

Abstract

The reaction of O6-benzyl-3’,5’-bis-O-(tert-butyldimethylsilyl)-2’-deoxyxanthosine with 1H-benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorophosphate (BOP) yielded the nucleoside C-2 tris(dimethylamino)phosphonium hexafluorophosphate salt as a stable, isolable species. This is in contrast to reactions of inosine nucleosides with BOP, where the in situ formed phosphonium salts undergo subsequent reaction to yield O6-(benzotriazol-1-yl)inosine derivatives. The phosphonium salt obtained from the 2’-deoxyxanthosine derivative can be effectively used to synthesize N2-modified 2’-deoxyguanosine analogues. Using this salt, a new synthesis of an acrolein-2’-deoxyguanosine adduct has also been accomplished.
The ability to modify natural nucleosides translates to novel applications in biochemistry, biology, and medicine.1 A classical method for nucleoside modification is via displacement chemistry. For modification at the C-2 position various protected or unproteced 2-halo-2’-deoxyinosines, namely fluoro,2 bromo,3 and chloro4 derivatives, have been used. In addition, use of triflate5 and tosylate4a derivatives have also been reported.
Phosphonium salts have been proposed as intermediates in the reactions of inosine nucleosides with Ph3P·I26,7 or with 1H-benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorophosphate (BOP).8,9 These salts can be converted to adenine derivatives via reaction with various amines.6,8 In this context, we demonstrated that in reactions of hypoxanthine nucleosides with BOP, the inosine-derived phosphonium salts undergo reaction with BtO− that is released. This results in the formation of O6-(benzotriazol-1-yl)inosine derivatives.9 More recently, we demonstrated that the inosine-derived phoshonium salt formed via reaction with Ph3P·I2 can also be converted to O6-(benzotriazol-1-yl)inosine derivatives in good yields.7 These new O6-(benzotriazol-1-yl)inosine derivatives possess excellent reactivity for a variety of transformations, leading to modification at the C-6 position of the purine (Scheme 1).7,9
Scheme 1.

Synthesis of O6-(Benzotriazol-1-yl) Derivatives of Inosine and 2’-Deoxyinosine via Reaction with BOP or Ph3P/I2/HOBt
On the basis of our prior work on inosine nucleosides, we became interested in studying the reaction of O6-protected 2’-deoxyxanthosine with BOP. This paper describes our preliminary results on the reaction of O6- benzyl-3’,5’-bis-O-(tert-butyldimethylsilyl)-2’-deoxyxanthosine with BOP. In the course of these studies we have identified the nucleoside C-2 phosphonium salt as an isolable compound that can be readily utilized for SNAr displacement chemistry with a broad range of amines. Finally, the C-2 phosphonium salt has been utilized in a new synthesis of an acrolein adduct with 2’-deoxyguanosine.
O6-Benzyl-3’,5’-bis-O-(tert-butyldimethylsilyl)-2’-deoxyguanosine (1) can be readily synthesized on the multigram scale via a Mitsunobu etherification of 3’,5’-bis-O-(tert-butyldimethylsilyl)-2’-deoxyguanosine.2b,3a,10 Diazotization-hydrolysis of 1 as described4a,11 yielded O6-benzyl-3’,5’-bis-O-(tert-butyldimethylsilyl)-2’-deoxyxanthosine (2 in Scheme 2, 64% yield).
Scheme 2.

Synthesis of O6-Benzyl-3’,5’-bis-O-(tert-butyldimethylsilyl)-2’-deoxyxanthosine
Under conditions similar to those we have described previously,9 (2 molar equiv BOP/1.5–2.0 molar equiv (i-Pr)2NEt, anhydrous CH2Cl2, room temperature), the reaction of 2 with BOP was evaluated (Scheme 3). A fairly rapid reaction was observed (4–5 h at room temperature) with the predominant formation of a new material that was isolated by chromatography on silica gel.
Scheme 3.

Reaction of O6-benzyl-3’,5’-bis-O-(tert-butyldimethylsilyl)-2’-deoxyxanthosine with BOP
Analysis of this new product indicated that it was the phosphonium salt 3 and not the benzotriazol-1-yl compound 4. From this reaction, two noteworthy points emerged: (a) the greater difficulty in SNAr displacement of HMPA by BtO− from the C-2 position, in contrast to reactions at the C-6 of purines9 and (b) the relative stability of phosphonium salt 3, which could be readily obtained by chromatographic purification.
