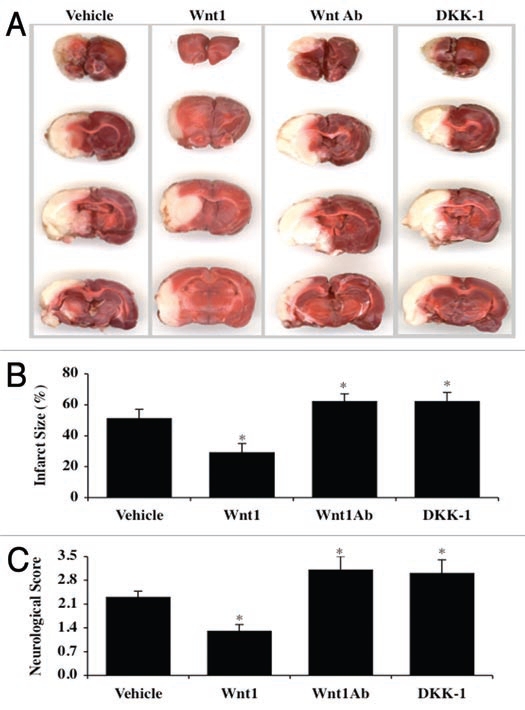
Wnt1 signaling is required for reduction in cerebral infarction following transient cerebral MCAO and maintains neurological recovery. In A, B and C, focal cerebral ischemia was induced by insertion of a monofilament thread (4-0) into the internal carotid artery and blockade of the origin of MCA. Reperfusion was performed following 90 minutes ischemia by withdrawal of the thread. In A, Wnt1 protein (24 µg/kg), Wnt1Ab (60 µg/kg), or DKK-1 (30 µg/kg) was injected into the internal carotid artery through the external carotid artery at 30 min prior to the onset of MCAO and at the onset of reperfusion. Animals were euthanized 24 hours following ischemia and infarct size was determined by 2,3,5-triphenytetrazolium (TTC) staining. Representative images illustrate that infarct size (white in color) was significantly reduced by treatment with Wnt1 protein. In contrast, infarct size was markedly increased by treatment with Wnt1Ab or DKK-1 during MCAO. In B, quantitative results demonstrate that infarct size was significantly decreased by Wnt1 (24 µg/kg) treatment following reperfusion after MCAO. However, infarct size was substantially increased with Wnt1Ab (60 µg/kg) or DKK-1 (30 µg/kg) administration during MCAO. The total infarct size was expressed as a percentage of the contralateral hemisphere (*p < 0.05 vs. vehicle). In C, the neurological deficit score was assessed in animals 24 hours following MCAO and reperfusion of a 90 minute period. Wnt1 (24 µg/kg) significantly lowered the neurological deficit score when compared to vehicle only treated animals. In contrast, Wnt1Ab (60 µg/kg) or DKK-1 (30 µg/kg) significantly increased the neurological deficit score (*p < 0.05 vs. vehicle). In all cases, each data point represents the mean and SEM from six experiments.
More importantly, cerebral protection by Wnt1 appears not only to be tied to reduction in cerebral infarct size following MCAO, but also is associated with improved neurological function. In Figure 8C, vehicle administration resulted in significant neurological score deficit of 2.3 ± 2%. Yet, Wnt1 (24 µg/kg) prior to the onset of MCAO and once again during the onset of reperfusion reduced the neurological deficit in animals to 1.3 ± 0.2%. In contrast, treatment with Wnt1Ab (60 µg/kg) or DKK-1 (30 µg/kg) prior to the onset of MCAO and once again during the onset of reperfusion not only significantly increased the neurological deficit score but to a greater degree than vehicle treated animals, further supporting that blockade of Wnt1 pathways prevents the ability of endogenous Wnt1 to offer cerebral protection during MCAO injury (Fig. 8B and C).
Discussion
Wnt proteins play a significant role in several biological functions that involve embryonic cell proliferation, cellular differentiation, and cell survival.4,7,31,47–57 Furthermore, Wnt signaling that involves β-catenin can promote renal cell survival during metabolic stress56 and may be involved in processes of transitional development such as muscle-to-fat cell conversion.55 Yet, the role of Wnt during cell injury, oxidative stress, and potential for drug platform development require further investigation.58 Wnt signaling has recently been reported to be downregulated in bone tissue during ethanol-induced oxidative stress,59 in endothelial cells during elevated glucose19 and in neurons during α-amyloid toxicity.27 We now show that Wnt1 expression is significantly diminished during oxidative stress in neurons in vitro and in vivo. Initially, Wnt1 expression is upregulated at 1 hour and 6 hours following OGD in primary hippocampal neurons and in the cerebral cortex following MCAO with reperfusion, suggesting that an endogenous cellular response occurs to increase Wnt1 expression during an oxidative stress insult. However, over a 24 hour course in both primary hippocampal neurons and in the cerebral cortex, endogenous Wnt1 expression is significantly lost that correlates with progressive neuronal injury.
Interestingly, we demonstrate that if expression of Wnt1 is maintained such as with transient overexpression of Wnt1 in primary neurons or with administration of the recombinant Wnt1 protein, both primary hippocampal neurons and cerebral cortical tissue are protected from oxidative stress. Prior studies have shown that co-culture of fibroblasts overexpressing Wnt1 can protect lymphoid cells from apoptosis60 and loss of β-catenin can increase infarct size during cerebral ischemia.61 In addition, overexpression of Wnt1 blocks β-amyloid toxicity in neuronal cells27 and administration of recombinant Wnt1 protects cells against apoptotic demise during elevated glucose in endothelial cells.19 We also demonstrate that Wnt1 has the ability to block both early apoptotic early PS externalization in neurons as well as subsequent genomic DNA degradation. This capacity of Wnt1 may be crucial for the therapeutic targeting of Wnt signaling for numerous disorders. Externalization of membrane PS residues during apoptosis can occur much earlier than nuclear DNA degradation and promote hypercoagulable states21,62 that lead to cardiovascular injury. PS exposure also can alert inflammatory cells to quickly phagocytize and eliminate injured cells with PS markings, potentially also removing functional cells and promoting further disability.36,43,63–67
Our in vivo and in vitro studies suggest that neuronal protection by Wnt1 is specific for Wnt1 signaling. With the MCAO experimental model, we initially observed that Wnt1 expression on the contralateral non-infarction hemisphere was not altered from control, but the ipsilateral ischemic hemisphere suffered from eventual loss of Wnt1 expression. This observation suggests that only cortical tissue subjected to oxidant stress is susceptible to injury as a result of the loss of endogenous Wnt1 expression. Our neuronal cell cultures studies also point to a course of cell demise over 24 hours following OGD that directly correlates with the loss of Wnt1 expression. Our subsequent studies employed the blockade of Wnt1 signaling with a Wnt1Ab or the antagonism of the Wnt/β-catenin pathway with DKK-1,7,32 an agent also considered for disorders with excessive Wnt activity,68 to illustrate that in primary neurons Wnt1Ab or DKK-1 can abrogate protection by Wnt1 during OGD, demonstrating that Wnt1 is necessary, at least as one potential component, to prevent cell injury and apoptotic PS exposure and DNA degradation. Furthermore, in the MCAO animal model, Wnt1Ab or DKK-1 worsened infarct size and increased the neurological deficit score, illustrating that endogenous Wnt1 in the brain offers significant protection against oxidative stress injury and functional recovery.
