

Abstract
There are few reports on the isolation, quantitative recovery, and relative purification of infectious particles that cause scrapie, Creutzfeldt-Jakob disease (CJD) and epidemic bovine spongiform encephalopathy (BSE). Because pure prion protein (PrP) has failed to show significant infectivity, it is critical to find other molecules that are integral agent components. Only complex diseased tissues such as degenerating brain have been fractionated, and agent recoveries have been quite low in concentrated abnormal prion protein (PrP-res) preparations. To simplify the purification of infectious particles, we evaluated a monotypic cell line that continuously produced high levels of the 22L scrapie agent (N2a-22L). A new rapid and accurate GT1 culture assay was used to titrate infectivity in six representative sucrose gradients. We developed a streamlined ~3-h procedure that yielded full recovery of starting infectivity in fractions with only a few selected protein bands (representing <1% of starting protein). Infectious particles reproducibly sedimented through >30% sucrose steps, whereas PrP and PrP-res sedimentation varied depending on the conditions used. Both normal and abnormal PrP could be largely separated from infectivity in a single short centrifugation. Because no foreign enzymes were added to achieve reasonably purified infectious particles, these preparations may be used to elicit diagnostic antibodies to foreign agent proteins.
Introduction
Transmissible spongiform encephalopathies (TSEs), such as human Creutzfeldt-Jakob disease (CJD), endemic sheep scrapie, epidemic bovine spongiform encephalopathy (BSE), and chronic wasting disease of cervids, are lethal neurodegenerative diseases caused by a group of related infectious agents that maintain their individual strain identities even after passage in different species (1). TSEs are therefore distinct from other late-onset neurodegenerative and amyloid-forming diseases that are not transmissible, such as common forms of Alzheimer's disease. It has been proposed that in TSEs a normal host membrane protein, known as the prion protein (PrP), becomes infectious through its own spontaneous or self-seeded amyloid misfolding (20,21). The prion hypothesis, that PrP is infectious, was first proposed in 1982 (22), and this notion has become widely accepted despite many experimental caveats (11). Most notably, intense efforts in many laboratories have failed to show reproducible evidence that PrP alone can convert into a form that encodes either significant infectivity or strain-specific properties. Although abnormal forms of PrP, such as aggregates that resist limited proteolysis (PrP-res), can be linked to infectious disease, they are not restricted to infectious samples, and thus are not specific for TSE infection (see Discussion). Because abnormal PrP is more likely to be a pathological product rather than the causal infectious agent (12,17), it has become critical to monitor non-PrP agent components for both diagnostic and preventive purposes. The recovery and relative purification of infectious particles with respect to starting material also needs to be addressed systematically with reference to standard cell numbers or grams of tissue. This type of quantitative purification analysis is rarely presented for most TSE agent preparations.
To the best of our knowledge there are no reports on the isolation or biophysical characteristics of a TSE agent in monotypic cultured cells. This may be because cultured cells generally show very low infectivity levels in animal bioassays, most often 100–10,000-fold less than that found in brain tissue (1). We have stably propagated a representative group of different TSE agent strains in murine culture models, and some of these models carry high brain-like levels of infectivity. These monotypic cells should be easy to fractionate. In contrast, it has been difficult to cleanly separate infectious particles from degenerating brain products and complex components such as collagen, myelin, vessels, and nuclei. Nevertheless, previous brain studies have shown a single homogeneous 120S peak of infectivity, as well as a virus-like density of 1.28 g/mL in sucrose gradient fractions (23,25). These infectious fractions, that have reduced PrP, also contain round virus-like particles of 25 nm diameter in size that do not bind to PrP antibodies (11). Furthermore, conditions that disrupt viral particles, and solubilize nucleic acids and protective nucleic acid binding proteins, lead to a marked reduction in TSE infectious titers (16,24). We evaluated several sucrose gradient conditions to find out if cytoplasmic supernatants from a highly infectious 22L scrapie-infected N2a cell line (N2a-22L) would yield high recoveries of 120S infectious particles with few residual proteins.
The reproducible sedimentation of infectivity in all gradients was verified by using a new, rapid and accurate GT1 culture assay for infectious components (8). Only infectious material induces continuous production of pathological PrP as well as infectivity in GT1 cells (1,8,11,19). These assays allowed us to define the most streamlined procedure for purifying infectious particles from nuclei, and from the vast majority of cytoplasmic proteins. N2a-22L infectious particles behaved comparably to those from brain, and they could be quantitatively recovered in concentrated sucrose fractions that contained very low amounts of protein. Only a few electrophoretic bands were detected in these infectious fractions. In contrast to infectious particles, PrP and PrP-res sedimentation was variable, and was dependent on both preparative and gradient conditions. The high yield of substantially purified infectious particles can be used to elucidate the structure and the essential molecular components that define TSE agent strains.
