

Abstract
Activating mutations in the Kit receptor tyrosine kinase are associated with Gastrointestinal Stromal Tumor (GIST). Imatinib inhibits Kit and is front-line therapy for GIST. However, imatinib most often elicits a partial response or stable disease and most GIST patients who initially respond to imatinib eventually acquire resistance. Thus improved treatment strategies for GIST are needed. We investigated the role of Src Family kinases (SFKs) in tumorigenesis in a mouse model of human GIST. Whereas the SFKs Src and Lyn were active in GIST, surprisingly imatinib treatment stimulated their phosphorylation/activation. We show that integrin signaling activates FAK and consequently SFKs in GIST and that imatinib enhanced integrin signaling implies a role for the extracellular matrix and integrin signaling in tumor maintenance and imatinib resistance. Dasatinib, an inhibitor of SFKs and Kit, inhibited SFKs and FAK activation in GIST but also inhibited Kit and Kit-dependent downstream signaling pathways including PI3-kinase, MAPK but not STAT signaling. While dasatinib and imatinib alone both produced a minimal histo-pathological response, combination therapy improved it leading to increased necrosis in GIST. These results highlight the importance of SFK and STAT signaling in GIST and suggest that stimulation of integrin signaling by imatinib may limit its clinical efficacy.
INTRODUCTION
Gastrointestinal Stromal Tumor (GIST) is the most common mesenchymal tumor of the gastrointestinal tract. GISTs express the Kit receptor tyrosine kinase (Kit) and are thought to derive from Kit+ or Kitlow interstitial cells of Cajal (ICC) or ICC progenitors (1–3). Kit gain-of-function mutations play a critical role in GIST development and maintenance and are found predominantly in the juxtamembrane domain of the Kit receptor. However mutations in the extracellular and kinase domains of Kit have also been described but are less frequent (4–6). We had produced a mouse model of GIST by a knock-in insertion in the mouse genome of a Kit activating mutation, KitV558Δ found in a case of human familial GIST syndrome (7). The KitV558Δ mutation is located in the juxtamembrane domain of Kit. Heterozygous mutant KitV558Δ/+ mice develop GIST with 100% penetrance and eventually die from complications of the disease.
Imatinib mesylate (Gleevec), an inhibitor of the Kit, PDGFR and BCR-ABL tyrosine kinases, is used successfully to treat patients with GIST and chronic myelogenous leukemia (CML) (8–9). In most GIST patients, imatinib elicits a partial response or stable disease (9). Treatment of GIST mice with imatinib produces a similar response (10). However, the molecular consequences of Kit inhibition by targeted therapy with imatinib are less well understood. Studies of Kit receptor signaling in cell lines and primary cell culture systems have identified several Kit ligand mediated signaling cascades including PI 3-kinase (11–14), Src family kinases (SFK) (14–15), tyrosine phosphatases (SHP-1 and SHP-2) (16) and phospholipase C γ-1 (11) as proximal signaling initiators. Furthermore, Kit activates the MAP kinase, Cbl and STAT signaling pathways.
We had used imatinib to block oncogenic Kit signaling in mouse GIST to identify downstream effectors of Kit signaling in vivo. Imatinib was shown to abrogate cell cycle progression concomitant with an increase in apoptosis in tumor lesions. Furthermore, biochemical analysis of tumor tissue from imatinib treated mice showed impairment of PI3-kinase and mTOR signaling (10). In addition, gene expression profiles showed close similarity between mouse and human GIST and revealed roles for cell cycle regulators and interferon inducible genes in GIST (10).
Today imatinib therapy is first-line treatment for advanced GIST patients. Unfortunately, half of the patients treated with imatinib develop disease progression after >2 years (17). The predominant mechanism of acquired resistance to imatinib is acquisition of second site mutations in the Kit kinase domain (17–18). Second-line therapy for imatinib-resistant GIST patients is sunitinib, a more broadly active receptor tyrosine kinase inhibitor, which inhibits some imatinib-resistant Kit mutants, but also PDGF-R, VEGF-R, RET, CSF1-R and flt3. However, sunitinib has been shown to delay disease progression and to extend the overall survival of GIST patient only by a median of six months (19). Therefore, the development of new strategies for the treatment of GIST is urgently needed.
Here the role of Src family kinase and integrin signaling in the mouse KitV558Δ/+ GIST model was investigated. We show that Src, Lyn and FAK are expressed and are active in GIST suggesting a role for integrin signaling in their activation. Whereas imatinib treatment stimulates SFK and FAK activation and thus integrin signaling. Furthermore, the usefulness of the Kit/SFK inhibitor dasatinib in GIST was investigated. Dasatinib inhibited Kit and some Kit-dependent downstream pathways as well as integrin signaling. Whereas dasatinib alone elicits a histologic response similar to imatinib, given in combination with imatinib it showed improved efficacy. Therefore, combination treatment of GIST with RTK inhibitors that target different effectors of Kit signaling may improve clinical efficacy in the treatment of GIST.
MATERIALS AND METHODS
Mice
Heterozygous KitV558Δ/+ mice have been described (7). The KitV558Δ/+ mice used in these experiments have been backcrossed to C57BL/6J for 11 to 13 generations. All procedures were approved by the Institutional Animal Care and Use Committee, IACUC, at MSKCC and conform to the legal mandates and federal guidelines for the care and maintenance of laboratory animals.
Drug treatment of heterozygous KitV558Δ/+ mice
Imatinib (Gleevec, STI571) was kindly provided by Novartis. Imatinib was dissolved in water as a 10 mM solution. Dasatinib was synthesized as in Shah et al., 2004 (20) and was dissolved in water as a 4 mg/ml solution. For 6 hours treatment regimens, heterozygous KitV558Δ/+ mice were treated by intraperitoneal injection, IP, with a single dose of 45 mg/kg imatinib or a single dose of 25 mg/kg dasatinib for monotherapy and for combination treatment with both imatinib and dasatinib, mice were treated with one dose of 45 mg/kg imatinib by intraperitoneal injection plus one dose of 25 mg/kg dasatinib by gavage (per OS). For longer treatments, KitV558Δ/+ mice were treated by intraperitoneal injection with 45 mg/kg imatinib twice daily for 7 days or with 25 mg/kg dasatinib twice daily for 10 days for monotherapy, and for combination treatment with both imatinib and dasatinib, imatinib was administered by intraperitoneal injection at 45 mg/kg and dasatinib by gavage (per OS) at 25 mg/kg or 35 mg/kg twice daily for 7 days.
After indicated treatment times mice were sacrificed 6h after the last administration of drug and the tumors quickly harvested and fixed in freshly prepared 4% paraformaldehyde for histology and immunohistochemistry or quickly frozen in liquid nitrogen for protein analyses. Seven to ten mice were used per treatment group. The histologic response to treatments was based on microscopic findings of necrosis and increased stromal fibrosis and was scored for each tumor as: minimal or no (<10% response), mild (10–50% response), moderate (50–90% response); or very good (≥90% response), as in (10).
