

Abstract
The kinetics, thermodynamics, and coordination dynamics for O2 and CO 1:1 binding to a series of pseudo-tetradentate ligand-copper(I)-complexes (DLCuI) to give CuI/O2 and CuI/CO product species are reported. The DLCuI series possess an identical tridentate core structure where the cuprous ion binds to the bispicolylamine (L) fragment. DL also contains a fourth variable N-donor moiety {D = benzyl (Bz); pyridyl (Py); imidazolyl (Im); dimethylamino (NMe2-); tert-butylphenyl pyridyl (TBP); quinolyl (Q)}. The structural characteristics of DLCuI-CO and DLCuI are detailed, with X-ray crystal structures reported for TBPLCuI-CO, BzLCuI-CO, and QLCuI. Infrared studies (solution and solid-state) confirm that DLCuI-CO possess the same four-coordinate core structure in solution with the variable D moiety ‘dangling’, i.e. not coordinated to the copper(I) ion. Other trends observed for the present series appear to derive from the degree to which the D-group interacts with the cuprous ion center. Electrochemical studies reveal close similarities of behavior for ImLCuI and NMe2LCuI (as well as for TBPLCuI and QLCuI), which relate to the O2-binding kinetics and thermodynamics. Equilibrium CO binding data (KCO, ΔH°, ΔS°) were obtained by conducting UV-visible spectrophotometric CO titrations, while CO binding kinetics and thermodynamics (kCO ; ΔH‡, ΔS‡) were measured through variable temperature (193 K – 293 K) transient absorbance laser flash photolysis experiments, λex = 355 nm. Carbon monoxide dissociation rate constants (k−CO) and corresponding activation parameters (ΔH‡, ΔS‡) have also been obtained. CO binding to DLCuI follows an associative mechanism with the increased donation from D leading to higher kCO values. Unlike that seen in previous work, the KCO values increased as the kCO and k−CO values declines; the latter decreased at a faster rate. By using the ‘flash-and-trap’ method (λex = 355 nm ; 188 K – 218 K), the kinetics and thermodynamics (kO2 ; ΔH‡, ΔS‡) for O2 binding to NMe2LCuI and ImLCuI were measured and compared to PyLCuI. A surprising change in the O2 binding mechanism was deduced from the thermodynamic ΔS‡ values observed, associative for PyLCuI but dissociative for NMe2LCuI and ImLCuI; these results are interpreted as arising from a difference in the timing of electron transfer from copper(I) to O2 as this molecule coordinates and a tetrahydrofuran (THF) solvent molecule dissociates. The change in mechanism was not simply related to alterations in DLCuII/I geometries or the order that O2/THF coordinate. The equilibrium O2 binding constant (KO2 ; ΔH°, ΔS°) and O2 dissociation rate constants (k−O2 ; ΔH‡, ΔS‡) were also determined. Overall the results demonstrate that subtle changes in the coordination environment, as occurs over time through evolution in nature or through controlled ligand design in synthetic systems, dictate to a critically detailed level the observed chemistry in terms of reaction kinetics, structure and reactivity, and thus function. Results reported here are also compared to relevant copper and/or iron biological systems and analogous synthetic ligand-copper systems.
Introduction
A detailed understanding of copper(II/I) protein active site properties and their 1:1 small molecule (CO, O2, NO) binding characteristics is of importance and has inspired a diverse array of research efforts for biochemists and synthetic bioinorganic chemists alike.1-6 Research advances through synthetic modeling have helped to gain important insights into copper coordination and reactivity through generation of ligands with precisely optimized formulations and through study of their ligand-copper/small-molecule chemistry. In this context, however, the ability to directly monitor the formation, structures, and subsequent reactivity of CuI/O2 derived primary species, i.e. mononuclear copper(II)-superoxo complexes formulated as (ligand)CuII-O2•−, is relatively rare.7-15 Indeed, such species are implicated as key active-site entities in the (bio)chemistry of dioxygen activating copper enzymes.16-19
Time resolved laser flash spectroscopy has been widely used to investigate small molecule interactions (CO, O2, NO, etc.) with natural and synthetic hemes.20-36 By taking advantage of the extremely short time scale resolution of such photolytic methods, detailed mechanistic information has been achieved. For example, a wealth of knowledge has been gained about both hemoglobin and myoglobin by monitoring photoinitiated CO dissociation from heme-CO in the presence of O2.37 Also, in the heme-copper protein cytochrome c oxidase (CcO; Figure 1), the characterization of photodriven CO transfer (as a surrogate for O2 and potentially NO38) from hemea3-CO to CuB suggested that the CuB site was a “doorway” for small molecules into and out of the heterobimetallic active site.3,27 Assuming a common O2 mechanistic pathway and with some indirect evidence existing,39-41 a CuIIB-O2•− intermediate is assumed to precede any hemea3/O2 interactions; a putative peroxo-bridged CcO intermediate that was crystallized is shown in Figure 1.
Figure 1.
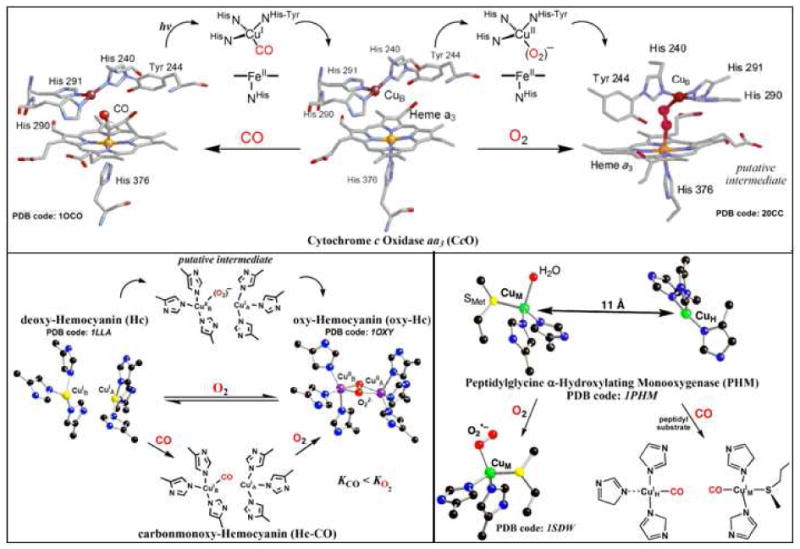
Overview of reduced copper containing protein active-site O2 and CO interactions, highlighting the hemea3-CuB active-site for O2 binding/reduction of mitochondrial cytochrome c oxidase (CcO), the arthropodal and molluscan hemolymph O2-carrier Hc, and the mammalian neuropeptide hormone producing monooxygenase PHM.
A key aspect for accomplishing such photoinitiated processes is the high binding affinity of heme systems for CO (KCO > KO2), as exploited by the “flash-and-trap” experiment pioneered by Gibson and coworkers.32,33 Similar studies involving CO and O2 have not been accomplished for the well-characterized type-3 binuclear copper proteins hemocyanin (Hc; Figure 1), tyrosinase (Tyr; widespread monooxygenase, converting phenols to o-catechols and o-quinones), and catechol oxidase (o-catechol → o-quinone), mainly due to the opposite behavior, a high stability (KO2 > KCO) of their 2:1 copper-dioxygen adduct, a dicopper(II)-μ-η2:η2-(side-on)-peroxo protein species.3,16,42,43
Early work established that only one small molecule, CO or O2, binds per dicopper active site, presumably to CuB, and that the close proximity of the two copper centers (CuA and CuB) enables fast electron-transfer (et) for the two-electron reduction of O2.3,16,42,43 However, even following photoexcitation and subsequent dissociation of O2 from oxy-Tyr44 and/or oxy-Hc,45,46 a CuIIB-O2•− adduct has not been detected and instead only implicated through theoretical modeling.16,47 Never-the-less, Hirota, Bubacco, and coworkers recently used flash photolysis and complementary K-edge X-ray absorption spectroscopy (XAS) measurements to show that the O2 binding rate constant (kO2) for a deoxy-Hc was dependent on the copper(I) coordination geometry.45 In addition, the thermodynamic data (ΔH‡, ΔS‡) they obtained were comparable to those reported for mononuclear synthetic model compounds, which emphasizes the important relationship between synthetic bioinorganic chemistry and metallobiochemistry.