Materials and Methods
Murine N2a58 neuroblastoma cells infected with the 22L scrapie agent (N2a-22L) and its derivative clones such as C2a-22L were split at 1:4 every 4 d as previously described (18). These N2a-22L cells display 25-nm dense particles in TE virus-like arrays, and have been titered for infectivity by both conventional mouse assays, as well as by a new rapid tissue culture assay as previously described (8). In this assay, hypothalamic neuronal GT1 indicator cells are exposed once to dilutions of infected test brain homogenate, or to culture subcellular fractions, and the resultant %PrP-res (of total GT1 PrP) determined by Western blotting at progressive passages (p) after exposure. Exposure to uninfected brain and cell preparations show no detectable PrP-res by Western blot as well as by in-situ analysis. Only infectious material verified by mouse bioassays can induce, by a single exposure, continuous and progressive increases in PrP-res production in subsequent cell passages. Moreover, all GT1 cells with PrP-res produce an infectious spongiform encephalopathy in mice (e.g., 1). Additionally, GT1 cells exposed to infectious material produce only GT1-specific PrP-res band patterns, whereas infected brain and N2a cells produce very different patterns [(8) and see Fig. 4]. GT1 cells sampled at both p3 and p5 after exposure yield reproducible tissue culture infectious doses (TC-ID) that directly relate to the progressively increasing percentage of PrP-res to total PrP produced. Test samples are applied at p0 to GT1 cells at several dilutions to ensure that the infectivity assay is in the linear range, as is done for animal incubation time assays (17). This assay can accurately discriminate the TC-ID over ≥3 logs, as shown by dilution curves and tests of different cell samples (8). GT1 cells can also be used to determine accurate end-points (TC-ID50). The C2a-S1 clone (8) was used for most of the >14 gradient studies here. Whole-cell lysates, as well as low-speed supernatants of these cells, sampled from passages 39–226, consistently yielded 3 × 107 TC-ID per 109 cells. This represents ~30% of the infectivity of brain infected with 22L scrapie at end-stage disease (108 TC-ID per 109 cells, where 109 cells = 1 g of brain). However, >80% of these N2a-22L cells displayed pathological PrP-res, suggesting >1 agent particle per 3 cells. For reference, the ~3 wk culture assay for infectivity also gives a ~3-fold lower estimate of end-point infectious doses (TC-ID50) than the >350-d mouse bioassay (LD50) of parallel samples (8).
FIG. 4.
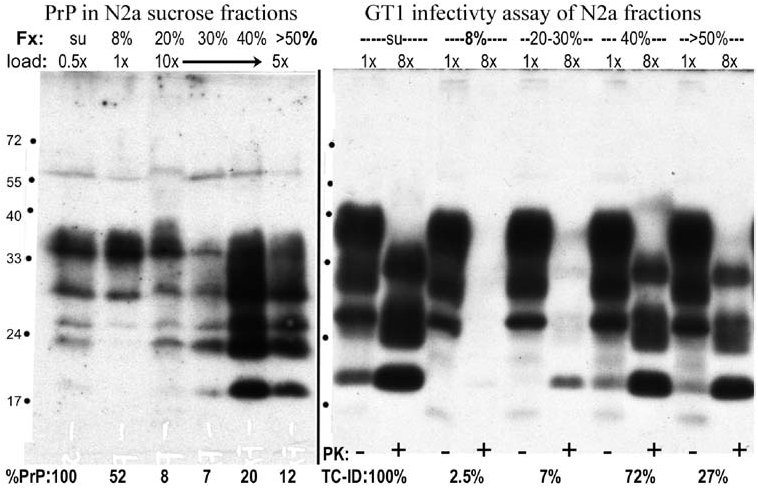
Infectivity assays of fractions from the other 1.5-h gradients showed that the vast majority of infectious particles again migrated to the bottom of the tube, whereas the majority of PrP remained in the top, as shown here for pooled fractions of a gradient loaded with unsonicated supernatant. The left panel shows the %PrP in each fraction, and the right panel shows aliquots of the same samples with equivalent cell equivalents applied to GT1 indicator cells for the infectivity assay; the average of four assay points (TC-ID) are shown beneath each GT1 lane. This blot is a representative sample from GT1 cells assayed at passage 3. The bottom fractions (>30% sucrose) contain virtually all the applied infectivity, whereas the top 8% sucrose fraction with high PrP contained <3% of the TC-ID; 108% of the loaded infectivity was recovered in these gradient fractions.
N2a-22L and N2a-mock infected cells grown in 75-cm flasks were washed with PBS, collected by scraping, and pelleted at 1500 rpm (450 g) × 10 min in an HS 4 Sorvall rotor at 4°C. The cell pellet (~108 cells) was gently suspended by tapping in the remaining ~0.5 mL of PBS, and 5 mL of lysis buffer [100 mM NaCl, 10 mM TrisCl pH 7.5, 5 mM EDTA pH 8.0, 0.5% Nonidet P40 (NP40), 0.5% sodium deoxycholate (DOC), and 5% or 8% sucrose] was added. An aliquot of the total lysate was removed after 5 min at 22°C for protein and other assays, and the lysate was then spun at 2300 rpm (1000 g) × 15 min at 4°C; 4′-6-diamidino-2-phenylindole (DAPI) was sometimes included at 5 μg/mL to monitor fluorescent nuclear DNA pelleting visualized with a hand-held UV light. The cytoplasmic supernatant was carefully removed, and 2 mL of lysis buffer without DAPI was added to resuspend the loose nuclear pellet. After a 3600-rpm (2500-g) × 10 min spin at 4°C, both post-nuclear supernatants were pooled (6–8 mL total per ~108 cells), and an aliquot of the sonicated nuclear pellet and cytoplasmic supernatant were taken for assays. Remaining supernatant was immediately frozen at −70°C, and after thawing was mixed by vortexing, or was vigorously bath sonicated (10 × 10 sec with 15 sec rests in ice) just before loading on sucrose step gradients. The supernatant sample was overlayered with TE (10 mM TrisCl, 1 mM EDTA, pH 8.0) buffer to fill the tube to the top, a procedure that helped minimize potential contamination of buckets. When sarkosyl was added (to 1% final), the supernatant was vortexed and could be cleared by an additional spin at 10,000 g × 20′ before loading on the gradient (see Fig. 5). Sucrose steps contained lysis buffer without detergents unless otherwise indicated, and sucrose step concentrations were verified by refractive index; detergent-free sucrose steps removed most detergents from the applied lysate. A 300-μL sucrose cushion of 60–65% was sequentially overlayered with five steps of lower sucrose concentrations (1–2 mL each) as indicated in the representative figures. Ultracentrifugation in a Beckman SW41 Ti rotor (Beckman-Coulter, Fullerton, CA) was done either at 41,000 rpm (207,000 g av) at 20°C for 1 h 15 min, or at 38,000 rpm (181,000 g av) × 2 h 30 min. Fractions were collected from the top by first removing the entire 7–8 mL supernatant layer, and then collecting 2–3 samples (~500 μL each) from underlying layers for assays. The sucrose steps, marked on the tube side, facilitated this collection, and the sucrose concentrations in the figures refer to the original sucrose layers to avoid contaminating the refractometer. When recovering the high-sucrose bottom-most fraction, although no pellet was visible, care was taken to include a ~200-μL buffer wash of the bottom of the tube.