Neuronal protection by Wnt1 also relies upon the activation of Akt1, a pathway with increasing prominence for consideration in Wnt signaling.7,29,30,32,69 Several studies have documented that Wnt can rely upon Akt activation to support cell survival.27,52,56,70–72 In addition, Akt is a primary pathway to promote cell survival and growth such as during inflammation,73,74 cardiomyocyte hypertrophy,75 ischemic-preconditioning,76 hypoxia,36 amyloid toxicity,27,37,77–79 excitotoxicity,80 vascular disease81 and oxidative stress.41–43 We show that Wnt1 in primary hippocampal neurons maintains the phosphorylation and activation of Akt1 that would otherwise be lost during OGD exposure. More importantly, our work illustrates that inhibition of the PI 3-K pathway or specific inhibition of Akt1 activity leads to the loss of protection by Wnt1 against cell injury, membrane PS exposure, and DNA degradation during oxidative stress with OGD, demonstrating that Akt1 is a vital downstream pathway for Wnt1 neuronal protection.
In addition to reliance upon Akt1, we show that Wnt1 also regulates mitochondrial membrane permeability and cytochrome c release in conjunction with Akt1. Mitochondrial membrane permeability plays a significant role during cell survival and the initiation of the apoptotic cascade,41,80,82 especially during oxidative stress.20,28,83–86 In addition, Wnt signaling pathways have been tied to apoptotic mitochondrial signaling.27,72 Given that Akt1 maintains mitochondrial membrane potential, prevent cytochrome c release87–89 and block caspase activity,36,87,89 the ability of Wnt1 to oversee mitochondrial permeability appears to be a direct extension of the control of Akt1. Our studies illustrate that Wnt1 is necessary to prevent mitochondrial membrane permeability and the release of cytochrome c during OGD, since blockade of Wnt1 signaling with Wnt1Ab or DKK-1 removes the ability of Wnt1 to perform this function. Furthermore, Wnt1 does employ Akt1 to modulate mitochondrial membrane permeability since specific inhibition of Akt1 with SH-5 prevents maintenance of mitochondrial membrane function and cytochrome c release during OGD.
Wnt signaling is a vital component for the modulation of cell and tissue growth in the nervous system, but the role of Wnt1 as a cytoprotectant against apoptotic neurodegeneration during oxidative stress is only recently being elucidated. Our studies provide further understanding of Wnt signaling in regards to Wnt1 during oxidative stress. Our work illustrates that Wnt1 not only enhances primary neuronal cell survival in both in vitro and in vivo experimental models of oxidative stress, but also prevents the onset of early and late apoptotic injury programs of PS membrane externalization and nuclear DNA degradation during OGD. Wnt1 is a critical component against apoptotic neurodegeneration since blockade of Wnt1 signaling leads to the abrogation of Wnt1 cytoprotection. Furthermore, the protection offered to primary neurons by Wnt1 translates into improved functional recovery, highlighting the potential of Wnt1 pathways for clinical utility. Ultimately, neuroprotection by Wnt1 is mediated through activation of Akt1 that can govern the apoptotic loss in mitochondrial permeability and the release of cytochrome c. Our work presents new avenues for Wnt1 and its downstream modulation of Akt1 and mitochondrial membrane permeability for the consideration of novel therapies for a multitude of disorders tied to oxidative stress.
Materials and Methods
Primary hippocampal neuronal cultures
Briefly per our prior protocols,90,91 the hippocampi were obtained from E-19 Sprague-Dawley rat pups and incubated in Hanks' balanced salt solution (HBBS) supplemented with 1 mM sodium pyruvate and 10 mM HEPES buffer solution (Invitrogen, Carlsbad, CA). The neurons were isolated by trituration with 10 repetitions and then centrifuged for 2 minutes at 200 g. The cells were washed in growth medium (Leibovitz's L-15 medium, Invitrogen, Carlsbad, CA) containing 6% sterile rat serum (ICN, Aurora, OH), 150 mM NaHCO3, 2.25 mg/ml of transferrin, 2.5 µg/ml of insulin, 10 nM progesterone, 90 µM putrescine, 15 nM selenium, 35 mM glucose, 1 mM L-glutamine, penicillin and streptomycin (50 µg/ml), and vitamins. The dissociated neurons were plated at a density of ∼1.5 x103 cells/mm2 in 35 mm polylysine/laminin-coated plates and were maintained in growth medium at 37°C in a humidified atmosphere of 5% CO2 and 95% room air for 10–14 days. All animal experimentation was conducted in accord with accepted standards of humane animal care and NIH guidelines.