In peptidylglycine α-hydroxylating monooxygenase (PHM), the active-site contains two uncoupled copper centers (Cu…Cu ∼ 11 Å; CuA ≡ CuH; CuB ≡ CuM). A copper(II)-superoxo species, which is suggested by some to be the key intermediate that initiates substrate oxidation reactions, has been characterized by X-ray crystallography.18 This “end-on” η1 -bound (to CuM) “precatalytic” copper-dioxygen adduct is shown in Figure 1. Interestingly, CO binds the catalytic (CuM) site as well as to the electron-transfer (CuH) center, the latter only if substrate is present.48,49 Key CuI/O2 1:1 species are also implicated in the enzymatic reactions carried out by copper amine oxidase (CAO)50-52 and galactose oxidase.19
As mentioned, CO heme protein active site interactions have been widely investigated. In addition, the use of CO for investigation of copper proteins provides tremendous insights and fundamental information.3 The occurrence of detectable and significant variations in active site copper ligation within a given protein has been noted and is also present in proteins from different species or organisms. The νCO values of carbonmonoxy-copper proteins are highly variable, even when equally coordinated by three imidazolyl N-donors. For example, νCO values corresponding to the CuI-CO adducts of CAO (2061 – 2085 cm-1),53 the CuH-CO site of PHM (2062 cm-1),48 and the CuB-CO sites of CcO (2053 – 2063 cm-1),54-56 nitrite reductase (NiR; 2050 cm-1),57 and Hc-CO (2043 – 2063 cm−1),58 exhibit large differences. Such variations in νCO for N3CuI -CO adducts indicates subtle changes in the local active site environment (e.g., coordination geometry changes, dielectric of the medium, etc.) that alter the copper ion's electron density and thus back-donation to the ligated carbon monoxide. This likely relates to or explains the diverse O2 and/or NO reactivity amongst various copper proteins.
With this background, one of our goals is to develop an understanding of the kinetics and thermodynamics of copper(I) mediated small molecule interactions, specifically in this report with CO and O2. Carbon monoxide is a good surrogate for dioxygen, even while possessing redox inactive binding characteristics. In addition, copper(I)-carbonyl adducts are typically more stable than their 1:1 copper-dioxygen counterparts and therefore easier to study. By gaining a deeper understanding of how the detailed nature of ligand environment affects CO binding to copper(I), as has been carried out in natural systems, reliable comparisons can be made to CuI/O2 and potentially NO (bio)chemistries.
We previously reported on the CO photodissociation chemistry of a number of cuprous pyridylalkylamine compounds.59 Effects upon systematic variations in the ligand framework were examined: (i) via changes in the electron-donating ability of TMPA {PyL ; tris(2-pyridylmethyl)amine} through addition of 4-pyridyl substituents (R-PyL ; R = N(CH3)2-, CH3O-); (ii) by an increase in the chelate ring-size from 5-membered to 6-membered ligand coordination to copper as in PMEA {bis[(3-pyridyl)methyl]-2-(2-pyridyl)ethylamine} vs PMAP {bis[2-(2-pyridyl)ethyl]-(2-pyridyl)methyl-amine}; and (iii) with a change in the donor group moieties as in BQPA {bis(2-quinolylmethyl)(2-pyridylmethyl)amine}. The effort advanced our understanding of the coordination environment required for ligand-copper(I)-carbonyl photodissociation and rebinding of carbon monoxide; tridentate coordination from the tetradentate ligand moieties to the copper(I)-carbonyl center (overall four-coordinate) was required (Chart 1), in which one N-donor moiety was ‘dangling’, i.e. uncoordinated. Observed variations in kinetics and thermodynamics were shown to derive from particular changes in the copper complex electron-releasing properties, coordination geometry, and/or steric effects.
Chart 1.

The subject and direction of the work discussed herein examines the effect of changes in the dangling N-donor moiety within various ligand-copper(I)-complexes (DLCuI) on both 1:1 CO and O2-binding kinetics and thermodynamics as well as CO and O2 coordination dynamics. Variable temperature transient absorbance (TA) laser flash photolysis in tetrahydrofuran (THF) solvent has been employed. The DL ligand series consists of potentially tetradentate ligands with the same PY1 {L ; bis(2-pyridylmethyl)amine} core, however with variable N-donor moieties (D), see Chart 1. For the studies of CO binding, a tridentate ligand species BzL was also examined as a “standard” for comparison.25 As will be described, drastic differences in CuI-CO and CuI-O2 chemistry result from seemingly minor changes in the DLCuI ligand framework.
Experimental
See Supporting Information for details concerning Materials and Methods, references to syntheses of previously published ligands and complexes along with synthetic procedures for new compounds.
Results
Synthesis of Ligand-Copper(I) (DLCuI) and Ligand-Copper(I)-Carbonyl (DLCuI-CO) Complexes
The ligands (DL) used in the present study were previously reported and characterized in detail (Chart 1).60-65 The ligand-copper(I) complexes as B(C6F5)4− salts (DLCuI) were straightforwardly prepared by addition of [CuI(MeCN)4]B(C6F5)4 to the appropriate ligand in deoxygenated Et2O and isolated by slow precipitation in dry air-free pentane under an argon atmosphere.8,60,64-66 The B(C6F5)4− counteranion was chosen to afford greater solubility in tetrahydrofuran (THF) solvent.
The presence of coordinated acetonitrile (CH3CN) in the isolated copper(I) complexes of PyLCuI, ImLCuI, NMe2LCuI, and BzLCuI was confirmed through elemental (C, H, N) combustion analysis and 1H-NMR spectroscopy studies in deuterated nitromethane (CD3NO2).8,60,64,65 The X-ray crystal structures of BzLCuI and the ClO4− salts of NMe2LCuI and PyLCuI were previously reported with each containing an exogenously derived CH3CN molecule.60,62,67 As expected for a tridentate ligand system, BzLCuI binds CH3CN the strongest as supported by the shorter Cu–Nnitrile bond distance of 1.900(4) Å versus a value of 2.038(2) Å for NMe2LCuI and 1.990(12) Å for PyLCuI, the complexes with tetradentate N4 ligation. Acetonitrile does not coordinate to TBPLCuI and QLCuI based on the characterization methods described above and X-ray structural characterization, vide infra.
Ligand-copper(I)-carbonyl complexes (DLCuI-CO) were formed in situ through vigorous CO bubbling into dry THF solutions. TBPLCuI-CO and BzLCuI-CO were isolated as overall four-coordinate species, i.e., possessing a dangling ligand donor arm (vide infra), following dissolution of TBPLCuI and BzLCuI in CO saturated Et2O and layering with pentane for slow diffusion.68 In a previous study, PyLCuI-CO was isolated as a five-coordinate species, i.e., with all ligand N donors coordinated.59
X-ray Crystallography of TBPLCuI-CO, BzLCuI-CO, QLCuI
ORTEP diagrams of TBPLCuI-CO (A) and BzLCuI-CO (B) are shown in Figure 2 and that for QLCuI is given in Figure 3; selected bond lengths and angles for all are provided in the figure caption.68 Both ligand-copper(I)-carbonyl structures display an overall four-coordinate geometry consisting of the copper(I) ion coordinated by the apical alkylamino nitrogen, two pyridyl donors, and the carbon from a coordinated CO molecule. The Npyridyl–Cu–Namine angles of ∼81° are rather severe in comparison to other structurally characterized four-coordinate copper(I) carbonyl complexes which have average N–Cu–N bond angles of ∼95°.59,69-75 The Cu-C bond length is shorter (stronger) for TBPLCuI-CO (1.801 Å) in comparison to that observed in BzLCuI-CO (1.815 Å), possibly due to an indirect increase in overall electron density in the former complex, with the difference being a tert-butylphenyl (tbp) substituted pyridine vs. benzyl substituent on the L tridentate moiety, see Chart 1.