FIG. 5.
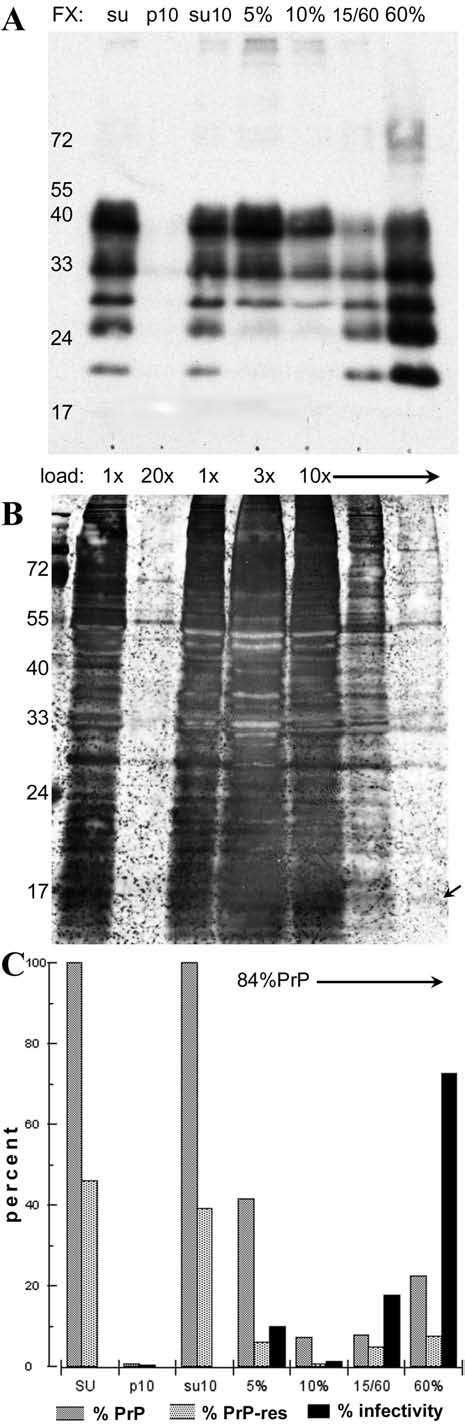
Streamlined and simplified step gradient for high recovery of infectivity with very low protein background. Supernatant lysates were sonicated in 1% sarkosyl, clarified at 10,000 g × 20 min to generate p10 and su10, and the su10 was loaded on a three-step gradient (9.3 mL of su10 in 5% sucrose with 108 cell equivalents was layered on 2 mL of a 10%, 0.5 mL of a 15%, and 0.1 mL of a 60% sucrose step), and spun for 1.25 h as above. Panel A shows the PrP blot, panel B shows the gold-stained blot, and panel C graphs the quantitative recovery of PrP (dark grey bars), PrP-res (light grey bars), and infectivity (black bars) in each fraction. The low-volume 60% sucrose step concentrates 75% of the infectivity and is highly purified with respect to starting protein. A few proteins, as at arrow, are also selectively enriched with little background.
Protein assays, Western blots for PrP and PrP-res, gold staining, and infectivity assays were performed and quantitated as previously described (8,25). For infectivity assays each fraction was diluted in feeding medium, and limiting dilutions (cell equivalents that induced the lowest %PrP-res in GT1 cells) were used to calculate the TC-ID. To avoid cell toxicity, final concentrations of NP40-DOC were <0.005%, sarkosyl was <0.01%, and sucrose was <3%. SDS could also be used successfully in this GT1 infectivity assay if diluted to <0.005%. Samples of supernatant and the corresponding high-detergent supernatant and gradient fractions could also be concentrated and washed with buffer at 500 g × 3 using a centrifugal microcon YM100 filter (Millipore, Billerica, MA) without losses (see Fig. 1). Each infectious assay here reflects the average titer determined from both p3 and p5 duplicate wells, where both input dilutions were in the validated TC-ID assay range.
FIG. 1.
Western blot of a N2a-22L cell lysate (LY), nuclear pellet (p), and cytoplasmic supernatant (su) fraction centrifuged on a sucrose step gradient for 2.5 h (453,000 g-h). The top panel shows PrP detected with antibody M20 before and after proteinase K digestion (PK − and +, respectively). The bottom panel shows gold-stained total proteins in the PK− lanes (stained after PrP detection). Loads on each lane were proportional and standardized for comparable cell equivalents (e.g., 10 μL of a 5-mL 5% sucrose layer is equivalent to 1 μL of a 0.5-mL 15% sucrose step layer). Negligible amounts of PrP or PrP-res are lost in the nuclear pellet (loaded with 20× and 30× of the starting lyate), whereas essentially all PrP and PrP-res are recovered in the supernatant. Total gold stained protein shows almost all the loaded lysate protein is recovered in the supernatant, with little protein in the nuclear pellet (p); this pellet showed high DAPI fluorescence, and also revealed a highly selective concentration of only a few protein bands (e.g., at arrow), while the supernatant contained little nuclear DNA (nucleic acid sequences in infectious fractions will be reported separately). To test if microcon concentration with buffer washing could be used to reduce detergent and sucrose concentrations without losses, aliquots of the microcon-filtered supernatant and several sucrose steps were compared (* samples); the PrP, PrP-res, and protein in both were comparable. Note that the bottom-most 60% sucrose fraction (loaded at 8× the supernatant) has very little protein, but a relatively high concentration of PrP and PrP-res. This gradient was loaded with 1.2 × 108 cell equivalents (CE) of supernatant. The cytoplasmic supernatant was brought to ~8.8 mL in 5% sucrose, and loaded onto a step gradient with 1 mL of 10%, 1 mL of 15%, 0.5 mL of 30%, and 0.1 mL of 60% sucrose in TE buffer.