The 1H NMR spectrum of 3 (CDCl3) showed a characteristic doublet at δ 2.83 ppm for the NMe2 resonance (JP-H = 10.7 Hz). The 31P NMR of 3 (CDCl3) showed a singlet at δ 34.11 ppm as well as a septet centered at δ −143.27 ppm (JP-F = 712.7 Hz) for the PF6 anion. The synthesis of phosphonium salt 3 is reproducible and scalable, usually returning product yields of 88–92%.12
Given the high isolated yield of phosphonium salt 3 and the relative simplicity of its synthesis, we were interested in evaluating its utility in displacement reactions with amines. Such reactions would involve HMPA as a neutral leaving group, and this would lead to a simple approach to N-modified 2’-deoxyguanosine analogues. A variety of amines were selected for this purpose (Table 1).
Table 1.
Synthesis of N2-Modified 2’-Deoxyguanosine Analogues from 3
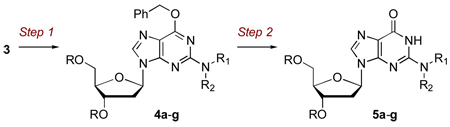 | |||
|---|---|---|---|
| entry | amine | Displacement (step 1) yield |
debenzylation (step 2) yielde |
| 1 |
4a: 65%a 4a: 90%b |
5a: 95% | |
| 2 | 4b: 96%b | 5b: 91% | |
| 3 | 4c: 100%b | 5c: 97% | |
| 4 | 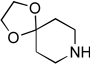 |
4d: 83%c | 5d: 97% |
| 5 | 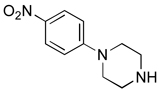 |
4e: 65%c | 5e: --f |
| 6 | 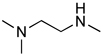 |
4f: 71%c | 5f: 66% |
| 7 | 4g: 78%d | 5g: 98% | |
Reaction using 5.7 molar equiv of amine, 2.0 molar equiv of Cs2CO3, DME, room temperature.
Reaction using 4 molar equiv of amine, DME, room temperature.
Reaction using 4 molar equiv of amine, DME, room temperature and then 85 °C.
Reaction using 7.5 molar equiv of amine, DME, room temperature and then 85 °C.
Debenzylation was performed using H2 (1 atm)/10% Pd-C, 1:1 THF-MeOH, room temperature.
Debenzylation was accompanied by nitro group reduction, no attempt was made at finding selective debenzylation conditions.
The displacement reactions on 3 were conducted in 1,2-dimethoxyethane (DME) at room temperature or at 85 °C when reactions were slow or incomplete at room temperature. Subsequent to the displacement, the O6-benzyl group was removed by catalytic hydrogenolysis at room temperature. The fact that the O6-protected derivative 3 could be used in these reactions makes 3 a substrate for SNAr displacement. This is different in comparison to the displacement reactions on 2-chloro-2’-deoxyinosine which were addition-elimination type processes on a conjugated system.4a Also, no degradation of 3 was observed with the primary amine (entry 7) and this contrasts to what has been reported in the reaction of O6-benzyl-3’,5’-bis-O-(tert-butyldimethylsilyl)-2-bromo-2’-deoxyinosine.13 All of these features bode well for the utility of 3 in SNAr displacement reactions.
With the simple displacement reactions completed, we then considered the use of 3 for the synthesis of a more complex, biologically relevant compound. Of several possibilites, we chose to evaluate the synthesis of the 2’-deoxyguanosine-acrolein adduct. This compound has been important in studies aimed at understanding the structure and biological implications of acrolein-induced DNA damage.
Typically compounds of this type have been synthesized by fluoride displacement from 2-fluoro-2’-deoxyinosine derivatives.14,15 However, this fluoro nucleoside requires a multistep synthesis and involves the use of HF-pyridine in the diazotization-fluorination step. In comparison, 3 offers significant advantages.
For our synthesis, we reasoned that ready access to the acrolein adduct with 2’-deoxyguanosine could be attained from commercially available 3-amino-1-propanol and 3. Initial experiments were therefore directed toward displacement of HMPA from 3 by 3-amino-1-propanol (Scheme 4). However, the yield of 6 via this approach was low (ca 30%).
Scheme 4.