Figure 2.

ORTEP diagrams of A. TBPLCuI-CO and B. BzLCuI-CO; hydrogen atoms and the B(C6F5)4 counteranion have been omitted for clarity. Selected bond lengths are: A. Cu–C 1.801(6) Å, C–O 1.124(6) Å, Cu–Nalkylamine 2.147(4) Å, Cu–Npyridine(avg) 2.051(5) Å; and B. Cu–C 1.815(1) Å, C–O 1.123(2) Å, Cu–Nalkylamine 2.158(6) Å, and Cu–Npyridine(avg) 2.048(3) Å. Selected bond angles are: A. C–Cu–Npyridine(avg) 121.4(2)°, C–Cu–Nalkylamine 131.0(0)°, Npyridine(avg)–Cu–Nalkylamine 81.0(9)°, Npyridyl–Cu–Npyridyl 109.6(0)°; and B. C–Cu–Npyridine(avg) 121.95(7)°, C–Cu–Nalkylamine 129.94(7)°, Npyridine(avg)–Cu–Nalkylamine 80.80(6)°, and Npyridyl–Cu–Npyridyl 108.94(6)°.68
Figure 3.

ORTEP diagram of QLCuI with hydrogen atoms and the B(C6F5)4 counteranion omitted for clarity. Selected bond lengths are: Cu–Npyridine(1) 1.988(2) Å, Cu–Npyridine(3) 2.006(2) Å, Cu–Nalkylamine(2) 2.185(2) Å, and Cu–Nquinoline(4) 2.012(2) Å. Selected bond angles are: Npyridyl(avg)–Cu–Nalkylamine(2) 82.84(8)°, Npyridyl(1)–Cu–Npyridyl(3) 123.63(8)°, Npyridyl(1)–Cu–Nquinolyl(4) 119.95(8)°, Namine(2)–Cu–Nquinolyl(4) 82.58(7)°, and Npyridyl(3)–Cu–Nquinolyl(4) 111.70(8)°.68
A tridentate (N3) coordination mode for TBPLCuI-CO is of interest due to the tetradentate (N4) nature of TBPL; the 6-tbp-substituted pyridyl arm dissociates, i.e. is dangling, due to the increased steric constraints. However in the absence of CO all three pyridyl donors as well as the bridgehead alkylamine coordinate to the copper(I) ion, as supported by the known X-ray crystal structure of TBPLCuI.64 The coordination sphere of TBPLCuI did not include a coordinated acetonitrile molecule as is generally observed for analogous ligand-copper(I) complexes, vide supra.
Similar structural characteristics have been reported based on X-ray crystallographic analysis of the copper(I) complex and copper(I)-carbonyl adduct of BQPA.59,63,76 For [CuI(bqpa)]B(C6F5)4, all four N-donor moieties coordinate to the copper(I) ion and one quinolyl donor arm dissociates upon coordination of CO or triphenylphosphine (PPh3).63 As shown in Figure 3, the X-ray crystal structure of QLCuI is four-coordinate with both pyridyl donors, the quinolyl donor, and the bridgehead alkylamine coordinated to the cuprous center. An X-ray crystal structure of QLCuI-CO has not been obtained but IR spectroscopic data supports a tridentate ligand coordination in which the quinolyl arm is dangling, vide infra.
Infrared Spectroscopy (νCO: Solution and Solid State)
The solution (THF) and solid-state (Nujol mull) νCO values of the ligand-copper(I)-carbonyl complexes (DLCuI -CO) were determined in order to gauge the ligand-N-donor ability of the variable D-moiety as well as to elucidate potential differences in coordination number and/or the overall geometry. As described above, in part, the CO stretching frequencies (νCO) of synthetic copper(I)-carbonyl complexes and carbonmonoxy- copper proteins are found to be in the range νCO = 2035 – 2137 cm-1. 3,73,77-82 Notably, the characteristic νCO values of copper(I)-carbonyl proteins coordinated by three imidazolyl N-donors are usually ΔνCO = 20 – 40 cm-1 lower than synthetic species with three N-donors. 3,79 This suggests that copper protein histidine imidazole groups are exceptionally strong donors and relative to most synthetic copper(I)-carbonyl complexes, this leads to weaker C–O (but stronger Cu-C) bonds.
Solution-State IR Spectroscopic and Structural Properties
As discussed in the Introduction (Chart 1), previous work established that an equilibrium mixture of four- (4 νCO = 2090 cm-1) and five- (5νCO = 2077 cm-1) coordinate copper(I)-carbonyl isomers exist for PyLCuI-CO in THF solvent, Table 1 and Chart 1. Similarly, an equilibrium mixture of ImLCuI-CO isomers exist in THF, 4νCO = 2087 cm-1 and 5νCO = 2063 cm-1. The lower energy shift of Δ5νCO = 14 cm-1 for ImLCuI-CO isomer in comparison to PyLCuI-CO indicates that the imidazole in ImL is a stronger donor for copper(I) than the pyridine in PyL. However, the hard aliphatic amine group within NMe2LCuI-CO does not ligate at all in THF solution, as indicated by the single CO stretch at 2090 cm-1.
Table 1.
Carbonyl Stretching Frequencies for DLCuI-CO Complexes and Cyclic Voltammetry Data for DLCuI Complexes.
| Compound | Neat solid | THF solution | E1/2b | ΔE b |
|---|---|---|---|---|
| ImLCuI-CO | 2097, 2035(sm) | 2087, 2063 | −445 | 110 |
| NMe2LCuI-CO | 2097 | 2090 | −445 | 85 |
| PyLCuI-CO | 2077 | 2091, 2074(sh) | −410 | 75 |
| QLCuI-CO | 2090 | 2091 | −325 | 110 |
| TBPLCuI-CO | 2091 | 2092 | −325 | 115 |
| BzLCuI-CO | 2093 | 2093 | −225 | 155 |
Abbreviations are as follows: sm = small, sh = shoulder.
Electrochemical measurements are of copper(I) complexes (not carbonyl) in deoxygenated CH3CN. Potentials are reported versus the Fc+/Fc redox couple and are rounded to the nearest 5 mV.
Consistent with their X-ray crystal structures, TBPLCuI-CO and BzLCuI-CO are four-coordinate structures in THF solution with 4νCO values of 2091 cm-1 and 2093 cm-1 respectively, see Table 1. THF solution data for QLCuI-CO reveals the same N3CuI-CO coordination based on the 4νCO value. Overall, the CO stretching frequencies for all four-coordinate DLCuI-CO solution complexes are approximately equal, 4νCO(avg) = 2091 cm-1, indicating that the dangling arm throughout the series must be the D-donor moiety, as depicted in Chart 1.
Solid-State IR Spectroscopic and Structural Properties
The 4νCO values of the isolated ImLCuI-CO and NMe2LCuI-CO species were both centered at 2097 cm-1 in Nujol mull spectra suggesting that the imidazole or aliphatic dimethylamine donor moieties do not coordinate in the static solid-state structure. Since these values are higher than the solution state 4νCO values, the DL ligation may be somewhat more two-coordinate in nature, i.e., perhaps the aliphatic amine nitrogen is less strongly bound than for the other cases (Chart 2). In the solid state, PyLCuI-CO has an overall five-coordinate structure.59
Chart 2.