Results
The stable propagation of high levels of infectivity in cultured cells combined with a new rapid culture assay for infectious titers (8) made it possible to systematically develop streamlined procedures for high-yield recoveries of infectious agent, as well as a substantial purification with respect to starting protein. These studies also tested if infected yet healthy monotypic cultures produced particles with the same virus-like sedimentation properties as those found in highly complex degenerating brain samples. In infected brain, the highest recoveries after differential centrifugation and/or sucrose gradient purification have been 15%, and published results of others typically show recoveries of only 0.1–1% of the starting brain infectivity in more purified preparations (23). Protein contamination can also be substantial, especially when obscured by proteinase K (PK) digestion to monitor PrP-res. We avoided PK digestion in order to directly visualize all the protein bands, including those of PrP, in highly infectious gradient fractions. Additionally, we chose N2a58 cells infected with the prototypic 22L scrapie agent (N2a-22L) because these cells 1) carry stable brain-like levels of infectivity, 2) display arrays of 25-nm virus-like particles, and 3) show different PrP and PrP-res bands than the target GT1 indicator cells used for agent titration (1,8,18). Although N2a-22L cells show no obvious degenerative changes, they do show large amounts of intracellular PrP amyloid fibrils that could compromise agent isolation. The results below are a fair representation of more than 14 independent N2a-22L gradient separations, six of which were analyzed for end-point infectivity.
We developed simplified sucrose step gradients that could be loaded with more material than continuous sucrose velocity sedimentation gradients previously used to define the 120S size of infectious particles. Because detergents often interfere with downstream enzymatic digestions, (e.g., by nucleases), supernatants were typically loaded on a sucrose step gradient without detergents. Additionally, detergents and sucrose can be toxic to the GT1 cells used for infectivity assays (see Materials and Methods). We therefore tested methods that might reduce toxic molecules without losses, and found microcon concentration with buffer washing could be useful. Parallel aliquots of microcon concentrated and untreated samples were assessed for PrP and protein recovery as shown in Fig. 1. The microcon-washed cytoplasmic supernatant (lane marked su*) as well as the concentrated and washed 5% and 10% sucrose gradient fractions (*) contained comparable amounts of PrP bands and gold-stained proteins as their untreated counterparts.
Fig. 1 also shows the starting whole-cell NP40-DOC lysate (LY), the derivative nuclear pellet (p), and all the sucrose steps. The cytoplasmic supernatant in this example was not sonicated prior to loading on the gradient, and was fractionated after an extended centrifugation time (2.5 h). Samples were analyzed on Western blots for both total PrP (PK-lanes) and PrP-res (PK+ lanes). These analyses demonstrate that the nuclear pellet, as compared to the starting lysate, contains little PrP or PrP-res (lanes loaded at 20× and 60×, respectively, of the lysate samples). The gold lane also shows that there is relatively little protein in the crude nuclear pellet, even at these high loads, with enrichment of a few specific selected protein bands (as at arrow). In contrast, the supernatant contains virtually the same amount of PrP, PrP-res, and total protein as the starting lysate. In this 2.5-h gradient, most of the PrP sedimented, whereas the bulk of the gold-stained protein stayed in the top 5% and 10% sucrose fractions. The 30% sucrose layer selected only very few gold-stained proteins (as at arrow), and the 60% fraction contained almost invisible amounts of protein. Even bands of PrP, that can be easily stained with colloidal gold (25), were not apparent in this bottom-most fraction.
Fig. 2A shows the quantitative results for this preparation and gradient. Less than 1% of PrP and PrP-res were lost in the nuclear pellet, and essentially all the starting PrP and PrP-res were found in the supernatant. Sucrose gradient fractions recovered 95% of the loaded PrP, ~⅔ of which sedimented into the 60% sucrose step. Only a small proportion of PrP-res remained at the top 5% sucrose step, and it accounted for <1% of the PrP in the 5% fraction. In contrast, PrP-res accounted for 21% of the 64% PrP recovered in the bottom 60% sucrose step. However, although the total PrP recovery was quantitative from gradients, the PrP-res recovered was <⅓ of that loaded on the gradient; in fact, a selective decrease in recovery of PrP-res was a consistent finding for all the gradients analyzed. One possibility for this selective “loss” of PrP-res may be due to disaggregation effects in the gradients. Complete disaggregation with loss of all PrP-res in hamster CJD brain can be induced by a change in pH with no reduction in infectivity (25). Several additional N2a-22L preparations for which gradients and fractions were made under the same conditions were analyzed. These were highly reproducible, even when sarkosyl was added to the cytoplasmic supernatant fraction to try to enhance disaggregation of the PrP and PrP-res (Fig. 2B). As in A, essentially all loaded PrP was recovered (98%), and the relative percentage of PrP-res was highest in the bottom 60% sucrose fraction. The PrP-res recovered in the gradient was again diminished, with no additional reduction due to sarkosyl. Protein assays on fractions from these two representative gradients are shown in Fig. 2C. These standard protein solution assays further confirm and quantify the gold blot results; almost all of the protein remained in the loaded 5% supernatant fraction, whereas <1% of the protein was found in the bottom fractions of ≥30% sucrose. Recoveries of total protein in the gradient were 92% and 100% (for gradients A and B, respectively).
FIG. 2.