Experimental treatments
Oxygen-glucose deprivation (OGD) in microglia was performed by replacing the media with glucose-free HBSS containing 116 mmol/l NaCl, 5.4 mmol/l KCl, 0.8 mmol/l MgSO4, 1 mmol/l NaH2PO4, 0.9 mmol/l CaCl2 and 10 mg/l phenol red (pH 7.4) and cultures were maintained in an anoxic environment (95% N2 and 5% CO2) at 37°C per the experimental paradigm. For treatments applied prior to OGD, human recombinant Wnt1 protein (R&D Systems, Minneapolis, MN) or mouse monoclonal anti body against Wnt1 (Wnt1Ab) (R&D Systems, Minneapolis, MN), the phosphatidylinositol 3-kinase (PI-3 K) inhibitor wortmannin (Calbiochem, La Jolla, CA), the recombinant Wnt antagonist dickkopf related protein 1 (DKK-1, 0.5 µg/ml, R&D Systems, Minneapolis, MN), or D-3-deoxy-2-O-methyl-myo inositol 1-(R)-2-methoxy-3-(octadecyloxy) propyl hydrogen phosphate (SH-5) (Alexis, San Diego, CA) were continuous.
Assessment of cell survival
Cell injury was determined by bright field microscopy using a 0.4% trypan blue dye exclusion method 24 hours following treatment with OGD per our previous protocols.42,43,63,92 The mean survival was determined by counting eight randomly selected non-overlapping fields with each containing approximately 10–20 cells (viable + non-viable). Each experiment was replicated six times independently with different cultures.
Assessment of DNA fragmentation
Genomic DNA fragmentation was determined by the terminal deoxynucleotidyl transferase nick end labeling (TUNEL) assay.36,42 Briefly, microglial cells were fixed in 4% paraformaldehyde/0.2% picric acid/0.05% glutaraldehyde and the 3′-hydroxy ends of cut DNA were labeled with biotinylated dUTP using the enzyme terminal deoxytransferase (Promega, Madison, WI) followed by streptavidin- peroxidase and visualized with 3,3′-diaminobenzidine (Vector Laboratories, Burlingame, CA).
Assessment of membrane phosphatidylserine (PS) residue externalization
Phosphatidylserine (PS) exposure was assessed through the established use of annexin V. Per our prior protocols41,42 a 30 µg/ml stock solution of annexin V conjugated to phycoerythrin (PE) (R&D Systems, Minneapolis, MN) was diluted to 3 µg/ml in warmed calcium containing binding buffer (10 mmol/l Hepes, pH 7.5, 150 mmol/l NaCl, 5 mmol/l KCl, 1 mmol/l MgCl2, 1.8 mmol/l CaCl2). Plates were incubated with 500 µl of diluted annexin V for 10 minutes. Images were acquired with “blinded” assessment with a Leitz DMIRB microscope (Leica, McHenry, IL) and a Fuji/Nikon Super CCD (6.1 megapixels) using transmitted light and fluorescent single excitation light at 490 nm and detected emission at 585 nm.
Transient transfection of Wnt1 cDNA construct in primary neurons
Overexpression of Wnt1 was generated by transfecting cells with Wnt1 cDNA construct under the control of a CMV promoter with HA-tagged Wnt1 cDNA (6.75 kb) inserted into the Xba I site of pUSEamp (+) vector (Upstate Biotechnology, Lake Placid, NY). Cells were seeded into 35 mm dishes at a concentration of 1–1.5 × 105 and transfected with 1 µg cDNA by using lipofection with Lipofectamine reagent (Invitrogen, Carlsbad, CA) according to the instructions of the manufacturer.
Expression of Wnt1, phosphorylated Akt1, and total Akt1
Cells were homogenized and each sample (50 µg/lane) was subjected to SDS-polyacrylamide gel electrophoresis (12.5% Wnt1, 7.5% Akt1). After transfer, the membranes were incubated with a rabbit polyclonal antibody against Wnt1 (1:1000, R&D Systems, Minneapolis, MN), a rabbit polyclonal antibody against phospho-Akt1 (Ser473, 1:1000, Cell Signaling, Beverly, MA), or a rabbit antibody against total Akt1. Following washing, the membranes were incubated with a horseradish peroxidase (HRP) conjugated secondary antibody goat anti-rabbit IgG (1:2000, Zymed Laboratories, Carlsbad, CA). The antibody-reactive bands were revealed by chemiluminescence (Amersham Pharmacia Biotech, Piscataway, NJ) and band density was performed using the public domain NIH Image program (developed at the US National Institutes of Health and available at http://rsb.info.nih.gov/nihimage/).
Assessment of mitochondrial membrane potential
The fluorescent probe JC-1 (Molecular Probes, Eugene, OR), a cationic membrane potential indicator, was used to assess the mitochondrial membrane potential. Microglia in 35 mm dishes were incubated with 2 µg/ml JC-1 in growth medium at 37°C for 30 min. The cultures were washed three times using fresh growth medium. Mitochondria were then analyzed immediately under a Leitz DMIRB microscope (Leica, McHenry, IL, USA) with a dual emission fluorescence filter with 515–545 nm for green fluorescence and emission at 585–615 nm for red fluorescence.42
Preparation of mitochondria for the analysis of cytochrome c release
After washing once with ice-cold PBS, cells were harvested, homogenized, and the supernatants were centrifuged at 10,000 g for 15 minutes at 4°C. The resulting pellet was resuspended in buffer A (20 mM HEPES, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol, 0.1 phenylmethylsulfonylfluoride) containing 250 mM sucrose and used as the mitochondrial fraction. The supernatant was subjected to ultracentrifugation at 50,000 g for 1 hour at 4°C with the resultant supernatant used as the cytosolic fraction.42. Western blot detection for cytochrome c was performed by using a rabbit polyclonal antibody against cytochrome c (1:1000, Cell Signaling, Beverly, MA).
Reversible middle cerebral artery occlusion (MCAO)
Focal cerebral ischemia was induced by occlusion of the origin of middle cerebral artery (MCA) through insertion of intraluminal suture into the external carotid artery and then into the internal carotid artery to pass the origin of MCA.93,94 Briefly, male Sprague-Dawley rats (280∼300 g) were anesthetized with chloral hydrate (350 mg/kg, intraperitoneal). A 2 cm midline incision was made in the neck to expose the left common carotid, external carotid, and internal carotid arteries. Dissection of the arteries was performed to free them from surrounding nerves and tissues. A monofilament thread (4-0 with coating at the tip, Doccol, Redlands, CA) was inserted from the external carotid artery into the lumen of internal carotid artery and advanced into the internal carotid artery for 22 mm (from the bifurcation of the common carotid artery). The suture surrounding the external carotid artery to prevent bleeding was tightened and secured. Reperfusion was performed by withdrawal of the monofilament suture and closure of the external carotid artery. For treatment protocols, Wnt1 protein (24 µg/kg) (R&D System, Minneapolis, MN), Wnt1 antibody (15 µg/ml) (R&D System, Minneapolis, MN), or Wnt1 antagonist DKK-1 (24 µg/kg) (R&D System, Minneapolis, MN) were injected into internal carotid artery (ICA) through external carotid artery (ECA) at 30 minutes prior to MCAO and at the onset of reperfusion. Rectal temperature was maintained at 36.5 to 37.5°C throughout the experiment with a thermal blanket. All animal experimentation was conducted in accord with accepted standards of humane animal care and NIH guidelines.