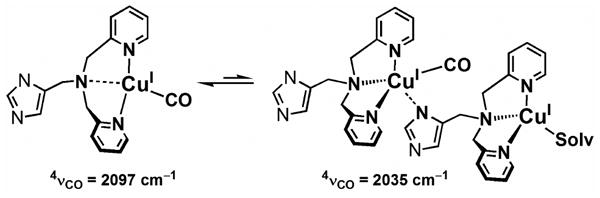
For ImLCuI-CO, an additional very low energy and intensity CO stretch at 2035 cm-1 is observed in the solid-state. Such a low νCO value suggests a coordination environment with anionic ligand donors, or three strong non-chelating N donors, as is known elsewhere.3,78 In previous work, the electron-donating ability of the four-coordinate R- PyL ligand series was increased by introduction of para-substituents such as R = N(CH3)2- and CH3O- (Chart 2)59. Here, νCO values shifted to lower energy by 14 to 28 cm-1, e.g., νCO = 2049 cm-1 for [CuI(NMe2- PyL)(CO)]+ in THF. To explain the very low 2035 cm-1 νCO value, we suggest that the dangling imidazolyl moiety of ImLCuI-CO may coordinate to a different ImLCuI complex, forming a dimeric structure, see Chart 2 below. Pyridyl π–π stacking may facilitate formation of such a structure, as has been observed elsewhere for copper(I) structures.83-85
Electrochemical Studies
Copper(II/I) redox chemistry was examined through cyclic voltammetry measurements carried out on the ligand-copper(I)-complexes (DLCuI). Quasi-reversible single electron-transfer processes (ipc/ipa ≈ 1) with peak-to-peak separations between ΔE = 75 – 155 mV were observed (Table 1). The CuII/CuI redox potentials, i.e. electron transfer, are largely affected by structural alterations prompted by changes in oxidation states as well as steric hindrance induced by ligand variations. Overall, the trend of CuII/I half-wave potentials (E1/2) were similar to the trend of the 4νCO values of DLCuI-CO species in THF, see Table 1. Ligand-copper-complexes with more negative E1/2 values favor a higher oxidation state (CuII) and suggest a more donating ligand system. For example, the formally tridentate BzLCuI, has the highest redox potential of −225 mV, hence favoring a lower oxidation state.
The same E1/2 values were measured for QLCuI and TBPLCuI at −325 mV and their peak-to-peak separation was also about the same, ΔEavg = 115 mV. These results suggest that the steric influence of the quinoline donor and 6-tbp-subsituent are very similar. The same E1/2 values were also measured for ImLCuI and NMe2LCuI at −445 mV, which was surprising because of their different static copper(II) structures. The X-ray crystal structure of [CuII(NMe2L)(Cl)]2+ reveals a distorted square pyramidal coordination geometry, τ = 0.26.62,86 The τ structural parameter is 0.00 for a perfect square pyramidal structure and l.00 for a perfect trigonal bipyramidal coordination geometry.86,87 By contrast, [CuII(IML)(CH3CN)]2+ displays a trigonal bipyramidal static structure, τ = 0.86.65,86
Since [CuII(PyL)(CH3CN)]2+ has an almost perfect trigonal bipyramidal static structure, τ = 0.96,67,86 the same degree of structural rearrangement would be expected to occur upon oxidation of ImLCuIand PyLCuI. Both of their cuprous-acetonitrile structures are assumed to be very similar based on their related DLCuI-CO 4νCO values. However, the E1/2 values are quite different for ImLCuI (107 mV) and PyLCuI (76 mV) suggesting that the in situ copper(II) structures are different. The E1/2 value of ImLCuI (and NMe2LCuI) in comparison to PyLCuI (−410 mV) is 35 mV more negative, therefore the latter is easier to reduce.
CO Equilibrium Binding and Thermodynamic Parameters (KCO: ΔH°, ΔS°)
The equilibrium CO binding constants (KCO, Scheme 1) and corresponding thermodynamic parameters (ΔH°; ΔS°) were determined by variable temperature monitoring of THF solvent reactions of CO with DLCuI to form DLCuI-CO.59 Throughout the entire series (Table 2), KCO increases are accompanied by decreasing (more favorable) enthalpies but less favorable (decreasing) reaction entropies. This behavior is typical, as enhanced binding can be described as being “tighter”. UV-visible spectral data representative of the conversion of TBPLCuI to TBPLCuI-CO are shown in Figure 4; data corresponding to all other species are found in the Supporting Information.
Scheme 1.

Table 2.
Kinetic and Thermodynamic Parameters for CO binding to DLCuI in THF; Constants Reported at 298 K.a
| Compound |
KCO (M-1) |
ΔH° | ΔS° |
kCO (M-1 s-1) |
ΔH‡ | ΔS‡ |
k-CO (s-1) |
ΔH‡ | ΔS‡ |
|---|---|---|---|---|---|---|---|---|---|
| ImLCuI-CO | (2.4 ± 0.04) × 103 | −31.4 | −40.6 | (2.8 ± 0.07) × 109 | 7.03 | −40.5 | (1.1 ± 0.04) × 106 | 38.5 | 0.1 |
| NMe2LCuI-CO | (5.0 ± 0.05) × 103 | −39.7 | −62.5 | (2.5 ± 0.05) × 109 | 9.07 | −34.7 | (5.0 ± 0.09) × 105 | 48.8 | 28 |
| PyLCuI-CO | (1.2 ± 0.05) × 104 | −35.9 | −42.6 | (1.9 ± 0.02) × 109 | 7.13 | −43.3 | (1.5 ± 0.04) × 105 | 43.0 | −0.7 |
| QLCuI-CO | (2.2 ± 0.2) × 104 | −39.2 | −48.5 | (9.7 ± 0.1) × 108 | 5.66 | −53.9 | (4.4 ± 0.01) × 104 | 44.9 | −5.3 |
| TBPLCuI-CO | (1.4 ± 0.03) × 104 | −40.4 | −56.2 | (5.2 ± 0.01) × 108 | 5.76 | −58.7 | (3.7 ± 0.01) × 104 | 46.2 | −2.5 |
| BzLCuI-CO | (5.6 ± 0.1) × 104 | −46.0 | −63.6 | (5.0 ± 0.01) × 108 | 0.74 | −75.9 | (9.0 ± 0.03) × 103 | 46.8 | −76 |
Units of kinetic and thermodynamic values are as follows: KCO, M-1; kCO, M-1 s-1; k–CO, s-1; ΔH, kJ mol-1; ΔS, J mol-1 K-1. See Supporting Information for Van't Hoff and Eyring plots.
Figure 4.

Spectrophotometric titration of CO to TBPLCuI in THF at room temperature. The inset is a Van't Hoff plot of the variable temperature KCO data collected from −30 °C to +20 °C.
CO Photodissociation and Rebinding Kinetics
The kinetics and thermodynamics of CO binding to DLCuI were measured through variable temperature (20 to −80°C) transient absorption (TA) laser flash photolysis experiments.15,59 Absorption difference spectra, Abs{[DLCuI] – [DLCuI-CO]}, calculated from steady-state absorption experiments were in agreement with observed transient data collected within the range λmon = 325 – 700 nm; see Figure 5 for ΔA spectra corresponding to photodissociation of CO from TBPLCuI-CO. The subsequent rebinding of CO followed a first-order kinetic model. Bimolecular rate constants (kCO) for DLCuI-CO formation were calculated based on [CO] dependence studies.59 Complementary activation (ΔH‡; ΔS‡) parameters were calculated by determination of kCO at variable temperatures and through Eyring analysis.59
Figure 5.

Transient absorption difference spectra observed after pulsed 355 nm excitation of TBPLCuI-CO in THF at room-temperature under 1 atm of CO. CO photodissociation and the formation of TBPLCuI followed by subsequent CO recombination is measured and shown at various delay times: 0 ns (black); 100 ns (red); 250 ns (blue); 500 ns (green); 1000 ns (purple). The inset is an Eyring analysis plot of the kCO data collected at variable temperature (−70 °C to 10 °C).
In all cases, CO (re)binding to DLCuI follows an associative mechanism as suggested by the kCO associated negative reaction entropies (ΔS‡) in the range −40 to −76 J mol-1 K-1, see Table 2. Furthermore, the nearly identical thermodynamic (ΔS°) and activation (ΔS‡) entropies along the DL series of complexes suggest a late transition state; the rate determining step involves an intermediate that is structurally similar to the final carbonylated product. Following CO photodissociation, the dangling N-donor arm of DLCuI-CO binds to the open coordination site of the cuprous ion and must dissociate before CO can rebind.