The graphs shows the quantitative recovery of PrP, PrP-res, protein, and infectivity in two representative gradients centrifuged for 2.5 h. Gradient A is from Fig. 1, and gradient B is a repeat preparation in which 1% sarkosyl was added in the supernatant, and was 0.1% in the sucrose steps. PrP in each fraction is shown by dark grey bars, and the percentage PrP-res of that fraction by light gray bars. The PrP and PrP-res profiles are the same in both gradients, with quantitative recovery of PrP in each gradient (denoted by arrow). Standard protein solution assays of both gradients in panel C, in accord with gold blot analyses, show that the vast majority of protein stays in the top fractions. Panel D shows the infectious titers (TC-ID) in the supernatant, and in each gradient fraction of A and B (without and with sarkosyl, respectively). Virtually all of the infectivity migrates to the bottom 60% sucrose fraction in both gradients, and no pellets were visible. The percentage of the loaded infectivity recovered from the gradient fractions is indicated by the arrows. Panel E shows one of the four GT1 assay cultures (passage 5 for gradient A) analyzed for infectivity; titers were calculated from the %PrP-res produced in infected GT1 indicator cells (8). The highest infectivity corresponds to the highest percentage of PrP-res that was evoked in GT1 indicator cells. Dilutions of sample fractions containing equal cell equivalents were applied to GT1 cells for comparison of fraction infectivity, and brain is a parallel 22L control homogenate with a known LD50 in mice assayed in parallel. Application of uninfected N2a cells at high cell equivalents gave no PrP-res signal, and microcon concentrated fractions were comparable to untreated fractions (data not shown).
According to the prion hypothesis, a loss of 66–75% of PrP-res, as found here with gradient centrifugation, should lead to reduction in infectivity. Both of these gradients were analyzed in detail for infectivity. As graphed in Fig. 2D, more than 80% of the tissue culture infectious doses (TC-IDs) were recovered in the bottom 60% sucrose fraction as shown, with 129% and 98% of the loaded TC-ID recovered (for gradients A and B, respectively). Based on its PrP-res content, the bottom 60% sucrose fraction should have had ⅓ to ¼ of the infectivity actually found. This indicated that PrP-res aggregation or conformation could not be precisely correlated with infectivity. The top 5% fraction with extremely low PrP-res should also have had negligible infectivity, but contained up to 9% of the infectivity loaded. Fig. 2E shows a representative example of one of the four TC-ID-limiting dilution GT1 cultures assayed for gradient A (aligned with its corresponding fraction above). In the TC-ID assay, the higher the %PrP-res (of total PrP) that is newly produced in the GT1 indicator cells, the higher the infectious titer. It is clear that the %PrP-res in the GT1 indicator cells is highest after exposure to the 60% sucrose fraction, and slightly higher than in the supernatant load control (reflecting the experimental 129% recovery in gradient A). A 22L brain dilution sample is also shown, included as a standard infectivity control in the GT1 assay (to the left of the supernatant fraction). The TC-ID of the microcon concentrated supernatant samples was the same as for parallel untreated samples (data not shown).
In summary, in step gradients loaded with low-speed total cytoplasmic supernatants, and ultracentrifuged for 2.5 h, there was a high degree of agent concentration relative to cytoplasmic protein. This purification was achieved without any enzymatic digestions, including those that can induce tight amyloid aggregates of PrP with possible trapping of non-PrP-agent components (17). Furthermore, all of the starting cellular infectivity was recovered, even though PrP-res was not. In brain preparations, ~75% of PrP and PrP-res are removed from sedimenting infectious particles that have more contaminating protein (25). The reasons for poor separation of PrP in the above culture preparations were not clear. However, the many microscopic intracellular amyloid fibril aggregates in the N2a-22L cells, as compared to CJD-infected hamster brain that does not produce large amyloid deposits, might underlie this difference. Another possibility was that the sedimentation conditions were not optimal for these shallow step gradients. We therefore tested a 1.25-h centrifugation at comparable g forces, as well as other conditions that might better separate or free infectious particles from cytoplasmic PrP amyloid fibrils.
Fig. 3A shows the distribution of PrP and gold-stained proteins after centrifugation for 1.25 h. In this gradient, the supernatant was also sonicated before loading, and the cell equivalents applied were reduced by half (to 5 × 107), to find if this improved the separation of infectivity from PrP. In contrast to the first set of gradients, in which ⅔ of the PrP migrated to the bottom of the gradient, more than 70% of the loaded PrP now remained at the top of the gradient, well separated from the 20% sedimenting PrP (the two bottommost fractions). Both bottom fractions (~0.8 mL total) showed lower Mr bands of PrP (arrow), indicating PrP-res again disproportionately sedimented when compared to the PrP in the top 8% sucrose step. The 65% sucrose fraction, loaded with 20× more sample than the 8% sucrose fraction on this blot, also again showed a high degree of purification with respect to total protein recovered (see corresponding gold-stained lanes). To precisely quantify the percentage of PrP-res in the various fractions, and to determine if this PrP separation was a consequence of reduced loads, another replicate gradient was run, and loaded with 108 cell equivalents made from a different N2a-22L preparation, with comparable results. Fig. 3B shows that <8% of the total PrP was present in the bottom two sucrose fractions, while >60% of the PrP remained at the top. Since 13% of the total PrP in the top 5% sucrose fraction was PrP-res (i.e., 7.8%), more PrP-res remained in the top as compared to the bottom fractions, that had <2.3% PrP-res. Notably, PrP-res again was reduced by gradient centrifugation, and only ¼ of the applied PrP-res was recovered. Despite this loss, as well as markedly reduced amounts of sedimenting PrP-res, essentially all the infectivity was consistently found in sucrose fractions at the bottom of all gradients assayed.
FIG. 3.
Sonicated supernatant samples loaded on more concentrated sucrose steps were centrifuged for a reduced time of 1.25 h (259,000 g-h). The top gradient (A) was loaded with 5 × 107 cell equivalents, and the PrP Western blot with corresponding gold stain are shown. Unlike the 2.5-h gradients in Fig. 2, substantially more PrP (69%) remains at the top of the gradient (8% sucrose layer); 97% of the loaded PrP was recovered from this gradient. Even without proteinase K digestion, smaller PrP-res bands (as at arrow) are more prominent in the lower two gradient fractions that together contain 20% of the loaded PrP, as well as reduced protein by gold staining. Gradients B and C show PrP and PrP-res in a repeat gradient loaded with 108 cell equivalents in which the relative %PrP-res was rigorously quantified. Panel B shows the undigested PrP samples, and panel C shows aliquots of the same samples digested with proteinase K. Again, the majority of total PrP remains at the top of the gradient, with less than 10% in the bottom two fractions. Lane loads and the percentage of PrP and PrP-res in each fraction are indicated. In each of the gradients ~7 mL of sonicated supernatant in 8% sucrose was loaded on top of a step gradient made with (from top to bottom) 1 mL, 2 mL, 1 mL, 1 mL, and 0.3 mL, respectively, of the sucrose concentrations indicated. As in previous gradients, the entire supernatant was collected as a single fraction (up to the interface), and ~0.5-mL fractions collected from the 20% sucrose step downwards.