MCAO infarct size determination
Animals were euthanized 24 hours following ischemia by decapitation and the brains were sliced into 3 mm coronal sections. The slices were incubated in 2% 2,3,5-triphenytetrazolium (TTC) solution for 30 minutes at 30°C and then were fixed in 4% paraformaldehyde. Each slice was scanned and the infarct area and the area of the contralateral hemisphere were performed using the public domain NIH Image program (developed at the US National Institutes of Health and available at http://rsb.info.nih.gov/nih-image/). The total infarct size was expressed as percentage of contralateral hemisphere.
Post MCAO animal neurological evaluation
Neurological deficit scores following MCAO in rats were determined by the following modified criteria95,96: no apparent deficit observed, deficit score = 0; with the animal suspended, only contralateral limb flexion observed, deficit score = 1; with the animal placed on a grille and gently pulled by the tail, the animal shows decreased grip in the contralateral forelimb, deficit score = 2; with the animal held by the tail and allowed to move freely showing cycling towards the contralateral side, deficit score = 3; and with the animal displaying severe paralysis without movement, deficit score = 4.
Statistical analysis
For each experiment, the mean and standard error were determined. Statistical differences between groups were assessed by means of analysis of variance (ANOVA) from six replicate experiments with the post-hoc Dunnett's test. Statistical significance was considered at p < 0.05.
Acknowledgments
This research was supported by the following grants to K.M.: American Diabetes Association, American Heart Association (National), Bugher Foundation Award, Janssen Neuroscience Award, LEARN Foundation Award, MI Life Sciences Challenge Award, Nelson Foundation Award, NIH NIEHS (P30 ES06639), NIH NIA, NIH NINDS, and NIH ARRA.
Abbreviations
- SH-5
D-3-deoxy-2-O-methyl-myo inositol 1-(R)-2-methoxy-3-(octadecyloxy) propyl hydrogen phosphate
- DKK-1
dickkopf related protein 1
- MCAO
middle cerebral artery occlusion
- OGD
oxygen-glucose deprivation
- PI 3-K
phosphatidylinositol 3-kinase
- PS
phosphatidylserine
- Akt1
protein kinase B
- TUNEL
terminal deoxynucleotidyl transferase nick end labeling
- Wnt1Ab
Wnt1 antibody
Footnotes
Previously published online: www.landesbioscience.com/journals/oximed/article/11758
References
- 1.Adachi K, Mirzadeh Z, Sakaguchi M, Yamashita T, Nikolcheva T, Gotoh Y, et al. beta-Catenin signaling promotes proliferation of progenitor cells in the adult mouse subventricular zone. Stem Cells. 2007;25:2827–2836. doi: 10.1634/stemcells.2007-0177. [DOI] [PubMed] [Google Scholar]
- 2.Bakre MM, Hoi A, Mong JC, Koh YY, Wong KY, Stanton LW. Generation of multipotential mesendodermal progenitors from mouse embryonic stem cells via sustained Wnt pathway activation. J Biol Chem. 2007;282:31703–31712. doi: 10.1074/jbc.M704287200. [DOI] [PubMed] [Google Scholar]
- 3.Cheng CW, Yeh JC, Fan TP, Smith SK, Charnock-Jones DS. Wnt5a-mediated non-canonical Wnt signalling regulates human endothelial cell proliferation and migration. Biochem Biophys Res Commun. 2008;365:285–290. doi: 10.1016/j.bbrc.2007.10.166. [DOI] [PubMed] [Google Scholar]
- 4.Jozwiak J, Kotulska K, Grajkowska W, Jozwiak S, Zalewski W, Oldak M, et al. Upregulation of the WNT pathway in tuberous sclerosis-associated subependymal giant cell astrocytomas. Brain Dev. 2007;29:273–280. doi: 10.1016/j.braindev.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 5.Maiese K. Triple play: Promoting neurovascular longevity with nicotinamide, WNT, and erythropoietin in diabetes mellitus. Biomed Pharmacother. 2008;62:218–232. doi: 10.1016/j.biopha.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maiese K, Chong ZZ, Shang YC, Hou J. Rogue proliferation versus restorative protection: where do we draw the line for Wnt and forkhead signaling? Expert Op on Ther Tar. 2008;12:905–916. doi: 10.1517/14728222.12.7.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maiese K, Li F, Chong ZZ, Shang YC. The Wnt signaling pathway: Aging gracefully as a protectionist? Pharmacol Ther. 2008;118:58–81. doi: 10.1016/j.pharmthera.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Speese SD, Budnik V. Wnts: up-and-coming at the synapse. Trends Neurosci. 2007;30:268–275. doi: 10.1016/j.tins.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chong ZZ, Li F, Maiese K. Oxidative stress in the brain: Novel cellular targets that govern survival during neurodegenerative disease. Prog Neurobiol. 2005;75:207–246. doi: 10.1016/j.pneurobio.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 10.Maiese K, Morhan SD, Chong ZZ. Oxidative stress biology and cell injury during type 1 and type 2 diabetes mellitus. Curr Neurovasc Res. 2007;4:63–71. doi: 10.2174/156720207779940653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maiese K, Shang YC, Chong ZZ, Hou J. Diabetes mellitus: Channeling care through cellular discovery. Curr Neurovasc Res. 2010;7:59–74. doi: 10.2174/156720210790820217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grant SF, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, Sainz J, et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet. 2006;38:320–323. doi: 10.1038/ng1732. [DOI] [PubMed] [Google Scholar]