Histological, immunohistochemical and statistical analysis
Microscopic and immunohistochemical analysis were as described previously (10). 5 μm sections obtained from paraffin embedded tissue were stained with Hematoxilin & Eosin for histologic response or subjected to immunohistochemistry. The antibodies used for immunohistochemistry were from Cell Signaling for cleaved Caspase 3, phospho-ribosomal protein S6 (S235/236) and P-MAPK (ERK1/2), Novacastra for Ki67 and Santa Cruz Biotechnology for P-FAK. Statistical analysis was as described previously (10): To determine cell proliferation and apoptosis in tumors, Ki67 and cleaved caspase 3 positive cells were counted under microscope on 20 fields of at least 5 different tumors for each time point. The Student’s t test assuming unequal variances between the two samples was used to determine the significance of differences of proliferating and apoptotic cells between untreated, imatinib and dasatinib treated GISTs. Groups were judged to differ significantly at P < 0.05. To determine the intensity of fluorescence of P-FAK, fixed frozen tumor sections from 5 untreated, 5 imatinib and 3 dasatinib treated mice were subjected to immunofluorescence staining with the P-FAK tyr 576/577 antibody (Santa Cruz, CA, USA) followed by a secondary antibody conjugated to Alexa fluor 488. The fluorescence in every sample was captured by scanning the sections with Mirax Scan. Twenty five pictures at 20× magnification were selected from the scan for all untreated, imatinib and dasatinib treated samples and analyzed with the MetaMorph software to determine the intensity of fluorescence in each picture. The Student’s t test assuming unequal variances between two samples was used to determine the significance of differences of P-FAK fluorescence between untreated, imatinib and dasatinib treated GISTs. Groups were judged to differ significantly at P < 0.05. For Real-Time PCR, triplicate values from GIST of 3 untreated, imatinib and dasatinib treated mice were analyzed. The Student’s t test assuming unequal variances between the two samples was used to determine the significance of differences of integrins and integrin ligands expression between untreated, imatinib and dasatinib treated GISTs. Groups were judged to differ significantly at P < 0.05 and were annotated with the “***” symbol. Densitometry of western blots specific bands were measured with Image Gauge from 4 to 8 samples per condition. For each specific protein, the phosphorylated bands were adjusted to the total amount of the protein in each well. The data were then averaged and the untreated samples were assigned a 100% phosphorylation arbitrary value. Imatinib, dasatinib and imatinib plus dasatinib treated samples were compared to the untreated samples as a percentage of phosphorylation. The Student’s t test assuming unequal variances between the two samples was used to determine the significance of differences of protein phosphorylation between untreated, imatinib, dasatinib and imatinib plus dasatinib treated GISTs. Groups were judged to differ significantly at P < 0.05 and were annotated with the “***” symbol.
Immunoprecipitation and Western Blotting: tumor lysates were prepared as described previously with the following modifications: the snap-frozen tumor was first homogenized in the PowerGen 700 homogenizer (Fisher Scientific) and then dounce-homogenized 20 times and incubated on ice for 30 min. Lysates were cleared by centrifugation at 4°C for 30 min. and then fractionated by SDS/PAGE for Western Blotting. For immunoprecipitations, 100 ug lysate was incubated with 4 ul of antibody in a volume of 200 ul with gentle rocking overnight at 4°C. 30 ul of 50% bead slurry Gammabind G sepharose (GE healthcare) was added for 2h at 4°C and then centrifuged for 30 seconds and the pellet washed 5 times with lysis buffer. The antibodies used for Western blot and immunoprecipitation included: phospho-p44/42 MAPK (Thr202/Tyr204), p44/42 MAPKinase, phospho-S6 protein (Ser235/236), S6 protein, phospho-Akt (Thr 308 ), Akt, phospho-STAT3 (Tyr705), STAT3, phospho-STAT5 (Tyr694), STAT5, phospho-Kit (Y719), Src-PY416 (P-SFK), Src, Lck, phospho-p130 CAS (Tyr410) were obtained from Cell Signaling Technologies (Beverly, MA, USA), phospho-FAK (Tyr861) was obtained from Millipore (Billerica, MA, USA), p130 CAS was from BD Biosciences (San Jose, CA, USA) and phospho-FAK (Tyr576/577), FAK, Kit, Lyn (sc-15) and Integrin β5 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Real-Time PCR
2 ug of total RNA were reverse-transcribed at 42°C for 30 minutes using the iScript cDNA Synthesis kit (Biorad). 40 ng of resultant cDNA were used in a Q-PCR reaction using an iCycler (biorad) and pre-designed TaqMan Gene expression Assays (Assay ID for: Blk, Mm00432074_m1; Fyn, Mm00433373_m1; Src, Mm00436783_m1; Fgr, Mm00438949_m1; Hck, Mm00439302_m1; Yes1, Mm00501523_m1; Lck, Mm00802897_m1; Lyn, Mm00802933_m1; Itgb1, Mm01253227_m1; Itga2, Mm00434371_m1; Itga6, Mm00434375_m1; Itgav, Mm00434506_m1; Itgb5, Mm00439825_m1; Itga3, Mm00442890_m1; Itgb7, Mm00442916_m1; Itgb3, Mm00443980_m1; L1cam, Mm00493049_m1; Vtn, Mm00495976_m1; col1a2, Mm01165187_m1; fn1, Mm01256744_m1) Primers were chosen based on their ability to span the 3′ most exon-exon junction. Amplification was carried out for 40 cycles (95 C for 15 sec, 60C for 1 min). To calculate the efficiency of the PCR reaction and to assess the sensitivity of each assay 7 point standard curves were established (10; 3.3; 1.1; 0.37; 0.123; 0.041 and 0.015 ng). Triplicates CT values were averaged, amounts of target were interpolated from the standard curves and normalized to HPRT (hypoxanthine guanine phosphoribosyl transferase).
RESULTS
1. Imatinib inhibits Kit receptor signaling and activates SFKs in mouse KitV558Δ/+ GIST
The Kit receptor is known to bind and activate SFKs and their respective signaling cascades in several cell systems. To investigate the role of Kit mediated SFK signaling in mouse GIST we performed quantitative PCR for all SFKs expressed in tumor tissue. This demonstrated that Blk, Fgr, Fyn, Hck, Lck, Lyn, Src and Yes were expressed in total RNA (not shown). Next we analyzed the activation status of the SFKs. An excellent measure for Src activation is phosphorylation of Src tyrosine 416, Src-PY416, in the activation loop of the kinase. Using a polyclonal anti Src-PY416 antibody which recognizes Src-PY416 and which cross-reacts widely with activation loop phospho tyrosines of SFK members, P-SFK, we found that SFKs are expressed and activated in mouse GIST (Fig. 1A lane 1–4). To assess individual variability tumor extracts were prepared from several mice and analyzed individually as previously described (10). Immunoprecipitation analysis with individual SFK antibodies and Western blotting with the anti- Src-PY416 antibody identified Src and Lyn as expressed and activated in GIST (Fig. 1C lane 1,2). The finding that SFKs are expressed and active in GIST raised the question if their activation resulted from oncogenic Kit signaling. Since imatinib blocks oncogenic Kit signaling we sought to determine if imatinib treatment affected SFK activation and signaling in mouse GIST. KitV558Δ/+ GIST mice were treated with imatinib for 6h and tumor extracts analyzed as reported previously (10). Kit auto-phosphorylation was diminished by imatinib treatment indicating that the drug inhibited Kit signaling; but contrary to our expectation imatinib treatment increased the phosphorylation of SFK activation loop tyrosines (Fig. 1A). This was a surprising finding because we didn’t expect imatinib, an inhibitor of the Kit, PDGFR and Abl kinases, to activate SFKs in GIST. A dose response analysis showed that SFK phosphorylation increased whereas Kit tyrosine 719 autophosphorylation decreased in parallel with increasing imatinib dose, thus demonstrating that there was a good correlation between Kit inhibition and SFK activation in GIST upon imatinib treatment (Fig. 1B). Imatinib did not induce SFK activation by a transcriptional mechanism since the gene expression profile of all SFKs in imatinib treated GIST was unchanged compared to placebo treated GIST (10). These data suggested that post translational modifications were responsible for SFK activation by imatinib. To identify the SFKs whose activation was increased by imatinib treatment, tumor lysates from untreated and treated animals were subjected to an immunoprecipitation analysis. Tumor lysates were immunoprecipitated with different SFK specific antibodies, fractionated by SDS/PAGE and then the broadly reacting anti Src-PY416 antibody was used to identify activated SFKs by Western blot analysis. Whereas Src and Lyn were expressed and activated in GIST (Fig. 1C), Lck and Hck were expressed but not activated (data not shown). Following imatinib treatment P-Lyn and P-Src levels were increased. Taken together these results show that Src and Lyn are active in GIST and that imatinib treatment increased their activity.