Fig. 4 shows the infectivity assay of a similar gradient spun for 1.25 h where appropriate fractions were pooled for analysis. The supernatant was not sonicated in this experiment, and gradients were loaded with more material (2 × 108 cell equivalents), which probably led to a higher contamination with PrP in the bottom fractions (left panel). Nevertheless, although 99% of the applied PrP was recovered, only 20% of the PrP sedimented, and is seen in the combined 40% sucrose fractions here. The right panel shows a representative passage 3 GT1 culture of the limiting dilution assay of these fractions. Virtually all infectivity is in the bottom two fractions (40% to >50% sucrose), and 108% of the loaded infectivity was recovered from this gradient. It is also apparent that the PrP-res at passage 3 has been made de novo by the GT1 cells in this infectivity assay because the GT1 cells produce only their own more heavily glycosylated and higher Mr PrP top band, and also display a very different PrP-res band profile than that produced by N2a-22L cells. PrP-res carryover from test samples, rather than de novo production of PrP-res by infected cells, has been problematic in other culture infectivity assays (7,25).
With the above information, we further simplified gradients for rapid high recoveries of the infectious agent with little protein contamination. Fig. 5 shows the details of this 1.25-h gradient, loaded with supernatant that had been brought to 1% sarkosyl, and then clarified at 10,000 g × 20 min at 20°C to yield the p10 and su10 as shown. Practically invisible amounts of PrP and protein were lost in p10 (Fig. 5A and B, respectively). There was also an obvious and marked reduction of protein in the bottom-most 60% sucrose fraction as determined by gold staining (Fig. 5B). These gradient samples were analyzed for PrP, PrP-res, and infectivity, and the quantitative results are plotted in Fig. 5C. Of the PrP loaded, 84% was recovered in the gradient fractions. The majority of recovered PrP (60%) again remained in the topmost fractions, whereas only 22% of PrP was found in the bottom 60% sucrose step. Only 1/3 of this was PrP-res. This is the same amount of PrP-res as was found in the top 5% sucrose step. Nevertheless, 75% of the infectivity, as shown by the black bars, sedimented to the bottom 60% sucrose fraction. This fraction displayed very few and weakly stained protein bands with a high degree of selection from the starting protein. Some of these bands potentially could represent non-PrP constituents that are critical for agent transmission. It is also apparent that PrP bands are not the major component of this highly infectious fraction because they are relatively invisible by gold staining, More purified and concentrated PrP-res and amyloid enriched preparations are obvious with gold staining (25). In summary, the infectious agent in all the various gradients showed a constant and reproducible sucrose sedimentation pattern that was comparable to that found in partially purified infectious brain preparations. In contrast, PrP and PrP-res migrations were highly variable and did not predict the position or behavior of the infectious agent.
Discussion
The above experiments demonstrate that infectious TSE particles can be rapidly, reproducibly, and quantitatively recovered in a low protein fraction starting with a crude culture supernatant. Infectious particles can be separated from a large portion of normal host membrane PrP, as well as pathological aggregates of PrP (PrP-res and amyloid fibers), without any reductions in titer. Although it remains to be seen if extra purification steps, as in brain (16), can reduce all forms of PrP to almost invisible levels in these culture preparations, the complete recovery of starting infectivity in gradient fractions containing ≤1% of the total cellular proteins is remarkable. Only a specific and small set of very minor proteins purifies with infectious particles in the short 1.25-h sedimentation procedure. These appear to be more homogeneous than those made from degenerating and complex brain preparations that derive from many different cell types (16,24). Some of these selected N2a-22L proteins may be integral agent structures, and/or components essential for agent infection. Since only a small portion of one gradient tube was used for protein and infectivity analyses, the remaining pooled fractions from parallel gradient tubes can be used for further protein purification, microsequencing, and synthesis of selected peptide bands. Relevant antibodies may also be generated from culture gradient fractions to provide diagnostic markers that are more specific for the infectious TSE particle than PrP antibodies. Because PrP is a conserved host protein that is poorly antigenic, the presence of contaminating PrP in these samples would probably not interfere with antibody production to a less abundant foreign or covert integral agent component, such as one that may protect a viral nucleic acid. The generation of high levels of infectivity in several different strain-specific culture models here (1,19) can also simplify the selection of agent-specific proteins, because these should be common to all the different culture models. Furthermore, the culture fractions shown, although still incompletely purified, may also be sufficient to elicit a protective “vaccination” effect. Dramatic long-term protective effects and virus-like interference properties have been achieved with infectious TSE inocula in both animal and culture models (14,15,19), and it is possible that one or more components of the 60% sucrose step may participate in protection against a TSE infection. Synthesis of protein sequences of interest can be used to rapidly verify their effects in the superinfection culture models.