- 13.Lehman DM, Hunt KJ, Leach RJ, Hamlington J, Arya R, Abboud HE, et al. Haplotypes of Transcription Factor 7-Like 2 (TCF7L2) Gene and Its Upstream Region Are Associated With Type 2 Diabetes and Age of Onset in Mexican Americans. Diabetes. 2007;56:389–393. doi: 10.2337/db06-0860. [DOI] [PubMed] [Google Scholar]
- 14.Scott LJ, Bonnycastle LL, Willer CJ, Sprau AG, Jackson AU, Narisu N, et al. Association of transcription factor 7-like 2 (TCF7L2) variants with type 2 diabetes in a Finnish sample. Diabetes. 2006;55:2649–2653. doi: 10.2337/db06-0341. [DOI] [PubMed] [Google Scholar]
- 15.Guo YF, Xiong DH, Shen H, Zhao LJ, Xiao P, Guo Y, et al. Polymorphisms of the low-density lipoprotein receptor-related protein 5 (LRP5) gene are associated with obesity phenotypes in a large family-based association study. J Med Genet. 2006;43:798–803. doi: 10.1136/jmg.2006.041715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mani A, Radhakrishnan J, Wang H, Mani A, Mani MA, Nelson-Williams C, et al. LRP6 mutation in a family with early coronary disease and metabolic risk factors. Science. 2007;315:1278–1282. doi: 10.1126/science.1136370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wright WS, Longo KA, Dolinsky VW, Gerin I, Kang S, Bennett CN, et al. Wnt10b Inhibits Obesity in ob/ob and Agouti Mice. Diabetes. 2007;56:295–303. doi: 10.2337/db06-1339. [DOI] [PubMed] [Google Scholar]
- 18.Lin CL, Wang JY, Huang YT, Kuo YH, Surendran K, Wang FS. Wnt/beta-catenin signaling modulates survival of high glucose-stressed mesangial cells. J Am Soc Nephrol. 2006;17:2812–2820. doi: 10.1681/ASN.2005121355. [DOI] [PubMed] [Google Scholar]
- 19.Chong ZZ, Shang YC, Maiese K. Vascular injury during elevated glucose can be mitigated by erythropoietin and Wnt signaling. Curr Neurovasc Res. 2007;4:194–204. doi: 10.2174/156720207781387150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maiese K, Chong ZZ, Li F, Shang YC. Erythropoietin: Elucidating new cellular targets that broaden therapeutic strategies. Prog Neurobiol. 2008;85:194–213. doi: 10.1016/j.pneurobio.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maiese K, Chong ZZ, Shang YC. Raves and risks for erythropoietin. Cytokine Growth Factor Rev. 2008;19:145–155. doi: 10.1016/j.cytogfr.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wiedau-Pazos M, Wong E, Solomon E, Alarcon M, Geschwind DH. Wnt-pathway activation during the early stage of neurodegeneration in FTDP-17 mice. Neurobiol Aging. 2009;30:14–21. doi: 10.1016/j.neurobiolaging.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Ferrari GV, Chacon MA, Barria MI, Garrido JL, Godoy JA, Olivares G, et al. Activation of Wnt signaling rescues neurodegeneration and behavioral impairments induced by beta-amyloid fibrils. Mol Psychiatry. 2003;8:195–208. doi: 10.1038/sj.mp.4001208. [DOI] [PubMed] [Google Scholar]
- 24.De Ferrari GV, Papassotiropoulos A, Biechele T, Wavrant De-Vrieze F, Avila ME, Major MB, et al. Common genetic variation within the low-density lipoprotein receptor-related protein 6 and late-onset Alzheimer's disease. Proc Natl Acad Sci USA. 2007;104:9434–9439. doi: 10.1073/pnas.0603523104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Esposito G, De Filippis D, Carnuccio R, Izzo AA, Iuvone T. The marijuana component cannabidiol inhibits beta-amyloid-induced tau protein hyperphosphorylation through Wnt/beta-catenin pathway rescue in PC12 cells. J Mol Med. 2006;84:253–258. doi: 10.1007/s00109-005-0025-1. [DOI] [PubMed] [Google Scholar]
- 26.Salins P, Shawesh S, He Y, Dibrov A, Kashour T, Arthur G, et al. Lovastatin protects human neurons against Abeta-induced toxicity and causes activation of beta-catenin-TCF/LEF signaling. Neurosci Lett. 2007;412:211–216. doi: 10.1016/j.neulet.2006.07.045. [DOI] [PubMed] [Google Scholar]
- 27.Chong ZZ, Li F, Maiese K. Cellular demise and inflammatory microglial activation during beta-amyloid toxicity are governed by Wnt1 and canonical signaling pathways. Cell Signal. 2007;19:1150–1162. doi: 10.1016/j.cellsig.2006.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maiese K, Chong ZZ, Hou J, Shang YC. The vitamin nicotinamide: translating nutrition into clinical care. Molecules. 2009;14:3446–3485. doi: 10.3390/molecules14093446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maiese K, Chong ZZ, Hou J, Shang YC. New strategies for Alzheimer's disease and cognitive impairment. Oxid Med Cell Longev. 2009;2:279–290. doi: 10.4161/oxim.2.5.9990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maiese K, Chong ZZ, Hou J, Shang YC. Oxidative stress: Biomarkers and novel therapeutic pathways. Exp Gerontol. 2010;45:217–234. doi: 10.1016/j.exger.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maiese K, Hou J, Chong ZZ, Shang YC. Erythropoietin, forkhead proteins, and oxidative injury: biomarkers and biology. Scientific World Journal. 2009;9:1072–1104. doi: 10.1100/tsw.2009.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li F, Chong ZZ, Maiese K. Winding through the WNT pathway during cellular development and demise. Histol Histopathol. 2006;21:103–124. doi: 10.14670/hh-21.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Endo Y, Beauchamp E, Woods D, Taylor WG, Toretsky JA, Uren A, et al. Wnt-3a and Dickkopf-1 stimulate neurite outgrowth in Ewing tumor cells via a Frizzled3- and c-Jun N-terminal kinase-dependent mechanism. Mol Cell Biol. 2008;28:2368–2379. doi: 10.1128/MCB.01780-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anitha M, Gondha C, Sutliff R, Parsadanian A, Mwangi S, Sitaraman SV, et al. GDNF rescues hyperglycemia-induced diabetic enteric neuropathy through activation of the PI3K/Akt pathway. J Clin Invest. 2006;116:344–356. doi: 10.1172/JCI26295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hou J, Chong ZZ, Shang YC, Maiese K. FoxO3a governs early and late apoptotic endothelial programs during elevated glucose through mitochondrial and caspase signaling. Mol Cell Endocrinol. 2010;321:194–206. doi: 10.1016/j.mce.2010.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chong ZZ, Kang JQ, Maiese K. Erythropoietin is a novel vascular protectant through activation of Akt1 and mitochondrial modulation of cysteine proteases. Circulation. 2002;106:2973–2979. doi: 10.1161/01.cir.0000039103.58920.1f. [DOI] [PubMed] [Google Scholar]