Figure 1.

Imatinib stimulates the SFKs Src and Lyn in GIST. (A) immunoblot analysis of tumor extracts from four untreated (lane 1–4), and four 6h imatinib treated mice (lane 5–8) using anti P-Kit Y719, Kit, Src-PY416 (P-SFK) and Src antibodies. (B) Dose response analysis of the effect of imatinib on phosphorylation of Kit-Y719 and the SFK activation loop tyrosine. GIST mice were treated for 6h with 0, 5, 15 or 30 mg/kg imatinib and tumor extracts subjected to immunoblot analysis with P-Kit-Y719 and Src-PY416 antibodies. The percent of reduction in Kit-Y719 phosphorylation and the fold induction in SFK-Y416 phosphorylation were measured by scanning the respective bands with image gauge. The data are presented as means ± standard error of three independent experiments. (C) Tumor lysates from two untreated (lanes 1 and 2), two 6h imatinib (lanes 3 and 4) and two 6h dasatinib (lanes 5 and 6) treated mice were immunoprecipitated with Src and Lyn antibodies and immunoblotted with Src-PY416 (P-SFK), Src and Lyn antibodies. Protein extracts were prepared from 5 different tumors to assess individual variability in the response to drugs. Legend: UT, untreated; Mock, immunoprecipitation without antibody; WTL, whole tumor lysate; IP, immunoprecipitation; WB; Western blot; IM, imatinib; DAS, dasatinib.
2. Dasatinib inhibits both Kit receptor and SFK signaling in mouse KitV558Δ/+ GIST
The Src family kinase inhibitor dasatinib was originally evaluated for inhibition of imatinib resistant BCR-ABL. Dasatinib had been shown to have broad anti-proliferative effects and is known to bind to and inhibit multiple kinases including SRC, ABL as well as Kit (21). Therefore dasatinib might be effective in inhibiting imatinib resistant Kit mutants as well as downstream effectors of oncogenic Kit signaling. Particularly dasatinib might inhibit the consequences of imatinib induced SFK activation and thus show improved efficacy in GIST compared to imatinib. To investigate the effect of dasatinib on oncogenic signaling in GIST, KitV558Δ/+ mice were treated by intraperitoneal injection of dasatinib, at 25 mg/kg twice daily and treatment outcome was compared to imatinib treated mice at 45 mg/kg as previously described (10). After 10 days of treatment, dasatinib induced arrest of cell proliferation similar to imatinib (Fig. 6A). But in contrast to imatinib, dasatinib did not induce apoptosis in GIST (Fig. 6B). H&E staining of dasatinib treated tumors showed a minimal to mild pathologic response similar to imatinib treated GIST mice on the C57BL/6J background (table 1). To investigate the signaling pathways that are affected by dasatinib after a short treatment period, GIST mice were treated for 6 hours and tumor extracts were prepared from several mice and analyzed individually as previously described (10). Because the KitV558Δ/+ mice carry a gain of function Kit allele responsible for tumor development, we first analyzed Kit receptor activation by monitoring autophosphorylation of Kit-Y719 by Western blotting. A reduction of Kit-Y719 phosphorylation was observed in the dasatinib treated samples compared to non-treated controls indicating that dasatinib inhibits Kit receptor activation in KitV558Δ/+ mice (Fig. 2A). We then evaluated inactivation/activation of downstream effectors of Kit signaling in GIST mice upon treatment with dasatinib in comparison to treatment with imatinib. Previously we found that treatment with imatinib inhibits components of the PI3-kinase signaling cascade and STAT3 and STAT5 signaling (10). Recent results with KitV558Δ/+ mice show that in addition to PI3-kinase and STAT signaling, MAP kinase signaling was also inhibited by imatinib as determined by a reduction in phosphorylation of ERK1/ERK2 at T202/Y204 in tumor extracts of mice treated with imatinib (Fig. 2B). Immunohistochemistry of tumor sections with phospho-ribosomal protein S6, and P-ERK1/2 antibodies confirmed the inhibition of PI3-kinase and ERK signaling by imatinib (Fig. 3). Dasatinib treatment similarly inhibited the serine threonine kinase Akt and ribosomal protein S6 phosphorylation was found to be reduced as well (Fig. 2A and Fig. 3). ERK1/ERK2 phosphorylation was also reduced by dasatinib. But in contrast to imatinib, in dasatinib treated GIST mice STAT5 and STAT3a phosphorylation was not reduced (Fig. 2A,B and Fig. 7B). Only the phosphorylation of the STAT3b isoform was slightly reduced. Next we investigated the effect of dasatinib on SFKs and their state of activation in GIST. Using the polyclonal anti Src-PY416 antibody we found that dasatinib treatment abolished activation of SFKs in GIST (Fig. 2C). Immunoprecipitation analysis with the individual SFK antibodies and subsequent Western blotting with the anti Src-PY416 antibody showed that Src and Lyn phosphorylation was inhibited by dasatinib treatment (Fig. 1C and Fig. 2D). These results indicate that dasatinib affected the PI3-kinase and MAP kinase signaling cascades as well as SFK signaling in GIST, but not STAT signaling.
Figure 6.

Combination treatment with imatinib plus dasatinib has additive effects in KitV558Δ/+ mice GIST. Quantitation of proliferating (A) and apoptotic (B) cells in tumor sections of placebo (ctr), 7 days imatinib (45 mg/kg) (IM), 10 day dasatinib-IP (25 mg/kg) (DAS) and 7 days imatinib (45 mg/kg) plus dasatinib-OS (25 mg/kg) treated KitV558Δ/+ mice (DAS+IM). Groups were judged to differ significantly at P < 0.05 (see materials and methods). (C) H&E staining of GIST sections from untreated, 7 days imatinib (45 mg/kg), 10 days dasatinib-IP (25 mg/kg) and 7 days imatinib (45 mg/kg) plus dasatinib-OS (25 mg/kg or 35 mg/kg) treated mice. The arrows denote areas of moderate response with decrease in cellularity and increase in stroma, the arrowheads denote large areas of necrosis. The numbers on the bottom-left correspond to the tumor samples (see table 1).
Table 1.