The lack of a straightforward and short in vitro assay for infectivity has made it difficult for most investigators to independently evaluate the titers of their preparations, or to critically re-examine accepted prion claims. There are hundreds of papers on PrP, as well as diagrammatic and computer-generated models of its normal and amyloid structure. In contrast, there are very few papers describing the systematic fractionation, relative contamination, and quantitative recovery of infectious particles during purification. The common assumption, still experimentally unproven, is that some unknown tertiary form of PrP is infectious, and is also capable of encoding all the unique TSE agent strains. The ability to stably propagate a variety of TSE agents in mono-typic GT1 cells in culture (1,19) provides a straightforward way to compare the relative infectivity and cellular residence of different agent strains. The GT1 assay can also uncover essential morphological and molecular structures required for infection. Close experimental attention to infectious titers becomes even more critical now, given the continued failure to create reproducible animal infections from PrP itself, or from pure recombinant PrP amplified to its pathological form in a test tube [reviewed in (11)]. Therefore the fundamental prion belief, that this host protein is infectious, remains highly speculative. Indeed, even prion proponents recognize that PrP-res is not the infectious form of the protein, and thus have proposed that there is an infectious but PK-sensitive form of PrP whose structure remains to be revealed (3,29). To address this new suggestion we evaluated all detectable PrP forms. None of the data substantiated a direct correlation of PK-sensitive PrP with infectious particles.
In terms of yields, 22L-infected starting brain homogenate has ~10× theinfectious titer of the N2a-22L detergent cytoplasmic supernatants. However, more than 80% of this N2a-22L infectivity can be recovered, as compared to 0.1–10% recoveries from typical brain preparations. Hence these permanent cultures yield a reproducible and relatively inexpensive source of agent for structural and molecular studies. The high sucrose fractions used here also contain more than a million-fold more infectious doses than abundantly amplified abnormal PrPs. Some of the best of these test-tube amplifications are also now known to have no demonstrable infectivity despite their high PrP-res content (2), and thus studying the natural, biologically produced agent can be more fruitful. Interestingly, these non-infectious amplified preparations display many PrP fibrils by electron microscopy, but contain none of the 25-nm virus-like particles found in infected brain and high-yield cultures.
Although the above streamlined purification of agent does not yet address the electron-dense ~25-nm-diameter particles in membrane bound arrays of fixed cells (18), previous brain studies have shown that comparable particles fractionate with infectivity (11). These 25-nm particles are found only in infected material and do not bind PrP antibodies. They are of particular interest because recent experiments with PrP antibodies have shown that abnormal PrP aggregates in non-infectious brain samples can be immunoprecipitated with antibodies originally claimed to be specific for the infectious tertiary conformations of PrP (5). Moreover, a panel of rigorous criteria used to define “authentic” (infectious) PrP aggregates has now also been shown in non-infectious samples (4). Thus abnormal PrP (PrP-res, PrP amyloid, and aggregated PrP) is an unreliable diagnostic marker for isolating or for verifying the presence of TSE infectious particles. In fact, conformation-specific PrP antibodies could be useful for the separation of infectious particles from residual PrP aggregates.
PrP appears to be is an essential host receptor that binds the infectious agent, as originally proposed (10,17). This concept is also most consistent with both the resistance of PrP-null mice to infection (6), and the failure to render PrP infectious. Abnormal PrP may be initiated or converted during agent-host membrane interactions, and its late appearance indicates it is a pathological by-product (9,12). This view, in which changes in PrP are part of the resulting disease rather than the causal agent, is also consistent with recent PrP transgenic models that support very high levels of agent replication in the absence of abnormal PrP production (3). That abnormal PrP is a result, rather than the initiating cause of pathology, is also evident from PrP disease-producing gene mutation experiments in which animals and cells produce abnormal PrP, but no infectivity (4).
In addition to opening new avenues to determine the structural and essential molecular components of TSE agents, reasonably purified culture preparations can be used to re-examine the unusual resistance properties ascribed to TSE agents. Components of degenerating brain with complex lipids may mask TSE particle sensitivity, and the range of resistance of TSE strains to inactivation reported can be highly variable. For example, it has been shown in several laboratories that reasonably conventional temperatures can inactivate the vast majority of infectious particles in Chandler (RML) murine scrapie fractions, whereas BSE agents appear to be more resistant. The more typical scrapie inactivation conditions are in line with the heat inactivation of latent and resistant viruses such as hepatitis B and endogenous retroviral particles (23), and are significantly lower than the temperatures at which complex thermophilic bacteria thrive (26). Interestingly, heated 263K hamster brain fractions enriched for abnormal PrP, are completely susceptible to proteolytic digestion by PK (so that no detectable PrP-res remains), but show no significant loss of titer after digestion (27). A better understanding of the resistance of various TSE agent particles, such as those causing bovine BSE and human CJD, are of practical importance for effective decontamination, as well as prevention of epidemic and iatrogenic spread (10,13).
Acknowledgments
This work was supported by National Institutes of Health grant NS 12674 and a gift from the William Prusoff fund. No competing financial interests exist.