Approaches to 3’,5’-Bis-O-(tert-butyldimethylsilyl)-N-(3-hydroxypropyl)-2’-deoxyguanosine
By analysis of the byproducts formed in the synthesis of 6, protection of the hydroxyl group in 3-amino-1-propanol was deemed necessary to suppress the undesired side reactions. Based upon a literature procedure,16 3-amino-1-propanol was selectively converted to the O-benzyl ether. The reaction of 3 with this benzyl-protected 3-amino-1-propanol (Scheme 4) proceeded smoothly at 85 °C in DME to provide the bis-benzyl ether protected nucleoside 7 in 82% yield.
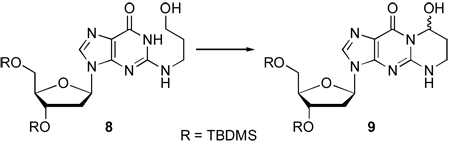
At this stage, removal of the two benzyl protecting groups in 7 followed by mild oxidation of the primary hydroxyl, should result in the requisite cyclized acrolein-2’-deoxyguanine adduct as its bis-TBDMS ether. Along these lines, exposure of 7 to 1 atm H2 and 10% Pd-C in 1:1 THF-MeOH resulted in the debenzylated product 8 (89% yield). Upon monitoring this reduction carefully, it was observed that the nucleoside benzyl ether underwent rapid deprotection (within 4 h) whereas the alkyl benzyl ether required prolonged exposure to the reductive conditions (23 h).
With 8 in hand, the final oxidative cyclization to 9 was explored. This proved to be nontrivial and both TPAP/NMO17,18 as well as PCC19,20 gave modest to low yields of 9 (Table 2). In the presence of silica gel, 4-acetylamino-2,2,6,6-tetramethylpiperidine-1-oxoammonium tetrafluoroborate has been shown to be an excellent mild oxidant.21,22 Application of this reagent resulted in successful synthesis of the desired 9 in 69% yield.
Table 2.
Conditions Tested for the Oxidative Cyclization of 8 as Well as the Yields of 9 in These Reactions
 | ||
|---|---|---|
| entry | conditions | resulta |
| 1 | TPAP (0.16 molar equiv), NMO (1.9 molar equiv), 4 Å molecular sieves, CH2Cl2, room temperature, 8 h | Incomplete reaction, 41% yield |
| 2 | PCC (3.0 molar equiv), 4 Å molecular sieves, CH2Cl2, room temperature, 16 h | 23% yield |
| 3 | 4-acetylamino-2,2,6,6-tetramethylpiperidine-1-oxoammonium tetrafluoroborate (1.2 molar equiv), silica gel, CH2Cl2, room temperature, 16 h | 69% yield |
Yield of isolated, purified product.
The in situ formation of phosphonium salts in the reactions of peptide coupling agents with amide and urea functionalities have been reported.23 However, in this letter we have shown that the C-2 tris(dimethylamino)phosphonium hexafluorophosphate salt 3 is formed in a high-yield reaction of O6-benzyl-3’,5’-bis-O-(tert-butyldimethylsilyl)-2’-deoxyxanthosine (2) with BOP, and is a readily isolated species. This reactivity contrasts to that of inosine nucleosides with BOP, where the final products are the O6-(benzotriazol-1-yl) derivatives.9
Salt 3 is a good substrate for SNAr displacement reactions with primary and secondary amines, providing a facile approach to N2-modified 2’-deoxyguanosine analogues. As demonstrated with the synthesis of the acrolein-2’-deoxyguanosine adduct 9, it appears that 3 can be used for the synthesis of other biologically important compounds. Thus, these C-2 nucleoside phosphonium salts can be considered as a new family of reactive nucleosides. Given the simplicity in synthesis, a variety of O6 protecting groups can be readily utilized in order to accommodate for a wide range of reactions. Other reactions of the C-2 tris(dimethyl)phosphonium hexafluorophosphate salt 3 and related compounds are currently under investigation in our laboratories
Supplementary Material
Acknowledgment
Support of this work by NSF Grant CHE-0640417 and a PSC CUNY-38 award are gratefully acknowledged. Acquisition of a mass spectrometer was funded by NSF Grant CHE-0520963. Infrastructural support at CCNY was provided by NIH RCMI Grant G12 RR03060. We thank Prof. James. M. Bobbitt (University of Connecticut) for a generous sample of 4-acetylamino-2,2,6,6-tetramethylpiperidine-1-oxoammonium tetrafluoroborate.