Individual histologic response of GIST lesions in KitV558Δ/+ mice to treatment with imatinib or dasatinib alone or imatinib/dasatinib combinations.
| imatinib | dasatinib | imatinib + dasatinib (25 mg/kg) | imatinib + dasatinib (35 mg/kg) |
|---|---|---|---|
| mild (#469) | mild (#317) | mild to moderate (#537) | moderate (#571) |
| very minimal (#350) | mild (#354) | mild (#538) | moderate (#573) |
| no (#339) | very minimal (#358) | moderate (#529) | moderate (#575) |
| no (#336) | focal mild (#362) | moderate with focal necrosis (#540) | moderate with focal necrosis (#572) |
| mild (#239) | focal mild (#365) | moderate (#541) | moderate (#585) |
| no (#465) | no (#368) | mild (#542) | moderate (#586) |
| no (#471) | no (#371) | mild to moderate (#539) | moderate with focal necrosis (#574) |
Seven mice in each group were treated for 7 days with imatinib (45 mg/kg), 10 days with dasatinib (25 mg/kg) or 7 days with combinations of imatinib (45 mg/kg) and dasatinib at 2 different doses of dasatinib (25 or 35 mg/kg). The histologic response to treatments was assessed based on microscopic findings of necrosis and increased stromal fibrosis and was scored for each tumor as: minimal or no (<10% response), mild (10–50% response), moderate (50–90% response); or very good (≥90% response), as in (10). Individual tumor samples are identified by # and the observed histologic response is indicated.
Figure 2.

Dasatinib down regulates the translational response, the MAPK and Src signaling pathways in GIST. Immunoblot analysis of tumor lysates from four untreated (lane 1–4) (A–C) and four 6h dasatinib treated mice (A, C) or 6h imatinib treated mice (lane 5–8) (B). Antibodies used include: phospho-Kit (Y719), Kit, phospho-Akt (Thr 308 ), Akt, phospho-S6 protein (Ser235/236), S6 protein, phospho-p44/42 MAPK (Thr202/Tyr204), p44/42 MAPKinase, phospho-STAT3 (Tyr705), STAT3, phospho-STAT5 (Tyr694), STAT5, Src-PY416 (P-SFK) and Src. (D) Tumor lysates from two untreated (lanes 1 and 2), and two 6h dasatinib (lanes 3 and 4) treated mice were immunoprecipitated with Src and Lyn antibodies and immunoblotted with Src-PY416 (P-SFK), Src and Lyn antibodies. Blots shown are representative of at least three independent experiments. Legend as in figure 1.
Figure 3.
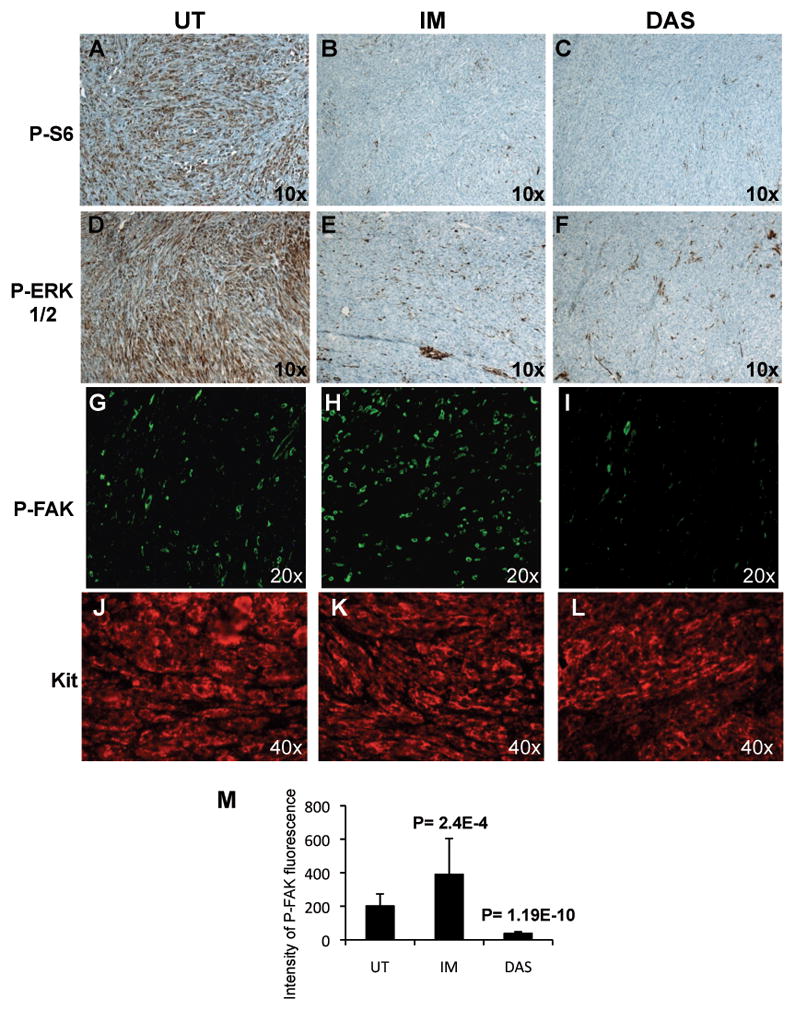
Comparison of Phospho ribosomal protein S6, P-ERK1/2, P-FAK and Kit expression by immunostaining of tumor sections from untreated, imatinib and dasatinib treated GIST mice. Immunohistochemical staining (A–F) of phospho ribosomal protein S6, P-S6 (A–C), P-ERK1/2 (Thr202/Tyr204) (D–F) and immunofluorescence staining (G–L) of P-FAK (Tyr576/577) (G–I) and Kit (J–L) from untreated (A,D,G,J), 6h imatinib treated (B,E,H,K) and 6h dasatinib (C,F,I,L) treated mice. (M) Intensity of fluorescence of P-FAK measured from tumor sections of untreated, imatinib and dasatinib treated mice as described in materials and methods. The intensity of fluorescence is represented in arbitrary unit. The student’s t test assuming unequal variances between two samples was used to determine the significance of differences of P-FAK fluorescence between untreated, imatinib and dasatinib treated GISTs. Groups were judged to differ significantly at P<0.05.
Figure 7.

Additive biochemical effects in tumors treated with imatinib and dasatinib. (A) Western blot analysis of tumor extracts from untreated, and 6h imatinib (45 mg/kg) plus dasatinib (25 mg/kg) co-treated mice. (B) densitometric tracing analysis of Western blots of tumor extracts from 6h imatinib (45 mg/kg), dasatinib (25 mg/kg) and imatinib plus dasatinib treated mice. The values are represented as a percentage of phosphorylation with untreated samples being assigned an arbitrary value of 100% phosphorylation.
3. Imatinib activates integrin signaling
Our results indicated that Src and Lyn are active in GIST and that their activation is increased by imatinib. The Kit autophosphorylation site Kit Y567 is a known docking site for SFKs and SFK docking is a mechanism of Kit mediated SFK signaling (14). Thus Kit could directly activate SFKs in GIST. However, our dose response analysis showed that SFK activation correlated with Kit inhibition, making direct activation of SFKs by Kit unlikely. To determine if Kit associates with SFKs in GIST and has a role in SFK activation, tumor lysates from untreated, imatinib and dasatinib treated GIST mice were subjected to a co-immunoprecipitation analysis. Western blots of fractionated Kit immunoprecipitates analyzed with the anti-Src-PY416 antibody did not reveal any bands indicating that Kit antibody did not precipitate any activated SFKs (Fig. 4A) suggesting that activation of SFKs in GIST is not directly mediated by the Kit receptor.