Thermal DLCuI-CO Dissociation Kinetics
With KCO and kCO values determined experimentally, the thermal CO dissociation rate constants (k−CO) and corresponding activation parameters (ΔH‡, ΔS‡) were calculated.59 As expected, the k−CO value increases with decreasing reaction enthalpies (ΔH‡) and increasing entropic (ΔS‡) values. The transition state largely correlates with the overall geometry change as related to the degree to which the dangling arm is or is not interacting with the cuprous-carbonyl upon Cu–C bond breakage. See below for further discussion.
Kinetics (kO2; ΔH‡, ΔS‡) in THF of 1:1 Dioxygen Binding to DLCuI following CO Photodissociation from DLCuI-CO; D = Py, Im, NMe2
The primary interaction (1:1) of O2 with PyLCuI, ImLCuI, and NMe2LCuI to form DLCuII-O2− was monitored and measured in THF solvent through the “flash-and-trap” method.15 The experiments were conducted in the presence of precisely determined concentrations of O2 and CO within the temperature range of −55 °C to −85 °C on nanosecond and longer time scales. The ΔA spectra, Abs{[NMe2LCuI/II-X] − [NMe2LCuI-CO]} X = THF or O2−, representing the two separate O2-binding processes (kfast, kslow) for formation of NMe2LCuII-O2− are shown in Figures 6A-B; corresponding data for formation of ImLCuII-O2− is given in the Supporting Information. As depicted in Scheme 6, the initial “fast” process (kfast; Figure 6A) involves the competitive binding of both CO and O2 with DLCuI, either regenerating DLCuI-CO or forming DLCuII-O2−, thus, kfast is a combination of two rate constants, kO2 and kCO. Following generation of DLCuII-O2−, since KCO ≫ KO2, CO subsequently displaces coordinated O2 to reform the initial DLCuI-CO species (kslow). The latter values, kO2 (and KO2 and k−O2) derived from kslow, are tabulated here and used for discussion.
Figure 6.
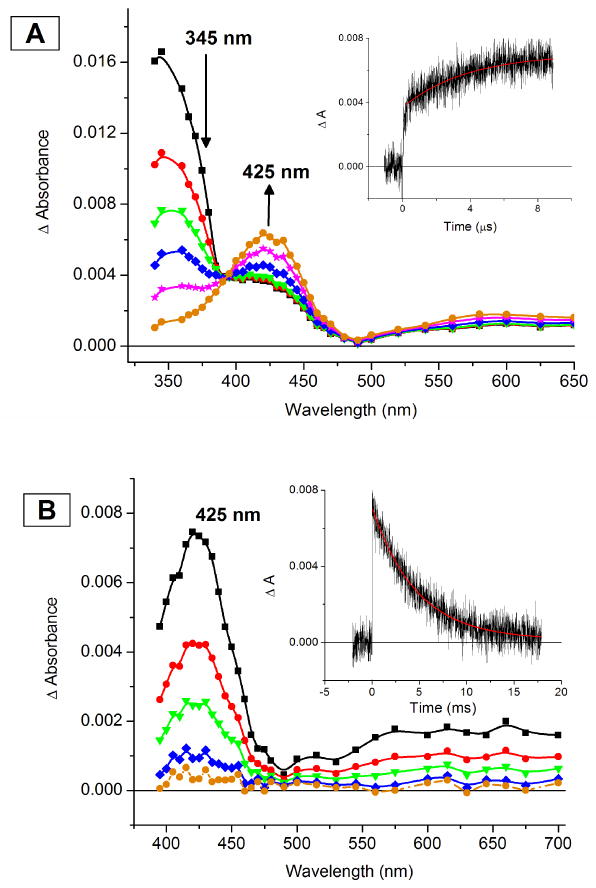
Absorption difference spectra [NMe2LCuI/II-X − NMe2LCuI-CO] recorded after pulsed λex =355 nm excitation of NMe2LCuI-CO in THF at 193 K under 1 atm O2:CO (1:1) mixture. The spectra were recorded at various delay times: (A) 0 to 5 μs representing the conversion of NMe2LCuI to a mixture of NMe2LCuI-CO and NMe2LCuII-O2−: black squares, 0 μs; red circles, 0.1 μs; green triangles, 0.25 μs; blue diamonds, 1 μs; magenta stars, 2.5 μs; orange circles, 5.0 μs. The inset is an absorption transient monitored at 425 nm with a superimposed first-order fit (in red), kobs = 2.92 × 105 s-1 and (B) 0 ms to 20 ms representing the conversion from NMe2LCuII-O2− to NMe2LCuI-CO: black squares, 0 ms; red circles, 2.5 ms; green triangles, 5.0 ms; blue diamonds, 10 ms; orange circles, 20 ms. The inset is an absorption transient monitored at 425 nm with a superimposed first-order fit (in red), kobs = 236 s-1.
Scheme 6.

A full analysis of the O2 binding kinetics and thermodynamics for formation of PyLCuII-O2− was reported previously in THF solvent via the “flash-and-trap” method,15 and in EtCN through stopped-flow UV-visible spectroscopy.8 In the present work, analogous data for formation of ImLCuII-O2− and NMe2LCuII-O2− in THF are discussed and compared to that for PyLCuII-O2−. Room temperature kO2 values were obtained by extrapolation from the activation parameters obtained at low temperatures where experimental data could be collected. All data here are also compared to those obtained by Schindler, Zuberbühler and coworkers on the closely related tripodal tetradentate ligand complex [CuI(Me6tren)]+ {tris(2-dimethylaminoethyl)amine},62 see Table 3. In fact, very fast reactions with kO2 > 107 M-1s-1 at 298 K have only been observed and determined for the DLCuI complexes described here, which includes the previously studied PyLCuI, and [CuI(Me6tren)]+.13-15
Table 3.
Comparison of the kinetics and thermodynamics for formation of DLCuII-O2− (D = Py, Im, NMe2) in THF and other ligand-copper(II)-η1-superoxo species.
| Parameter |
PyLCuI THF(a) |
PyLCuI EtCN(b) |
[CuI(Me6tren)]+ EtCN(b) |
ImLCuI THF(c) |
NMe2LCuI THF(c) |
|---|---|---|---|---|---|
| kO2, 193 K (M-1 s-1) | (1.5 ± 0.02) × 108 | (3.8 ± 0.01) × 104 | (1.8 ± 0.04) × 105 | (1.8 ± 0.03) × 108 | (6.9 ± 0.02) × 107 |
| kO2, 298 K (M-1 s-1) | (1.3 ± 0.02) × 109 | (5.8 ± 0.8) × 107 | (1.2 ± 0.1) × 107 | (3.4 ± 0.6) × 1010 | (2.3 ± 0.4) × 1011 |
| ΔH‡ (kJ mol-1) | 7.62 | 31.6 | 17.1 | 23.4 | 32.1 |
| ΔS‡ (J mol-1 K-1) | − 45.1 | 10 | − 52 | 35.1 | 80.1 |
| k-O2, 193 K (s-1) | 240 ± 6 | 130 ± 1 | 0.62 ± 0.01 | 1600 ± 0.05 | 470 ± 0.02 |
| k-O2, 298 K (s-1) | (1.3 ± 0.03) × 108 | (1.5 ± 0.2) × 108 | (7.7 ± 0.9) × 105 | (1.9 ± 0.2) × 109 | (2.3 ± 0.3) × 109 |
| ΔH‡ (kJ mol-1) | 58.0 | 61.5 | 62.0 | 64.5 | 66.5 |
| ΔS‡ (J mol-1 K-1) | 105 | 118 | 76 | 150 | 157 |
| KO2, 193 K (M-1) | (6.5 ± 0.02) × 105 | 260 ± 4 | (2.9 ± 0.04) × 105 | (1.1 ± 0.03) × 105 | (1.5 ± 0.06) × 105 |
| KO2, 298 K (M-1) | 15.4 ± 0.3 | 0.38 ± 0.02 | 15.5 ± 0.5 | 17.4 ± 0.8 | 100 ± 0.7 |
| ΔH° (kJ mol-1) | − 48.5 | − 29.8 | − 44.9 | − 41.1 | − 34.4 |
| ΔS° (J mol-1 K-1) | − 140 | − 108 | − 128 | − 114 | − 77.3 |
O2 binding data was previously published and collected through
flash-and-trap techniques in THF,15
Averaged values were determined from analysis of kslow using the flash-and-trap methods discussed herein, see text for more details. Bimolecular kO2 values calculated at additional temperatures are given in the Supporting Information, Table S2.