Footnotes
Supporting Information Available: Experimental procedures, 1H and 13C NMR spectra of 3, 4a–g, and 7–9. 1H NMR spectra of 5a–d, 5f, 5g and 31P{1H} NMR spectrum of 3. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Blackburn GM, Gait MJ, Loakes D, Williams DM, editors. Nucleic Acids in Chemistry and Biology. 3rd ed. Cambridge, UK: RSC Publishing; 2006. [Google Scholar]; (b) Simons C. Nucleoside Mimetics: Their Chemistry and Biological Properties. Amsterdam: Gordon and Breach; 2001. [Google Scholar]; (c) Kisakürek MV, Rosemeyer H, editors. Perspectives in Nucleoside and Nucleic Acid Chemistry. Weinheim: Verlag Helvetica Chimica Acta: Zürich and Wiley-VCH; 2000. [Google Scholar]; (d) Suhadolnik RJ. Nucleosides as Biological Probes. New York: Wiley; 1979. [Google Scholar]
- 2.Some examples for the synthesis of 2-fluoro-2’-deoxyinosine derivatives: Woo J, Sigurdsson ST, Hopkins PB. J. Am. Chem. Soc. 1993;115:3407–3415. Zajc B, Lakshman MK, Sayer JM, Jerina DM. Tetrahedron Lett. 1992;33:3409–3412. Kim SJ, Stone MP, Harris CM, Harris TM. J. Am. Chem. Soc. 1992;114:5480–5481.
- 3.Some examples for the synthesis of O6-protected and unprotected 2-bromo-2’-deoxyinosine derivatives: Harwood EA, Sigurdsson ST, Edfeldt NBF, Reid BR, Hopkins PB. J. Am. Chem. Soc. 1999;121:5081–5082. Jhingan AK, Meehan T. Synth. Commun. 1992;22:3129–3135.
- 4.Some examples for the synthesis of 2-chloro-2’-deoxyinosine derivatives: Pottabathini N, Bae S, Pradhan P, Hahn H-G, Mah H, Lakshman MK. J. Org. Chem. 2005;70:7188–7195. doi: 10.1021/jo050847j. Ramasamy KS, Zounes M, Gonzalez C, Freier SM, Lesnik EA, Cummins LL, Griffey RH, Monia BP, Cook PD. Tetrahedron Lett. 1994;35:215–218.
- 5.Some examples of an O6-protected C-2 triflate: Edwards C, Boche G, Steinbrecher T, Scheer S. J. Chem. Soc., Perkin Trans. 1. 1997:1887–1893. Steinbrecher T, Wameling C, Oesch F, Seidel A. Angew. Chem., Int. Ed. Engl. 1993;32:404–406.
- 6.Lin X, Robins MJ. Org. Lett. 2000;2:3497–3499. doi: 10.1021/ol000255h. [DOI] [PubMed] [Google Scholar]
- 7.Bae S, Lakshman MK. J. Org. Chem. 2008;73:1311–1319. doi: 10.1021/jo7021795. [DOI] [PubMed] [Google Scholar]
- 8.Wan Z-K, Binnun E, Wilson DP, Lee J. Org. Lett. 2005;7:5877–5880. doi: 10.1021/ol052424+. [DOI] [PubMed] [Google Scholar]
- 9.Bae S, Lakshman MK. J. Am. Chem. Soc. 2007;129:782–789. doi: 10.1021/ja064682n. [DOI] [PubMed] [Google Scholar]
- 10.Himmelsbach F, Schulz BS, Trichtinger T, Charubala R, Pfleiderer W. Tetrahedron. 1984;40:59–72. [Google Scholar]
- 11.(a) Jurczyk SC, Horlacher J, Devined KG, Benner SA, Battersby TR. Helv. Chim. Acta. 2000;83:1517–1524. [Google Scholar]; (b) Kodra JT, Benner SA. Synlett. 1997:939–940. [Google Scholar]