Figure 4.

Imatinib activates integrin signaling in GIST. (A) Tumor lysates from two untreated, two imatinib and two dasatinib treated mice were immunoprecipitated with Kit antibody and immunoblotted with Src-PY416 (P-SFK) and Kit antibodies. (B) Immunoblot analysis of tumor extracts from untreated, 6h imatinib and 6h dasatinib treated mice. Antibodies used included: anti-phospho-FAK (Tyr397), phospho-FAK (Tyr576/577), phospho-FAK (Tyr861) and FAK. The different P-FAK/FAK combinations shown represent separate experiments. (C) Tumor lysates were immunoprecipitated with FAK antibody and immunoblotted with Src-PY416 (P-SFK), Lyn and Src, and antibodies. Blots shown are representative of at least three independent experiments.
An alternative way to activate SFKs could be through integrin signaling. Focal Adhesion Kinase (FAK) is a critical mediator of integrin signaling in part through phosphorylation by Src. FAK-Src signaling may mediate cell survival, proliferation and motility in normal cells, and was found to be activated in many tumor cells and generating signals for tumor growth and metastasis (22–24). Therefore, it is possible that in GIST SFKs are activated via integrin signaling. To investigate this possibility, we determined the phosphorylation status of FAK. FAK Y397 is phosphorylated upon activation of FAK by integrin clustering. Subsequently SFKs bind to FAK PY397, become activated as a result and phosphorylate other tyrosine residues of FAK. FAK was found to be phosphorylated and imatinib treatment induced a slight but consistent increase in Y397 phosphorylation. In addition imatinib treatment leads to an increase in phosphorylation of FAK at Y576/577 in the activation loop and at Y861 (Fig. 4B). This indicated that FAK is active in GIST and that imatinib stimulated FAK signaling. In agreement with this, the downstream effector of FAK-Src signaling, the adaptor protein p130 CAS, was phosphorylated in GIST at Y410 and phosphorylation was increased by imatinib treatment (Fig. 4B). Dasatinib treatment also induced an increase in Y397 phosphorylation but in contrast to imatinib, phosphorylation of FAK Y576/577 and Y861 was inhibited by dasatinib, in keeping with dasatinib’s activity as an SFK and a FAK inhibitor (Fig. 4B) (25). In addition, immunohistochemistry of GIST sections with anti-phospho-FAK antibody confirmed FAK activation by imatinib and inhibition by dasatinib in the tumor (Fig. 3G–I). The tumor cells were found to be strongly positive for staining with anti-Kit antibody (Fig. 3J–L). This indicated that imatinib stimulated FAK activation in Kit expressing tumor cells rather than in cells of the tumor microenvironment which is Kit negative.
To determine whether FAK is in a complex with SFKs in GIST a co-immunoprecipitation analysis using anti-FAK antibody was done next. Fractionation of FAK immunoprecipitates and Western blotting with SFK antibodies identified Src and Lyn in tumor extract. Western blot analysis with the anti-Src-PY416 antibody indicated that the Src and/or Lyn proteins which are associated with FAK were active in untreated mice. Furthermore, in the imatinib treated samples phosphorylation of P-SFK appeared to be increased while no phosphorylation was observed upon treatment with dasatinib (Fig. 4C). This indicated that FAK-Src and/or FAK-Lyn complexes were stimulated by imatinib and inhibited by dasatinib.
Integrins are heterodimeric transmembrane receptors composed of an α and a β subunit. We assessed the expression of different integrin subunits in GIST using quantitative PCR analysis in order to determine if the level of integrin expression was affected by imatinib or dasatinib treatment (Fig. 5A). This analysis showed high expression of integrins αV and β1, in untreated GIST although integrins α3, β3 and β5 were present as well. Imatinib and dasatinib treatment didn’t change the level of expression of integrins αV and β1, but increased the level of expression of integrins α3, β3, α2 and α6. We furthermore examined the levels of expression of different integrin ligands. Integrin ligands known to activate FAK like Collagen, fibronectin and vitronectin were expressed in GIST but the level of expression was not altered by drug treatment. Expression of L1 cam, a ligand for integrins αVβ1, αVβ3 and αVβ5, has been reported in GIST patients (26). Expression of L1 cam was found in untreated GIST, and surprisingly the level of expression of L1 cam was dramatically increased after imatinib and dasatinib treatment (Fig. 5B). These results imply that imatinib and dasatinib treatment activates integrin signaling by elevating mRNA expression of some integrins and/or one integrin ligand L1 cam. Taken together these results indicate that integrins activate FAK in GIST and that imatinib and dasatinib treatments stimulate the first step in FAK activation, the phosphorylation of Y397. FAK-Lyn and FAK-Src complexes are formed in GIST and are active and imatinib further activated the dual kinase complexes. Interestingly FAK-SFK complexes are also formed in dasatinib treated mice; however they are not active in keeping with dasatinib’s activity as an SFK inhibitor (25).
Figure 5.
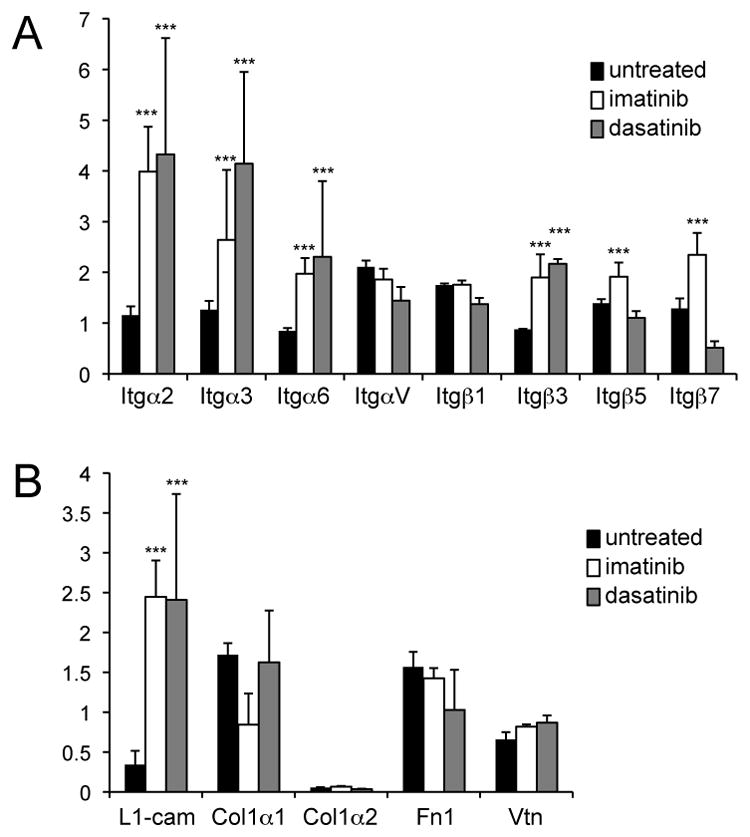
Validation of expression of integrins and integrin ligands in untreated and treated GIST by quantitative PCR. (A comparison of integrin subunit expression including Itgα2, Itgα3, Itgα6, ItgαV, Itgβ7, Itgβ1, Itgβ3 and Itgβ5, and (B) comparison of integrin ligands L1cam, collagen 1 (chains α1 and α2) fibronectin (Fn1) and vitronectin (Vtn) expression in untreated, imatinib and dasatinib treated (6 h)GIST. The student’s t test assuming unequal variances between two samples was used to determine the significance of differences of integrin and integrin ligand expression between untreated, imatinib and dasatinib treated GISTs. Groups were judged to differ significantly at P<0.05.