References
- 1.Arjona A. Simarro L. Islinger F. Nishida N. Manuelidis L. Two Creutzfeldt-Jakob disease agents reproduce prion protein-independent identities in cell cultures. Proc Natl Acad Sci USA. 2004;101:8768–8773. doi: 10.1073/pnas.0400158101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Atarashi R. Moore RA. Sim VL, et al. Ultrasensitive detection of scrapie prion protein using seeded conversion of recombinant prion protein. Nat Methods. 2007;4:645–650. doi: 10.1038/nmeth1066. [DOI] [PubMed] [Google Scholar]
- 3.Barron RM. Campbell SL. King D. Bellon A. Chapman KE. Williamson RA. Manson JC. High titers of transmissible spongiform encephalopathy infectivity associated with extremely low levels of PrPSc in vivo. J Biol Chem. 2007;282:35878–35886. doi: 10.1074/jbc.M704329200. [DOI] [PubMed] [Google Scholar]
- 4.Biasini E. Medrano AZ. Thellung S. Chiesa R. Harris DA. Multiple biochemical similarities between infectious and non-infectious aggregates of a prion protein carrying an octapeptide insertion. J Neurochem. 2008;104:1293–1308. doi: 10.1111/j.1471-4159.2007.05082.x. [DOI] [PubMed] [Google Scholar]
- 5.Biasini E. Seegulam ME. Patti BN, et al. Non-infectious aggregates of the prion protein react with several PrP(Sc)-directed antibodies. J Neurochem. 2008 doi: 10.1111/j.1471-4159.2008.05306.x. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 6.Büeler H. Aguzzi A. Sailer A. Greiner R-A. Autenried P. Auget M. Weissmann C. Mice devoid of PrP are resistant to scrapie. Cell. 1993;73:1339–1347. doi: 10.1016/0092-8674(93)90360-3. [DOI] [PubMed] [Google Scholar]
- 7.Klohn P. Stoltze L. Enari FEM. Weissmann C. A quantitative, highly sensitive cell-based infectivity assay for mouse scrapie prions. Proc Natl Acad Sci USA. 2003:11666–11671. doi: 10.1073/pnas.1834432100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Y. Sun R. Chakrabarty T. Manuelidis L. A rapid accurate culture assay for infectivity in transmissible encephalopathies. J Neurovirol. 2008;14:1–9. doi: 10.1080/13550280802105283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu ZH. Baker C. Manuelidis L. New molecular markers of early and progressive CJD brain infection. J Cell Biochem. 2004;93:644–652. doi: 10.1002/jcb.20220. [DOI] [PubMed] [Google Scholar]
- 10.Manuelidis L. The dimensions of Creutzfeldt-Jakob disease. Transfusion. 1994;34:915–928. doi: 10.1046/j.1537-2995.1994.341095026981.x. [DOI] [PubMed] [Google Scholar]
- 11.Manuelidis L. A 25 nm virion is the likely cause of transmissible spongiform encephalopathies. J Cell Biochem. 2007;100:897–915. doi: 10.1002/jcb.21090. [DOI] [PubMed] [Google Scholar]
- 12.Manuelidis L. Fritch W. Infectivity and host responses in Creutzfeldt-Jakob disease. Virology. 1996;215:46–59. doi: 10.1006/viro.1996.0033. [DOI] [PubMed] [Google Scholar]
- 13.Manuelidis L. Decontamination of Creutzfeldt-Jakob disease and other transmissable agents. J Neurovirol. 1997;3:62–65. doi: 10.3109/13550289709015793. [DOI] [PubMed] [Google Scholar]
- 14.Manuelidis L. Lu ZY. Attenuated Creutzfeldt-Jakob disease agents can hide more virulent infections. Neurosci Lett. 2000;293:163–166. doi: 10.1016/s0304-3940(00)01514-7. [DOI] [PubMed] [Google Scholar]
- 15.Manuelidis L. Lu ZY. Virus-like interference in the latency and prevention of Creutzfeldt-Jakob disease. Proc Natl Acad Sci USA. 2003;100:5360–5365. doi: 10.1073/pnas.0931192100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manuelidis L. Sklaviadis T. Akowitz A. Fritch W. Viral particles are required for infection in neurodegenerative Creutzfeldt-Jakob disease. Proc Natl Acad Sci USA. 1995;92:5124–5128. doi: 10.1073/pnas.92.11.5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manuelidis L. Sklaviadis T. Manuelidis EE. Evidence suggesting that PrP is not the infectious agent in Creutzfeldt-Jakob disease. EMBO J. 1987;6:341–347. doi: 10.1002/j.1460-2075.1987.tb04760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manuelidis L. Yu Z-X. Barquero N. Mullins B. Cells infected with scrapie and Creutzfeldt-Jakob disease agents produce intracellular 25-nm virus-like particles. Proc Natl Acad Sci USA. 2007;104:1965–1970. doi: 10.1073/pnas.0610999104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nishida N. Katamine S. Manuelidis L. Reciprocal interference between specific CJD and scrapie agents in neural cell cultures. Science. 2005;310:493–496. doi: 10.1126/science.1118155. [DOI] [PubMed] [Google Scholar]
- 20.Prusiner S. Prions. Proc Natl Acad Sci USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prusiner S. Development of the prion concept. In: Prusiner S., editor. Prion Biology and Diseases. Cold Spring Harbor Press; Cold Spring Harbor, NY: 1999. pp. 67–112. (cf. p. 81). [Google Scholar]
- 22.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 23.Sklaviadis T. Akowitz A. Manuelidis EE. Manuelidis L. Nuclease treatment results in high specific purification of Creutzfeldt-Jakob disease infectivity with a density characteristic of nucleic acid-protein complexes. Arch Virol. 1990;112:215–229. doi: 10.1007/BF01323166. [DOI] [PubMed] [Google Scholar]
- 24.Sklaviadis T. Akowitz A. Manuelidis EE. Manuelidis L. Nucleic acid binding proteins in highly purified Creutzfeldt-Jakob disease preparations. Proc Natl Acad Sci. 1993;90:5713–5717. doi: 10.1073/pnas.90.12.5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sklaviadis TL. Manuelidis EE. Manuelidis L. Physical properties of the Creutzfeldt-Jakob disease agent. J Virol. 1989;63:1212–1222. doi: 10.1128/jvi.63.3.1212-1222.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Somerville R. Oberthur R. Havekost U. MacDonald F. Taylor D. Dickinson A. Characterization of thermodynamic diversity between transmissible spongiform encephalopathy agent strains and its theoretical implications. J Biol Chem. 2002;277:11084–11089. doi: 10.1074/jbc.M111766200. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki SY. Takata M. Teruya K. Shinagawa M. Mohri S. Yokoyama T. Conformational change in hamster scrapie prion protein (PrP27-30) associated with proteinase K resistance and prion infectivity. J Vet Med Sci. 2008;70:159–165. doi: 10.1292/jvms.70.159. [DOI] [PubMed] [Google Scholar]
- 28.Vorberg I. Raines A. Priola S. Acute formation of protease-resistant prion protein does not always lead to persistent scrapie infection in vitro. J Biol Chem. 2004;279:29218–29225. doi: 10.1074/jbc.M402576200. [DOI] [PubMed] [Google Scholar]
- 29.Weissmann C. Birth of a prion: Spontaneous generation revisited. Cell. 2005;122:165–168. doi: 10.1016/j.cell.2005.07.001. [DOI] [PubMed] [Google Scholar]