Discussion
CO Equilibrium Binding and Thermodynamic Parameters (KCO: ΔH°, ΔS°)
The KCO values for formation of DLCuI-CO increase as the E1/2 values become more positive and the 4νCO values shift to higher energy (Table 1 and Table 2). In previous work that examined the CO binding properties of copper(I)-carbonyl adducts of the R-PyL ligand series where only electronic effects are present, the opposite trend was observed; thus, a ligand having better donor properties led to enhanced CO binding.59 This suggests that steric constraints or structural differences induced by changing the dangling “D” N-donor moiety of DL significantly affect the KCO values in the present systems.
We suggest that the differences in ΔS° values or degree of disorder in the reaction are affiliated in part with the extent that the dangling “D” N-donor moiety is interacting with the final DLCuI-CO adduct. For example, the CuI-CO adduct of the purely tridentate BzL has the highest KCO value of 5.6 × 104 M-1. This clearly derives from BzLCuI-CO having the most favorable reaction enthalpy in the series, ΔH° = −46.0 kJ mol-1 K-1, since the reaction is the most unfavored entropically, ΔS° = −63.6 kJ mol-1 K-1. In more depth, formation of BzLCuI-CO is the most ordered reaction, meaning the benzyl D-group does not coordinate or donate any significant electron density to the cuprous center and also does not hinder coordination of CO to BzLCuI.
The KCO values for CO binding to QLCuI and TBPLCuI are the highest of the tetradentate ligands studied, 2.2 × 104 M-1 and 1.4 × 104 M-1 respectively. Based on their solution and solid state 4νCO values, the ligand-copper(I)-carbonyl complexes of both are tridentate in nature, consistent with the X-ray crystal structure of TBPLCuI-CO (vide supra). However, in comparison to BzLCuI-CO, the coordination of CO to the cuprous ion of QLCuI and TBPLCuI must be somewhat hindered by their bulky “D” N-donor groups (i.e., quinolyl or tBu-pyridyl arms), resulting in their slightly lower KCO values (Table 2).
At the low end of the range, reaction of ImLCuI with CO results in the smallest KCO value of 2.4 × 103 M-1 with the most favorable entropy, ΔS° = −40.6 kJ mol-1 K-1. We suggest that the dangling imidazole is more interacting with the cuprous-carbonyl center resulting in increased disorder. The bond enthalpy for formation of ImLCuI-CO is the highest (ΔH° = −31.4 kJ mol-1), consistent with the weakest Cu–C bond of the series.
In comparison to the reaction of CO with PyLCuI, the KCO value of 1.2 × 104 M-1 is approximately five times higher as a result of the slightly lower thermodynamic values, ΔH° = −35.9 kJ mol-1, ΔS° = −42.6 kJ mol-1 K-1.59 Such small differences in the thermodynamic parameters, yet large effect on the equilibrium CO binding constant may suggest that the dangling pyridyl arm of PyL is less interacting with the cuprous ion than is the imidazolyl moiety of ImL.
By contrast, the equilibrium binding constant of KCO = 5.0 × 103 M-1 for reaction of CO with NMe2LCuI is lower. However, the highly negative ΔS° value of −62.5 kJ mol-1 K-1 is similar to that of BzLCuI-CO suggesting that the aliphatic dimethylamino arm is barely interacting if at all with the cuprous ion of NMe2LCuI in situ. Such a characteristic is consistent with the absence of an observable 5νCO for NMe2LCuI-CO.
CO Photodissociation and Rebinding Kinetics
The rate constant for formation of DLCuI-CO (kCO) appears to be controlled by the degree to which the dangling “D” arm interacts with the cuprous ion during Cu–C bond formation. This conclusion comes about from the observation that decreasing 4νCO frequencies for DLCuI-CO along with decreased complex E1/2 values for DLCuI, which both indicate a better donor ligand, correlate with an increase in kCO values (Table 1 and Table 2). For example, ImLCuI-CO is comprised of the most electron-donating N-donor system for copper(I) due to the presence of the imidazolyl donor moiety and this complex gives the highest rate constant, kCO = 2.8 × 109 M-1 s-1. The corresponding activation parameters for formation of ImLCuI-CO, ΔH‡ = 7.03 kJ mol-1 and ΔS‡ = −40.5 kJ mol-1 K-1, are very similar to those measured for PyLCuI-CO, suggesting little differences in the CO coordination dynamics. The coordination of CO to ImLCuI is depicted in Scheme 2.
Scheme 2.

Consistent with the near equivalent E1/2 values determined for ImLCuI and NMe2LCuI, NMe2LCuI-CO has a similar CO rebinding rate of 2.5 × 109 M-1 s-1. However, the ΔS‡ value affiliated with CO binding to NMe2LCuI is much more positive than the ΔS° value suggesting that the course of reaction is more disordered as a result of the dangling or very weakly interacting donor moiety. Unlike for ImLCuI-CO and PyLCuI-CO, a five-coordinate carbonyl species is not structurally favorable for NMe2LCuI-CO; in support of this supposition, note that only a 4νCO value was observed, vide supra. As a result, dissociation of the fourth ligand N-donor arm of NMe2LCuI followed by subsequent CO rebinding likely involves significant structural changes resulting in a higher ΔS‡ value.
The parallel thermodynamic and activation parameters associated with CO binding to QLCuI (9.7 × 108 M-1 s-1) and TBPLCuI (5.2 × 108 M-1 s-1) suggests that the mechanism of reaction is the same for both (Scheme 3). Such similarities are consistent with their analogous 4νCO values and identical E1/2 values. Since solvent (THF) does not coordinate to QLCuI and TBPLCuI, only the “D” N-donor must dissociate from the cuprous ion to form their respective ligand-copper(I)-carbonyl adducts.
Scheme 3.

Carbon monoxide rebinding to BzLCuI, where a fourth endogenous N-donor is not present, results in the lowest kCO value of 5.0 × 108 M-1 s-1; note that the CuII/CuI compound exhibits the most positive E1/2 value. The negligible enthalpy (ΔH‡ = 0.74 kJ mol-1) and highly negative entropy (ΔS‡ = −75.9 J mol-1 K-1) of activation implies that the tridentate nature of BzL minimizes geometric changes in the reaction of CO with BzLCuI and that the ‘dangling’ benzyl group does not hamper CO rebinding to the cuprous center. However, since the standard entropy (ΔS°) for formation of BzLCuI-CO is greater than the activation entropy (ΔS‡), it is possible that the CO molecule rebinds to the naked [CuI(BzL)]+ species before THF transiently coordinates, resulting in a more ordered reaction and negligible enthalpy of activation (ΔH‡) as shown in Scheme 3.
Thermal DLCuI-CO Dissociation Kinetics
As shown in Scheme 2 for CO dissociation from ImLCuI-CO, the dangling arm ligates to the copper(I) ion following CO dissociation. The negligible activation entropy (ΔS‡ ∼ 0 J mol-1 K-1) suggests that there is little overall geometry differences between the initial carbonylated complex and the transition state. Therefore, the increased donation offered by the slightly interacting imidazole in ImLCuI-CO results in an overall decrease in the structural changes as well as a decrease in the activation barrier to cleave the Cu–C bond. ImLCuI-CO has a k−CO value of 1.1 × 106 s-1 with a corresponding reaction enthalpy of ΔH‡ = 38.5 kJ mol-1. Similar characteristics were exhibited by PyLCuI-CO,59 QLCuI-CO, and TBPLCuI-CO for CO dissociation, all with near zero ΔS‡ values (Table 2).