4. Treatment of KitV558Δ/+ GIST mice with imatinib plus dasatinib produces an increased clinical response
Given the differing biochemical consequences of imatinib and dasatinib treatment in GIST, we investigated whether treatment with both drugs together would be beneficial. GIST mice were treated simultaneously for seven days with imatinib (45 mg/kg) and dasatinib (25 mg/kg) twice daily, both doses representing close to a maximal tolerated dose in mice. Immunohistochemical analysis of tumor tissue indicated a complete arrest of tumor cell proliferation determined by Ki67 staining, consistent with the effect seen with each drug individually (Fig. 6A). However a net increase in apoptosis was observed in comparison with imatinib mono-therapy (Fig. 6B). Furthermore, H&E staining of tumor sections demonstrated a consistently improved histological response compared to treatment with imatinib or dasatinib alone, assessed as a moderate response with focal areas of necrosis (Fig. 6C and table 1). The histological response of combination treatment was even stronger if the dasatinib dose was increased to 35 mg/kg. These results indicate that combination treatment of imatinib plus dasatinib is beneficial compared to imatinib mono-therapy in GIST mice.
Finally we investigated the consequences of combination therapy on oncogenic signaling pathways in GIST. As expected, phosphorylation of Kit, Akt, ribosomal protein S6 and ERK1/2 was reduced by the combination treatment as it is the case for each drug individually. Phosphorylation of SFK tyrosine 416 was also reduced by the double treatment, indicating that dasatinib can overcome the activation of SFKs seen with imatinib treatment (Fig. 7A,B). Furthermore, phosphorylation of the STAT3a, STAT3b and STAT5 transcription factors was diminished as well. Taken together these results indicate that simultaneous treatment with imatinib and dasatinib improved treatment outcome in an additive manner and down-regulated four oncogenic cascades in GIST: the PI3-kinase, SFK, MAPK and STAT pathways. Furthermore, these results imply that the simultaneous down regulation of several pathways is required in order to achieve a good histologic/clinical response.
DISCUSSION
Mice carrying a KitV558Δ germline mutation in the juxtamembrane domain of Kit had been generated based on a case of human familial GIST with the same mutation. Since juxtamembrane domain mutations in Kit represent a majority of somatic Kit mutations in GIST, the KitV558Δ/+ mouse is an excellent tool to assess new treatment strategies to enhance imatinib efficacy. By using the KitV558Δ/+ GIST mouse we have previously shown that imatinib inhibits cell cycle progression and induces apoptosis, inhibits the translational response, MAP kinase signaling as well as STAT3 and STAT5 activation (10). As Kit is known to activate SFK signaling we investigated the effect of imatinib treatment on SFK signaling in mouse GIST. Interestingly and surprisingly we found that imatinib an inhibitor of the Kit, PDGFR and Abl kinases induces increased activation of SFKs in particular Lyn and Src, as well as FAK in mouse GIST. We have investigated mechanisms of SFK activation in GIST and its stimulation upon imatinib treatment. SFKs are known to associate with and act as effectors of different cell membrane receptors including receptor tyrosine kinases, cytokine receptors and integrins. Since Kit -PY567 is a docking site for SFKs it was possible Kit may activate SFKs even in the presence of imatinib. However, this is unlikely because imatinib binds Kit in its inactive conformation and inhibits Kit autophosphorylation and kinase activity. In fact, in Kit immunoprecipitates of tumor lysates both from untreated and treated mice no active SFKs could be identified.
In the tumor microenvironment integrins and their ligands are thought to provide signals for tumor cell survival and proliferation as well as have roles in tumor progression and metastasis (27–28). Integrin signaling is bidirectional; whereas inside-out signaling mediated by growth factors such as VEGF, IGF and Kit ligand may increase the affinity of integrins to extra cellular ligands in the extracellular matrix, outside-in signaling mediates the cellular responses to cell adhesion. In mouse GIST several integrin subunits are highly expressed and are thought to have roles in tumor progression and metastasis. It is likely that integrin signaling in GIST is also bidirectional and that both the integrin ligands and Kit ligand are agonists in this signaling network and the question arises how integrin signaling is affected by inhibition of Kit with imatinib in the mouse GIST model. Different integrin ligands including fibronectin, laminin, Vcam vitronectin and L1cam are expressed in mouse GIST. Interestingly, L1cam expression was up-regulated by both imatinib and dasatinib treatment in our gene expression profile and quantitative PCR analysis in contrast to the other integrin ligands. Although we don’t know the molecular mechanisms of increased L1cam expression by imatinib treatment, since L1cam is a ligand for integrins αVβ1, αVβ3 and αVβ5, which are expressed in GIST, this could explain the up-regulation of integrin signaling by imatinib (29–30). Furthermore, upregulation of expression of different integrin subunits, i.e. integrins α3, β3, α2 and α6 by imatinib and dasatinib could result in increased integrin signaling.
The cytoplasmic tyrosine kinase FAK associates with integrins in focal adhesions and is critical in signaling mediated by integrin clustering. Upon integrin clustering, FAK is auto-phosphorylated on FAK Y397 creating an SH2 binding site for SFKs, and consequent SFK activation. Maximal FAK activity is attained by Src mediated phosphorylation of FAK tyrosines Y576/Y577 in the activation loop of the FAK kinase (22–23, 31). In turn, maximal Src kinase activity is attained upon phosphorylation of Src Y416 in the activation loop of the Src kinase by FAK (32). Whereas in untreated GIST, FAK Y397 is phosphorylated, imatinib treatment increased FAK Y397 phosphorylation indicating increased integrin clustering and signaling. Furthermore, FAK Y576/Y577 were phosphorylated in untreated GIST and phosphorylation was increased upon imatinib treatment, implying a role for FAK signaling in SFK activation in GIST. In addition a protein complex was identified that includes FAK-Src/and Lyn. Both the Src and Lyn tyrosine kinases are known to interact with FAK upon integrin stimulation (31, 33). This complex was active because SFK Y416 was phosphorylated. Therefore, integrin-mediated compensatory FAK-SFK signaling activated by imatinib is independent of oncogenic Kit signaling. In agreement with this, tyrosine phosphorylation of SFK, FAK and paxilin, were found to be resistant to Kit inhibition in vitro in GIST cell lines (34).
Our results imply a role for integrin-FAK mediated signaling in tumor maintenance and a role as a possible negative regulator in imatinib treatment. Interestingly, FAK is highly expressed in patients with malignant GIST (35). In a large fraction of breast cancers the FAK gene is amplified and mechanistic studies showed that FAK sustains tumorigenesis by mediating SFK mediated phosphorylation of p130 Cas (36). Furthermore, a ligand of integrin alphaV-β5, L1cam (CD171), is highly expressed in GIST and high L1cam expression may correlate with reduced survival (26). Therefore, targeting of FAK and SFKs or their downstream effectors should be beneficial for GIST treatment.