- 37.Chong ZZ, Li F, Maiese K. Erythropoietin requires NF-kappaB and its nuclear translocation to prevent early and late apoptotic neuronal injury during betaamyloid toxicity. Curr Neurovasc Res. 2005;2:387–399. doi: 10.2174/156720205774962683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tajes M, Yeste-Velasco M, Zhu X, Chou SP, Smith MA, Pallas M, et al. Activation of Akt by lithium: Prosurvival pathways in aging. Mech Ageing Dev. 2009;130:253–261. doi: 10.1016/j.mad.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 39.Morissette M, Al Sweidi S, Callier S, Di Paolo T. Estrogen and SERM neuroprotection in animal models of Parkinson's disease. Mol Cell Endocrinol. 2008;290:60–69. doi: 10.1016/j.mce.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 40.Morissette M, Le Saux M, D'Astous M, Jourdain S, Al Sweidi S, Morin N, et al. Contribution of estrogen receptors alpha and beta to the effects of estradiol in the brain. J Steroid Biochem Mol Biol. 2008;108:327–338. doi: 10.1016/j.jsbmb.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 41.Chong ZZ, Kang JQ, Maiese K. Akt1 drives endothelial cell membrane asymmetry and microglial activation through Bcl-x(L) and caspase 1, 3, and 9. Exp Cell Res. 2004;296:196–207. doi: 10.1016/j.yexcr.2004.01.021. [DOI] [PubMed] [Google Scholar]
- 42.Kang JQ, Chong ZZ, Maiese K. Critical role for Akt1 in the modulation of apoptotic phosphatidylserine exposure and microglial activation. Mol Pharmacol. 2003;64:557–569. doi: 10.1124/mol.64.3.557. [DOI] [PubMed] [Google Scholar]
- 43.Kang JQ, Chong ZZ, Maiese K. Akt1 protects against inflammatory microglial activation through maintenance of membrane asymmetry and modulation of cysteine protease activity. J Neurosci Res. 2003;74:37–51. doi: 10.1002/jnr.10740. [DOI] [PubMed] [Google Scholar]
- 44.Bellacosa A, Chan TO, Ahmed NN, Datta K, Malstrom S, Stokoe D, et al. Akt activation by growth factors is a multiple-step process: the role of the PH domain. Oncogene. 1998;17:313–325. doi: 10.1038/sj.onc.1201947. [DOI] [PubMed] [Google Scholar]
- 45.Wymann MP, Bulgarelli-Leva G, Zvelebil MJ, Pirola L, Vanhaesebroeck B, Waterfield MD, et al. Wortmannin inactivates phosphoinositide 3-kinase by covalent modification of Lys-802, a residue involved in the phosphate transfer reaction. Mol Cell Biol. 1996;16:1722–1733. doi: 10.1128/mcb.16.4.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kozikowski AP, Sun H, Brognard J, Dennis PA. Novel PI analogues selectively block activation of the prosurvival serine/threonine kinase Akt. J Am Chem Soc. 2003;125:1144–1145. doi: 10.1021/ja0285159. [DOI] [PubMed] [Google Scholar]
- 47.Espada J, Calvo MB, Diaz-Prado S, Medina V. Wnt signalling and cancer stem cells. Clin Transl Oncol. 2009;11:411–427. doi: 10.1007/s12094-009-0380-4. [DOI] [PubMed] [Google Scholar]
- 48.Jozwiak J, Wlodarski P. Hamartin and tuberin modulate gene transcription via beta-catenin. J Neurooncol. 2006;79:229–234. doi: 10.1007/s11060-006-9134-0. [DOI] [PubMed] [Google Scholar]
- 49.Kikuchi A, Yamamoto H, Sato A. Selective activation mechanisms of Wnt signaling pathways. Trends Cell Biol. 2009;19:119–129. doi: 10.1016/j.tcb.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 50.Lee JH, Lee EO, Kang JL, Chong YH. Concomitant degradation of beta-catenin and GSK-3 beta potently contributes to glutamate-induced neurotoxicity in rat hippocampal slice cultures. J Neurochem. 2008;106:1066–1077. doi: 10.1111/j.1471-4159.2008.05444.x. [DOI] [PubMed] [Google Scholar]
- 51.Luo JM, Dai CF, Lin SY, Huang PQ. Asymmetric syntheses and Wnt signal inhibitory activity of melleumin A and four analogues of melleumins A and B. Chem Asian J. 2009;4:328–335. doi: 10.1002/asia.200800355. [DOI] [PubMed] [Google Scholar]
- 52.Mercado-Gomez O, Hernandez-Fonseca K, Villavicencio-Queijeiro A, Massieu L, Chimal-Monroy J, Arias C. Inhibition of Wnt and PI3K signaling modulates GSK-3beta activity and induces morphological changes in cortical neurons: role of tau phosphorylation. Neurochem Res. 2008;33:1599–1609. doi: 10.1007/s11064-008-9714-9. [DOI] [PubMed] [Google Scholar]
- 53.Muruganandan S, Roman AA, Sinal CJ. Adipocyte differentiation of bone marrow-derived mesenchymal stem cells: cross talk with the osteoblastogenic program. Cell Mol Life Sci. 2009;66:236–253. doi: 10.1007/s00018-008-8429-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sutton LP, Honardoust D, Mouyal J, Rajakumar N, Rushlow WJ. Activation of the canonical Wnt pathway by the antipsychotics haloperidol and clozapine involves dishevelled-3. J Neurochem. 2007;102:153–169. doi: 10.1111/j.1471-4159.2007.04527.x. [DOI] [PubMed] [Google Scholar]