Dissociation of CO from NMe2LCuI-CO is characterized by ΔS‡ = 28 J mol-1 K-1, a more favorable value than is observed for any other in the series (Table 2). Thus, Cu–C bond cleavage involves considerable structural rearrangement and the high ΔS‡ value implies an early transition state structurally similar to the terminal copper(I)-carbonyl species. Such a mechanism would require the dimethylamino donor arm of NMe2L to be predominantly in the unbound state when the Cu–C bond is broken and further suggests that coordination of solvent occurs prior to or concomitant with CO release, see Scheme 4. As already stated, it is unfavorable for the hard dimethylamino arm to interact with the soft cuprous-carbonyl center, as evidenced or supported by a lack of observation of 5νCO for NMe2LCuI-CO.
Scheme 4.

Conversely, dissociation of CO from BzLCuI-CO is accompanied by a strikingly unique unfavorable activation entropy of ΔS‡ = −76 J mol-1 K-1 that leads to the smallest k−CO value (9.0 × 103 s-1) in the series. The almost identical ΔS‡ values for the forward and reverse reaction of BzLCuI/CO suggest that virtually no geometry change results and therefore the reaction is highly ordered, i.e. there is a late transition state.
Kinetics (kO2; ΔH‡, ΔS‡) in THF of 1:1 Dioxygen Binding to DLCuI following CO Photodissociation from DLCuI-CO; D = Py, Im, NMe2
An examination of activation parameters (ΔH‡, ΔS‡; Table 3) indicates there is a striking difference between those for formation of PyLCuII-O2− compared to the other two copper(II) η1-superoxo complexes, ImLCuII-O2− and NMe2LCuII-O2−. The binding of O2 to PyLCuI occurs with a relatively large and negative activation entropy (−45.1 J mol-1 K-1, Table 3) indicating an associative reaction mechanism is involved.15 This suggests O2 binds prior to loss of the bound solvent molecule and then THF solvent de-ligates. Or, solvent may not even coordinate to the cuprous center, accounting for a simple (unhindered) association of dioxygen. As an example of the latter point, the tetradentate ligand-copper(I)-complex [CuI(Me6-tren)]+ does not coordinate an exogenously derived nitrile ligand.14 As a result, an associative O2 binding mechanism (ΔS‡ = −52 J mol-1 K-1) resulted for formation of [CuII(Me6-tren)(O2−)]+ in EtCN, see Table 3. By extreme contrast, the positive activation entropies measured for NMe2LCuII-O2− (ΔS‡ = 80.1 J mol-1 K-1) and ImLCuII-O2− (ΔS‡ = 35.1 J mol-1 K-1) indicate that the O2-binding reaction follows a dissociative mechanism where de-ligation of solvent (THF) occurs prior to O2 binding. Throughout the remainder of the text, this latter reaction mechanism is referred to as a dissociative process, however, we note that a purely dissociative mechanism would result in first-order kinetics. Therefore, it is perhaps more appropriate to describe this process as a dissociative interchange reaction, in which bond breakage dominates over bond formation. More detailed explanation and analysis of the dioxygen binding mechanisms is given below.
As discussed earlier in the text, the structurally characterized cupric-chloride species [NMe2LCuII(Cl)]+ has a square pyramidal geometry (τ = 0.26),62 different than the trigonal bipyramidal structures exhibited by [PyLCuII(Cl)]+ (τ = 1.00),88 [PyLCuII(CH3CN)]2+ (τ = 0.96),67 and [ImLCuII(CH3CN)]2+ (τ = 0.86)65.86 Also, the complementary ligand-copper(I)-CH3CN structures of the three aforementioned species DLCuI have analogous trigonal bipyramidal geometries. Therefore, based on the assumption that the cupric-superoxo species are analogous in structure to their X-ray crystallographically characterized copper(II) adducts, the structural rearrangement that results upon redox changes in the NMe2L ligand system would be much greater. Consistent with this supposition is the observed highly positive activation entropy ΔS‡ = 80.1 kJ mol-1 for the reaction of O2 with NMe2LCuI. Also, the overall redox process, i.e. O2-binding, results in a higher enthalpy of activation of ΔH‡ = 32.1 kJ mol-1. The more favorable activation enthalpy for O2 binding to ImLCuI (ΔH‡ = 23.4 kJ mol-1) and less favorable ΔS‡ (35.1 kJ mol-1) in comparison to NMe2LCuI is consistent with a smaller geometry change with copper redox state due to O2-binding.
Solvent dependence (EtCN vs THF) of 1:1 Copper-Dioxygen Adducts
The dynamics of the O2 binding reaction of PyLCuI are largely influenced by solvent medium based on the analogous O2 binding data collected through the “flash-and-trap” method in THF15 and stopped-flow UV-visible spectroscopy in EtCN,8 see Table 3. The entropy of activation (ΔS‡) for formation of PyLCuII-O2− in EtCN (ΔS‡ = 10 J mol-1 K-1) versus THF (ΔS‡ = −45.1 J mol-1 K-1) indicates that O2 binding to PyLCuI follows a dissociative versus associative pathway, respectively, depending on the solvent. To observe such a solvent effect is not an unreasonable expectation since nitrile solvents are strong Lewis bases and soft donors, therefore strong ligands for copper(I) ions. In some cases, nitrile solvents severely inhibit binding of dioxygen such as in the reaction of O2 with BzLCuI, where EtCN solvent prevents formation of the commonly observed O2 adduct, characterized as a dicopper(III) bis-μ-oxo species in less coordinating solvents (THF, acetone, toluene).60
Previously, Schindler and coworkers attempted to time-resolve the formation of the cupric superoxo species NMe2LCuII-O2−.62 However, the formation was too fast to be measured by stopped-flow UV-visible spectroscopy, even in EtCN (coordinating solvent) at 183 K. Similarly, we have unsuccessfully attempted to measure the formation of ImLCuII-O2− by stopped-flow methods in EtCN.65 Conversely, the kO2 value for O2 binding to PyLCuI in EtCN was easily measured (Table 3). These comparisons suggest that like the kO2 measurements in THF, a much higher kO2 value in EtCN is expected for formation of NMe2LCuII-O2− and ImLCuII-O2− compared to PyLCuII-O2−. Such measurements for formation of NMe2LCuII-O2− and ImLCuII-O2− in EtCN were not carried out, but the values of kO2 reported here in THF through the present “flash-and-trap” experiment, indeed are very large (> 6.9 × 107 M−1 s−1 at 193 K; see Table 3), above the limit of stopped-flow UV-visible spectroscopic methods (ms timescale).
THF is usually considered a weakly coordinating solvent for ligand-copper-species and has even been described as a non-coordinating solvent. However, THF contains an electron pair centered on the oxygen-atom potentially capable of binding to copper(I) or any Lewis acid. A series of ligand-copper(II)-peroxo complexes utilizing the tridentate ligand R-MePY2 {bis[2-(2-pyridyl)ethyl]methylamine; R = Cl-, H-, MeO-, NMe2-} with 4-pyridyl substituents were previously reported to efficiently oxidize THF to 2-hydroxy tetrahydrofuran (THF-OH).85 Substrate (THF) oxidation was proposed as an “inner-sphere oxidation” because of a likely pre-equilibrium step involving THF-binding to the copper-dioxygen adduct.89 Also, in a detailed study conducted by Tolman and coworkers on the mechanism of formation for the β-diketiminate CuIII-η2-O22− adduct, an associative oxygenation pathway (Scheme 7) involving THF coordination (pathway B) was proposed based on comparisons to the same reaction in THF/MeCN mixtures (pathway A).13 Complementary theoretical calculations led to a proposed transition state that involved dioxygen and solvent bound to copper simultaneously, either THF or MeCN accordingly.