We have used the SFK/FAK and Kit inhibitor dasatinib as a single agent and in combination with imatinib to investigate efficacy in mouse GIST. As a single agent, dasatinib inhibits cell proliferation, Kit autophosphorylation/activation and Kit downstream targets, the PI3-kinase and MAPK signaling cascades in the GIST mouse. This is consistent with studies in mast cells and hematopoietic cell lines demonstrating inhibition by dasatinib of wild type, juxtamembrane mutant isoforms of Kit, and imatinib-resistant activation loop mutant Kit isoforms (37–38). In a study of kinase inhibitor selectivity, dasatinib was also found to have a better affinity for Kit WT and KitV559D than imatinib (39). However, dasatinib’s activity in GIST differs from imatinib as follows: first, whereas dasatinib inhibits cell cycle progression it does not induce apoptosis; second whereas imatinib inhibits STAT3 and STAT5 activation, dasatinib does not; and third, in contrast to imatinib, dasatinib inhibits SFKs whereas imatinib activates SFKs. These results are consistent with dasatinib’s high affinity for SFKs (25, 39).
In a recent study dasatinib was shown to inhibit the active conformation of FAK (25). In agreement with this dasatinib was shown to inhibit phosphorylation of the activation loop tyrosines of FAK Y576/Y577, but not the phosphorylation of FAK tyrosine 397, of the kinase. The failure of dasatinib to inhibit Y397 phosphorylation implies that the phosphorylation of this tyrosine residue does not require a fully activated FAK kinase, or alternatively inhibition of Src/Lyn blocks the complete activation of FAK. Therefore, these results provided a rationale for the use of dasatinib in combination with imatinib in the treatment of GIST.
The question then arises why combination of imatinib and dasatinib produced a better histological response than dasatinib alone, since dasatinib inhibits Kit, SFKs and FAK. One possibility is that STAT signaling has an important but not sufficient role in GIST maintenance. We previously showed that imatinib inhibits STAT signaling in GIST. Whereas dasatinib does not inhibit STAT3 and STAT5 phosphorylation, combination treatment diminishes phosphorylation of STAT3 and STAT5. This may indicate that inhibition of STAT signaling results in an enhanced histological response. In agreement with a critical role for STAT3 signaling in GIST, in colitis associated cancer STAT3 was shown to have critical roles in mediating cell proliferation and cell survival (40). The observation of a failure of dasatinib to inhibit STAT3/5 signaling is of interest but an investigation of the role of STAT3/5 signaling in mouse GIST will be a challenging next step. For combination therapy both imatinib and dasatinib were used at close to maximally tolerated doses to optimally inhibit the drug sensitive signaling cascades. The downside of this strategy was that only additive effects of the combination therapy were identified. Clearly, careful dose response studies will need to be done in the future to further understand the benefits of combination imatinib/dasatinib combination therapy.
Previously, we demonstrated that imatinib inhibits PI3-kinase, MAP kinase and STAT signaling in our mouse GIST model. Now we demonstrate that imatinib also activates SFKs by compensatory integrin signaling and furthermore we demonstrate that dasatinib treatment inhibits Kit activation, SFK, FAK, PI3-kinase and MAP kinase signaling but not STAT signaling. Therefore, the increased clinical response by combination treatment with imatinib and dasatinib appears to result from the simultaneous inhibition of PI3-kinase, MAP kinase, STAT and SFK signaling. It is known that tyrosine kinase inhibitors can be active against “off-targets” and we cannot exclude that such ‘off-target’ effects may increase or decrease their therapeutic potential in GIST (25). However it has been shown that combination of two tyrosine kinase inhibitors that target Bcr-Abl can preempt in vitro resistance (41). Similarly, a combination of different tyrosine kinase inhibitors could preempt imatinib resistance in GIST. Therefore, these results provide a rationale for the development of combined targeted therapies for the treatment of human GIST.
Acknowledgments
The authors thank Volodia Gueorguiev, Mesruh Turkekul, Afsar Barlas, Tao Tong and Sho Fujisawa from the Molecular Cytology Facility, Huiyong Zhao from the Antitumor Assessment Facility, Yuliya Pylayeva-Gupta and Kelly M. Gillen for discussion and help with providing integrin and FAK antibodies, the Transgenic Mouse Facility and the Research Animal Resource Center at Memorial Sloan Kettering Cancer Center for expert help. We also would like thank David Cobrinik for comments on the manuscript. Laura Debatin, Narsi Agaram and Tianhua Guo for discussion and Zachary Oberzan and John Burrowes for secretarial assistance. This work was supported by NIH grant R01-CA-102774, RO1-HL55748, the LifeRaft Group and the Starr Cancer Consortium.
References
- 1.Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577–80. doi: 10.1126/science.279.5350.577. [DOI] [PubMed] [Google Scholar]
- 2.Maeda H, Yamagata A, Nishikawa S, Yoshinaga K, Kobayashi S, Nishi K. Requirement of c-kit for development of intestinal pacemaker system. Development. 1992;116:369–75. doi: 10.1242/dev.116.2.369. [DOI] [PubMed] [Google Scholar]
- 3.Ward SM, Burns AJ, Torihashi S, Sanders KM. Mutation of the proto-oncogene c-kit blocks development of interstitial cells and electrical rhythmicity in murine intestine. J Physiol. 1994;480:91–7. doi: 10.1113/jphysiol.1994.sp020343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antonescu CR, Sommer G, Sarran L, et al. Association of KIT exon 9 mutations with nongastric primary site and aggressive behavior: KIT mutation analysis and clinical correlates of 120 gastrointestinal stromal tumors. Clin Cancer Res. 2003;9:3329–37. [PubMed] [Google Scholar]
- 5.Lasota J, Wozniak A, Sarlomo-Rikala M, et al. Mutations in exons 9 and 13 of KIT gene are rare events in gastrointestinal stromal tumors. A study of 200 cases. Am J Pathol. 2000;157:1091–5. doi: 10.1016/S0002-9440(10)64623-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rubin BP, Singer S, Tsao C, et al. KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res. 2001;61:8118–21. [PubMed] [Google Scholar]
- 7.Sommer G, Agosti V, Ehlers I, et al. Gastrointestinal stromal tumors in a mouse model by targeted mutation of the Kit receptor tyrosine kinase. Proc Natl Acad Sci U S A. 2003;100:6706–11. doi: 10.1073/pnas.1037763100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Druker BJ, Sawyers CL, Kantarjian H, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344:1038–42. doi: 10.1056/NEJM200104053441402. [DOI] [PubMed] [Google Scholar]