- 55.Vettor R, Milan G, Franzin C, Sanna M, De Coppi P, Rizzuto R, et al. The origin of intermuscular adipose tissue and its pathophysiological implications. Am J Physiol Endocrinol Metab. 2009 doi: 10.1152/ajpendo.00229.2009. In press. [DOI] [PubMed] [Google Scholar]
- 56.Wang Z, Havasi A, Gall JM, Mao H, Schwartz JH, Borkan SC. Beta-catenin promotes survival of renal epithelial cells by inhibiting Bax. J Am Soc Nephrol. 2009;20:1919–1928. doi: 10.1681/ASN.2009030253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wilusz M, Majka M. Role of the Wnt/beta-catenin network in regulating hematopoiesis. Arch Immunol Ther Exp. 2008;56:257–266. doi: 10.1007/s00005-008-0029-y. [DOI] [PubMed] [Google Scholar]
- 58.Bolognesi ML, Cavalli A, Bergamini C, Fato R, Lenaz G, Rosini M, et al. Toward a rational design of multitarget-directed antioxidants: merging memoquin and lipoic acid molecular frameworks. J Med Chem. 2009;52:7883–7886. doi: 10.1021/jm901123n. [DOI] [PubMed] [Google Scholar]
- 59.Chen JR, Lazarenko OP, Shankar K, Blackburn ML, Badger TM, Ronis MJ. A role for ethanol-induced oxidative stress in controlling lineage commitment of mesenchymal stromal cells through inhibition of Wnt/beta-catenin signaling. J Bone Miner Res. 2010 doi: 10.1002/jbmr.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Doubravska L, Simova S, Cermak L, Valenta T, Korinek V, Andera L. Wnt-expressing rat embryonic fibroblasts suppress Apo2L/TRAIL-induced apoptosis of human leukemia cells. Apoptosis. 2008;13:573–587. doi: 10.1007/s10495-008-0191-z. [DOI] [PubMed] [Google Scholar]
- 61.Lei ZN, Zhang LM, Sun FY. Beta-catenin siRNA inhibits ischemia-induced striatal neurogenesis in adult rat brain following a transient middle cerebral artery occlusion. Neurosci Lett. 2008;435:108–112. doi: 10.1016/j.neulet.2008.02.031. [DOI] [PubMed] [Google Scholar]
- 62.Dombroski D, Balasubramanian K, Schroit AJ. Phosphatidylserine expression on cell surfaces promotes antibody- dependent aggregation and thrombosis in beta2-glycoprotein I-immune mice. J Autoimmun. 2000;14:221–229. doi: 10.1006/jaut.2000.0365. [DOI] [PubMed] [Google Scholar]
- 63.Chong ZZ, Li F, Maiese K. The pro-survival pathways of mTOR and protein kinase B target glycogen synthase kinase-3beta and nuclear factor-kappaB to foster endogenous microglial cell protection. Int J Mol Med. 2007;19:263–272. [PMC free article] [PubMed] [Google Scholar]
- 64.Maiese K, Li F, Chong ZZ. New avenues of exploration for erythropoietin. JAMA. 2005;293:90–95. doi: 10.1001/jama.293.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mallat M, Marin-Teva JL, Cheret C. Phagocytosis in the developing CNS: more than clearing the corpses. Curr Opin Neurobiol. 2005;15:101–107. doi: 10.1016/j.conb.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 66.Shang YC, Chong ZZ, Hou J, Maiese K. The forkhead transcription factor FoxO3a controls microglial inflammatory activation and eventual apoptotic injury through caspase 3. Curr Neurovasc Res. 2009;6:20–31. doi: 10.2174/156720209787466064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shang YC, Chong ZZ, Hou J, Maiese K. FoxO3a Governs Early Microglial Proliferation and Employs Mitochondrial Depolarization with Caspase 3, 8, and 9 Cleavage During Oxidant Induced Apoptosis. Curr Neurovasc Res. 2009;6:223–238. doi: 10.2174/156720209789630302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McCarthy HS, Marshall MJ. Dickkopf-1 as a potential therapeutic target in Paget's disease of bone. Expert Op Ther Tar. 2010;14:221–230. doi: 10.1517/14728220903525720. [DOI] [PubMed] [Google Scholar]
- 69.Binet R, Ythier D, Robles AI, Collado M, Larrieu D, Fonti C, et al. WNT16B is a new marker of cellular senescence that regulates p53 activity and the phosphoinositide 3-kinase/AKT pathway. Cancer Res. 2009;69:9183–9191. doi: 10.1158/0008-5472.CAN-09-1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Balaraman Y, Limaye AR, Levey AI, Srinivasan S. Glycogen synthase kinase 3beta and Alzheimer's disease: pathophysiological and therapeutic significance. Cell Mol Life Sci. 2006;63:1226–1235. doi: 10.1007/s00018-005-5597-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chong ZZ, Li F, Maiese K. Stress in the brain: novel cellular mechanisms of injury linked to Alzheimer's disease. Brain Res Brain Res Rev. 2005;49:1–21. doi: 10.1016/j.brainresrev.2004.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Venkatesan B, Prabhu SD, Venkatachalam K, Mummidi S, Valente AJ, Clark RA, et al. WNT1-inducible signaling pathway protein-1 activates diverse cell survival pathways and blocks doxorubicin-induced cardiomyocyte death. Cell Signal. 2010;22:809–820. doi: 10.1016/j.cellsig.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Slaets H, Dumont D, Vanderlocht J, Noben JP, Leprince P, Robben J, et al. Leukemia inhibitory factor induces an antiapoptotic response in oligodendrocytes through Akt-phosphorylation and up-regulation of 14-3-3. Proteomics. 2008;8:1237–1247. doi: 10.1002/pmic.200700641. [DOI] [PubMed] [Google Scholar]
- 74.Williams R, Dhillon NK, Hegde ST, Yao H, Peng F, Callen S, et al. Proinflammatory cytokines and HIV-1 synergistically enhance CXCL10 expression in human astrocytes. Glia. 2009;57:734–743. doi: 10.1002/glia.20801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Munoz JP, Collao A, Chiong M, Maldonado C, Adasme T, Carrasco L, et al. The transcription factor MEF2C mediates cardiomyocyte hypertrophy induced by IGF-1 signaling. Biochem Biophys Res Commun. 2009;388:155–160. doi: 10.1016/j.bbrc.2009.07.147. [DOI] [PubMed] [Google Scholar]