- 12.Synthesis of O6-benzyl-3’,5’-bis-O-(tert-butyldimethylsilyl)-O2-tris(dimethylamino)phosphonium-2’-deoxyxanthosine hexafluorophosphate (3). In a clean, dry flask equipped with stirring bar were placed O6-benzyl-3’,5’-bis-O-(tert-butyldimethylsilyl)-2’-deoxy-xanthosine (2) (0.588 g, 1.00 mmol) and BOP (0.885 g, 2.00 mmol). CH2Cl2 (10.0 mL) and (i-Pr)2NEt (0.35 mL, 2.01 mmol) were added. The mixture was flushed with nitrogen gas and allowed to stir at room temperature. After 5 h, the reaction was complete and the mixture was concentrated. Chromatographic purification (SiO2, eluted with 50% EtOAc in hexanes followed by 30% acetone in CH2Cl2) afforded 0.785 g (88% yield) of compound 3 as a beige foam. Rf (5% MeOH in CH2Cl2) = 0.40. 1H NMR (500 MHz, CDCl3): δ 8.36 (s, 1H, H–8), 7.46 (d, 2H, Ar–H, J = 6.8), 7.38–7.31 (m, 3H, Ar–H), 6.38 (t, 1H, H–1’, J = 6.4), 5.67 (s, 2H, OCH2), 4.58 (app q, 1H, H–3’, J ~ 4.2), 4.02 (br q, 1H, H–4’, J = 2.9), 3.85 (dd, 1H, H–5’, J = 11.7, 3.2), 3.78 (dd, 1H, H–5’, J = 11.7, 2.4), 2.83 (d, 18H, NCH3, JH–P = 10.7), 2.44 (t, 2H, H–2’, J = 5.9), 0.91 (s, 18H, t–Bu), 0.10 (br s, 12H, SiCH3). 13C NMR (125 MHz, CDCl3): δ 161.9, 152.7, 152.6, 141.5, 135.3, 128.6, 128.5, 127.8, 120.2, 88.0, 84.0, 71.6, 69.7, 62.6, 41.9, 37.0 (d, JC–P = 4.5), 26.0, 25.7, 18.4, 17.9, –4.7, –4.8, –5.4, –5.5. 31P{1H} NMR (202 MHz, CDCl3): δ 34.11 (s, P[N(CH3)2]3),– 143.27 (septet, PF6, JP–F = 712.7). ESI HRMS calcd for C35H63N7O5PSi2+ 748.4161, found 748.4151.
- 13.Harwood EA, Hopkins PB, Sigurdsson ST. J. Org. Chem. 2000;65:2959–2964. doi: 10.1021/jo991501+. [DOI] [PubMed] [Google Scholar]
- 14.Acrolein: Khullar S, Varaprasad CV, Johnson F. J. Med. Chem. 1999;42:947–950. doi: 10.1021/jm980605u. Nechev LV, Harris CM, Harris TM. Chem. Res. Toxicol. 2000;13:421–429. doi: 10.1021/tx990167+.
- 15.Cinnamaldehyde: Rezaei M, Harris TM, Rizzo CM. Tetrahedron Lett. 2003;44:7513–7516.
- 16.Hu XE, Cassady JM. Synth. Commun. 1995;25:907–913. [Google Scholar]
- 17.Griffith WP, Ley SV. Aldrichimica Acta. 1990;23:13–19. [Google Scholar]
- 18.Manaviazar S, Frigerio M, Bhatia GS, Hummersone MG, Aliev AE, Hale KJ. Org. Lett. 2006;8:4477–4480. doi: 10.1021/ol061626i. [DOI] [PubMed] [Google Scholar]
- 19.Corey EJ, Suggs JW. Tetrahedron Lett. 1975;16:2647–2650. [Google Scholar]
- 20.Su Q, Panek JS. Angew. Chem., Int. Ed. 2005;44:1223–1225. doi: 10.1002/anie.200462408. [DOI] [PubMed] [Google Scholar]
- 21.(a) Zakrzewski J, Grodner J, Bobbitt JM, Karpińska M. Synthesis. 2007:2491–2494. [Google Scholar]; (b) Bobbitt JM, Merbouh N. Org. Synth. 2005;82:80–86. [Google Scholar]
- 22.(a) For a mechanism, see: Bailey WF, Bobbitt JM, Wiberg KB. J. Org. Chem. 2007;72:4504–4509. doi: 10.1021/jo0704614. Bobbitt JM. J. Org. Chem. 1998;63:9367–9374.
- 23.For examples, see: Wan Z-K, Wacharasindhu S, Levins CG, Lin M, Tabei K, Mansour TS. J. Org. Chem. 2007;72:10194–10210. doi: 10.1021/jo7020373. Kang F-A, Kodah J, Guan Q, Li X, Murray WV. J. Org. Chem. 2005;70:1957–1960. doi: 10.1021/jo040281j.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.