- 9.Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–80. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 10.Rossi F, Ehlers I, Agosti V, et al. Oncogenic Kit signaling and therapeutic intervention in a mouse model of gastrointestinal stromal tumor. Proc Natl Acad Sci U S A. 2006;103:12843–8. doi: 10.1073/pnas.0511076103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rottapel R, Reedijk M, Williams DE, et al. The Steel/W transduction pathway: kit autophosphorylation and its association with a unique subset of cytoplasmic signaling proteins is induced by the Steel factor. Mol Cell Biol. 1991;11:3043–51. doi: 10.1128/mcb.11.6.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serve H, Hsu YC, Besmer P. Tyrosine residue 719 of the c-kit receptor is essential for binding of the P85 subunit of phosphatidylinositol (PI) 3-kinase and for c-kit- associated PI 3-kinase activity in COS-1 cells. J Biol Chem. 1994;269:6026–30. [PubMed] [Google Scholar]
- 13.Serve H, Yee NS, Stella G, Sepp-Lorenzino L, Tan JC, Besmer P. Differential roles of PI3-kinase and Kit tyrosine 821 in Kit receptor- mediated proliferation, survival and cell adhesion in mast cells. Embo J. 1995;14:473–83. doi: 10.1002/j.1460-2075.1995.tb07023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Timokhina I, Kissel H, Stella G, Besmer P. Kit signaling through PI 3-kinase and Src kinase pathways: an essential role for Rac1 and JNK activation in mast cell proliferation. Embo J. 1998;17:6250–62. doi: 10.1093/emboj/17.21.6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Linnekin D, DeBerry CS, Mou S. Lyn associates with the juxtamembrane region of c-Kit and is activated by stem cell factor in hematopoietic cell lines and normal progenitor cells. J Biol Chem. 1997;272:27450–5. doi: 10.1074/jbc.272.43.27450. [DOI] [PubMed] [Google Scholar]
- 16.Kozlowski M, Larose L, Lee F, Le DM, Rottapel R, Siminovitch KA. SHP-1 binds and negatively modulates the c-Kit receptor by interaction with tyrosine 569 in the c-Kit juxtamembrane domain. Mol Cell Biol. 1998;18:2089–99. doi: 10.1128/mcb.18.4.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Antonescu CR, Besmer P, Guo T, et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res. 2005;11:4182–90. doi: 10.1158/1078-0432.CCR-04-2245. [DOI] [PubMed] [Google Scholar]
- 18.Debiec-Rychter M, Cools J, Dumez H, et al. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology. 2005;128:270–9. doi: 10.1053/j.gastro.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 19.Joensuu H. Second line therapies for the treatment of gastrointestinal stromal tumor. Curr Opin Oncol. 2007;19:353–8. doi: 10.1097/CCO.0b013e3281338885. [DOI] [PubMed] [Google Scholar]
- 20.Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305:399–401. doi: 10.1126/science.1099480. [DOI] [PubMed] [Google Scholar]
- 21.Carter TA, Wodicka LM, Shah NP, et al. Inhibition of drug-resistant mutants of ABL, KIT, and EGF receptor kinases. Proc Natl Acad Sci U S A. 2005;102:11011–6. doi: 10.1073/pnas.0504952102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol. 2006;18:516–23. doi: 10.1016/j.ceb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 23.Parsons JT, Slack-Davis J, Tilghman R, Roberts WG. Focal adhesion kinase: targeting adhesion signaling pathways for therapeutic intervention. Clin Cancer Res. 2008;14:627–32. doi: 10.1158/1078-0432.CCR-07-2220. [DOI] [PubMed] [Google Scholar]
- 24.Shattil SJ. Integrins and Src: dynamic duo of adhesion signaling. Trends Cell Biol. 2005;15:399–403. doi: 10.1016/j.tcb.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 25.Bantscheff M, Eberhard D, Abraham Y, et al. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat Biotechnol. 2007;25:1035–44. doi: 10.1038/nbt1328. [DOI] [PubMed] [Google Scholar]
- 26.Kaifi JT, Strelow A, Schurr PG, et al. L1 (CD171) is highly expressed in gastrointestinal stromal tumors. Mod Pathol. 2006;19:399–406. doi: 10.1038/modpathol.3800547. [DOI] [PubMed] [Google Scholar]
- 27.Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol. 2004;5:816–26. doi: 10.1038/nrm1490. [DOI] [PubMed] [Google Scholar]
- 28.Hehlgans S, Haase M, Cordes N. Signalling via integrins: implications for cell survival and anticancer strategies. Biochim Biophys Acta. 2007;1775:163–80. doi: 10.1016/j.bbcan.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 29.Felding-Habermann B, Silletti S, Mei F, et al. A single immunoglobulin-like domain of the human neural cell adhesion molecule L1 supports adhesion by multiple vascular and platelet integrins. J Cell Biol. 1997;139:1567–81. doi: 10.1083/jcb.139.6.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mechtersheimer S, Gutwein P, Agmon-Levin N, et al. Ectodomain shedding of L1 adhesion molecule promotes cell migration by autocrine binding to integrins. J Cell Biol. 2001;155:661–73. doi: 10.1083/jcb.200101099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parsons JT. Focal adhesion kinase: the first ten years. Journal of cell science. 2003;116:1409–16. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- 32.Wu L, Bernard-Trifilo JA, Lim Y, et al. Distinct FAK-Src activation events promote alpha5beta1 and alpha4beta1 integrin-stimulated neuroblastoma cell motility. Oncogene. 2008;27:1439–48. doi: 10.1038/sj.onc.1210770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chung J, Wang XQ, Lindberg FP, Frazier WA. Thrombospondin-1 acts via IAP/CD47 to synergize with collagen in alpha2beta1-mediated platelet activation. Blood. 1999;94:642–8. [PubMed] [Google Scholar]
- 34.Zhu MJ, Ou WB, Fletcher CD, Cohen PS, Demetri GD, Fletcher JA. KIT oncoprotein interactions in gastrointestinal stromal tumors: therapeutic relevance. Oncogene. 2007;26:6386–95. doi: 10.1038/sj.onc.1210464. [DOI] [PubMed] [Google Scholar]
- 35.Koon N, Schneider-Stock R, Sarlomo-Rikala M, et al. Molecular targets for tumour progression in gastrointestinal stromal tumours. Gut. 2004;53:235–40. doi: 10.1136/gut.2003.021238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pylayeva Y, Gillen KM, Gerald W, Beggs HE, Reichardt LF, Giancotti FG. Ras-and PI3K-dependent breast tumorigenesis in mice and humans requires focal adhesion kinase signaling. The Journal of clinical investigation. 2009;119:252–66. doi: 10.1172/JCI37160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo T, Agaram NP, Wong GC, et al. Sorafenib inhibits the imatinib-resistant KITT670I gatekeeper mutation in gastrointestinal stromal tumor. Clin Cancer Res. 2007;13:4874–81. doi: 10.1158/1078-0432.CCR-07-0484. [DOI] [PubMed] [Google Scholar]
- 38.Schittenhelm MM, Shiraga S, Schroeder A, et al. Dasatinib (BMS-354825), a dual SRC/ABL kinase inhibitor, inhibits the kinase activity of wild-type, juxtamembrane, and activation loop mutant KIT isoforms associated with human malignancies. Cancer Res. 2006;66:473–81. doi: 10.1158/0008-5472.CAN-05-2050. [DOI] [PubMed] [Google Scholar]
- 39.Karaman MW, Herrgard S, Treiber DK, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–32. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 40.Grivennikov S, Karin E, Terzic J, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer cell. 2009;15:103–13. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O’Hare T, Eide CA, Tyner JW, et al. SGX393 inhibits the CML mutant Bcr-AblT315I and preempts in vitro resistance when combined with nilotinib or dasatinib. Proc Natl Acad Sci U S A. 2008;105:5507–12. doi: 10.1073/pnas.0800587105. [DOI] [PMC free article] [PubMed] [Google Scholar]