- 76.Dreixler JC, Hemmert JW, Shenoy SK, Shen Y, Lee HT, Shaikh AR, et al. The role of Akt/protein kinase B subtypes in retinal ischemic preconditioning. Exp Eye Res. 2009;88:512–521. doi: 10.1016/j.exer.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Burgos-Ramos E, Martos-Moreno GA, Lopez MG, Herranz R, Aguado-Llera D, Egea J, et al. The N-terminal tripeptide of insulin-like growth factor-I protects against beta-amyloid-induced somatostatin depletion by calcium and glycogen synthase kinase 3 beta modulation. J Neurochem. 2009;109:360–370. doi: 10.1111/j.1471-4159.2009.05980.x. [DOI] [PubMed] [Google Scholar]
- 78.Burgos-Ramos E, Puebla-Jimenez L, Arilla-Ferreiro E. Minocycline provides protection against beta-amyloid(25–35)-induced alterations of the somatostatin signaling pathway in the rat temporal cortex. Neuroscience. 2008;154:1458–1466. doi: 10.1016/j.neuroscience.2008.04.036. [DOI] [PubMed] [Google Scholar]
- 79.Burgos-Ramos E, Puebla-Jimenez L, Arilla-Ferreiro E. Minocycline prevents Abeta(25-35)-induced reduction of somatostatin and neprilysin content in rat temporal cortex. Life Sci. 2009;84:205–210. doi: 10.1016/j.lfs.2008.11.019. [DOI] [PubMed] [Google Scholar]
- 80.Campos-Esparza MR, Sanchez-Gomez MV, Matute C. Molecular mechanisms of neuroprotection by two natural antioxidant polyphenols. Cell Calcium. 2009;45:358–368. doi: 10.1016/j.ceca.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 81.Vogiatzi G, Briasoulis A, Tousoulis D, Papageorgiou N, Stefanadis C. Is there a role for erythropoietin in cardiovascular disease? Expert Opin Biol Ther. 2010;10:251–264. doi: 10.1517/14712590903547819. [DOI] [PubMed] [Google Scholar]
- 82.Astiz M, de Alaniz MJ, Marra CA. Effect of pesticides on cell survival in liver and brain rat tissues. Ecotoxicol Environ Saf. 2009;72:2025–2032. doi: 10.1016/j.ecoenv.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 83.Lee I, Pecinova A, Pecina P, Neel BG, Araki T, Kucherlapati R, et al. A suggested role for mitochondria in Noonan syndrome. Biochim Biophys Acta. 2010;1802:275–283. doi: 10.1016/j.bbadis.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liang JH, Du J, Xu LD, Jiang T, Hao S, Bi J, et al. Catalpol protects primary cultured cortical neurons induced by Abeta(1-42) through a mitochondrialdependent caspase pathway. Neurochem Int. 2009;55:741–746. doi: 10.1016/j.neuint.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 85.Maiese K, Hou J, Chong ZZ, Shang YC. A fork in the path: Developing therapeutic inroads with FoxO proteins. Oxid Med Cell Longev. 2009;2:119–126. doi: 10.4161/oxim.2.3.8916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Su Y, Sun H, Fang J, Hu G, Xiao M. Brain mitochondrial dysfunction in ovariectomized mice injected with D-galactose. Neurochem Res. 2010;35:399–404. doi: 10.1007/s11064-009-0068-8. [DOI] [PubMed] [Google Scholar]
- 87.Chong ZZ, Kang JQ, Maiese K. Apaf-1, Bcl-xL, Cytochrome c, and Caspase-9 Form the Critical Elements for Cerebral Vascular Protection by Erythropoietin. J Cereb Blood Flow Metab. 2003;23:320–330. doi: 10.1097/01.WCB.0000050061.57184.AE. [DOI] [PubMed] [Google Scholar]
- 88.Chong ZZ, Lin SH, Kang JQ, Maiese K. Erythropoietin prevents early and late neuronal demise through modulation of Akt1 and induction of caspase 1, 3, and 8. J Neurosci Res. 2003;71:659–669. doi: 10.1002/jnr.10528. [DOI] [PubMed] [Google Scholar]
- 89.Chong ZZ, Kang JQ, Maiese K. Erythropoietin fosters both intrinsic and extrinsic neuronal protection through modulation of microglia, Akt1, Bad, and caspase-mediated pathways. Br J Pharmacol. 2003;138:1107–1118. doi: 10.1038/sj.bjp.0705161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chong ZZ, Li F, Maiese K. Attempted Cell Cycle Induction in Post-Mitotic Neurons Occurs in Early and Late Apoptotic Programs Through Rb, E2F1, and Caspase 3. Curr Neurovasc Res. 2006;3:25–39. doi: 10.2174/156720206775541741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chong ZZ, Lin SH, Maiese K. The NAD+ precursor nicotinamide governs neuronal survival during oxidative stress through protein kinase B coupled to FOXO3a and mitochondrial membrane potential. J Cereb Blood Flow Metab. 2004;24:728–743. doi: 10.1097/01.WCB.0000122746.72175.0E. [DOI] [PubMed] [Google Scholar]
- 92.Chong ZZ, Maiese K. Erythropoietin involves the phosphatidylinositol 3-kinase pathway, 14-3-3 protein and FOXO3a nuclear trafficking to preserve endothelial cell integrity. Br J Pharmacol. 2007;150:839–850. doi: 10.1038/sj.bjp.0707161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 94.Maiese K, Pek L, Berger SB, Reis DJ. Reduction in focal cerebral ischemia by agents acting at imidazole receptors. J Cereb Blood Flow Metab. 1992;12:53–63. doi: 10.1038/jcbfm.1992.7. [DOI] [PubMed] [Google Scholar]
- 95.Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472–476. doi: 10.1161/01.str.17.3.472. [DOI] [PubMed] [Google Scholar]
- 96.Menzies SA, Hoff JT, Betz AL. Middle cerebral artery occlusion in rats: a neurological and pathological evaluation of a reproducible model. Neurosurgery. 1992;31:100–106. doi: 10.1227/00006123-199207000-00014. discussion 6–7. [DOI] [PubMed] [Google Scholar]