Scheme 7.

Proposed Mechanisms for O2-Binding to the DLCuI Complexes (D = NMe2, Im, Py)
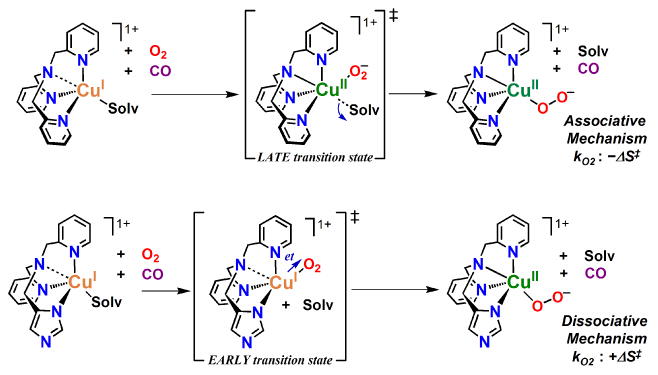
In the present work and as described above, positive ΔS‡ values were measured for O2 binding to ImLCuI (ΔS‡ = 35.1 J mol-1 K-1) and NMe2LCuI (ΔS‡ = 80.1 J mol-1 K-1) in THF solvent, to the first approximation similar to that found for PyLCuI in EtCN, but very different to that for PyLCuI in THF (and [CuI(Me6tren)]+ in EtCN), see Table 3. Such a drastic difference in the O2-binding mechanism depending on the variable N-donor moiety of DL is surprising. We suggest changes in the electron distribution and/or coordination variations within the species upon O2 ligation to CuI are responsible and it is not simply related to the order in which O2 or solvent coordinate. More specifically, the transition state differences may be largely based on whether or not CuI → O2 electron transfer (et) occurs before or after solvent dissociation with the coordination of O2 always being the initial step.
As shown in Scheme 8, the negative kO2 affiliated ΔS‡ value (associative mechanism) for PyLCuII-O2− in THF suggests a late transition state in which et occurs before or concomitant with solvent release. Conversely, the positive kO2 affiliated ΔS‡ value (dissociative mechanism) for O2 binding to ImLCuI in THF, NMe2LCuI in THF, and PyLCuI in EtCN, suggests an early transition state in which et can occur only after solvent is released, see Scheme 8. Note that a computational analysis of the proposed mechanism has not been conducted and therefore the proposed internal electron density distributions or solvent positioning (ligated or not) cannot be conclusively supported. However, the similar dissociative processes for formation of the square pyramidal NMe2LCuII-O2− species in THF and the trigonal bipyramidal ImLCuII-O2− and PyLCuII-O2− species in THF and EtCN (respectively) again points out that the reaction mechanism is not solely dependent on geometry or solvent medium, but rather the degree of solvent and/or O2-fragment interaction, i.e. electronic distribution within a given species.
Scheme 8.
Smirnov and Roth have proposed a two-step, inner-sphere electron-transfer mechanism for the oxidation of O2•− to O2 by the copper(II)-complexes of PyL and TEPA (tris(2-pyridylethyl)amine) in DMF/THF mixtures (DMF = dimethylformamide).90 Based on low-temperature stopped-flow kinetic experiments and competitive 18O2 isotope effects, a pre-equilibrium step with formation of a Cu-O2 species was suggested to form prior to O2 release, see Scheme 9. Formal oxidation states were not emphasized (CuII-O2− versus CuI-O2) for the intermediate species; therefore an electrostatic versus covalent Cu–O2 interaction was not designated. However, the release of O2 was designated as the rate determining step which would complete the overall et process, i.e. reduction of the copper(II) ion. Formation of a pre-equilibrium complex is analogous to our proposal of a pre-electron transfer intermediate step upon formation of DLCuII-O2− (Scheme 9).
Scheme 9.
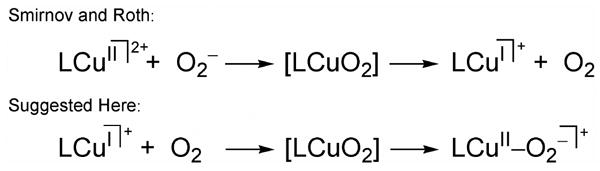
Temperature dependence (193 K vs. 298 K) of equilibrium O2 binding constants (KO2; ΔH°, ΔS°) and dissociation rates (k−O2; ΔH‡, ΔS‡)
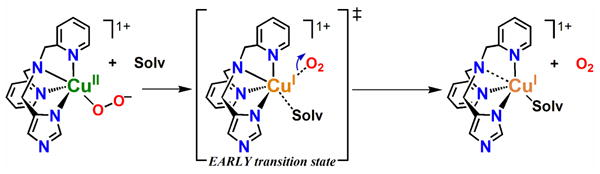
Dissociation of O2 from DLCuII-O2− has an early transition state (+ΔS‡) for all species studied that involves the transfer of an electron (et) from O2•− to copper(II) followed by coordination of solvent (THF) and concomitant release of O2. This Cu-O2 bond breakage process is depicted for ImLCuII-O2− in Scheme 10. The k−O2 activation parameters corresponding to ImLCuII-O2− are almost the same as those for NMe2LCuII-O2− suggesting the same O2 release mechanism. However, for O2 dissociation from PyLCuII-O2−, the corresponding activation parameters (Table 3) are of slightly higher values than the aforementioned species suggesting a larger geometric change upon dioxygen loss.
Scheme 10.
The O2 binding constants (KO2) for all DLCuII-O2− complexes are of approximately the same magnitude (105 M-1) at 193 K, overall favoring formation of the cupric-superoxo species. As expected, the equilibrium shifts more towards formation of the solvento species at 298 K. The KO2 values clearly show the solvent effect where as compared to THF, EtCN significantly inhibits O2 binding to PyLCuI. However, Me6TREN is a more Lewis basic ligand than is PyL such that O2 binding to [CuI(Me6tren)]+ seems unaffected.
Summary of 1:1 Cu/CO and Cu/O2 binding to DLCuI and Related Cu/O2 = 2:1 Complexes
In the present study, CO, as a redox-inactive O2-surrogate, was examined in its interactions with ligand-copper(I)-complexes (DLCuI). Data obtained concerning the CO binding kinetics and thermodynamics in THF were compared to those for 1:1 O2 binding to PyLCuI, ImLCuI, and NMe2LCuI, where end-on (η1) coordination occurs. The formation of 1:1 Cu/O2 adducts were not observed for QLCuI, TBPLCuI, and BzLCuI under the same solution reaction conditions by laser flash photolysis of the CO adducts.91 Since these cuprous complexes in fact do react with dioxygen to form 2:1 Cu/O2 species under steady state conditions,60,63,64 it is likely that primary 1:1 Cu/O2 binding does not compete kinetically (e.g., Δabsorbance is too small) under the conditions employed, i.e., those used for all of the complexes examined in these studies.
In our previous reports,15,59 the CO and O2 binding kinetics of PyLCuI in THF solvent were detailed, with one finding being that kCO ≈ kO2 (Table 2). A look at the highlighted portion of Table 4, with data added in from the present study on ImLCuI and NMe2LCuI (i.e., with one altered N-donor arm), gives the impression that these latter complexes have a very similar behavior. However, a significant finding in this report (Tables 3 and 4) is that there is a major change in the mechanism of O2-binding to ImLCuI and NMe2LCuI in THF; the ligand binding occurs via dissociative rather than associative processes. Recall that the dissociative mechanism does occur for both CO and O2 binding to PyLCuI in the strong competing solvent EtCN. Electron-transfer (et) from copper(I) to O2 seems the only factor possible to explain a change in mechanism. The CO binding behavior to the reduced copper(I) species for the DL series is essentially the same (Tables 1, 2). The (electrochemical study) derived E1/2 values for ImLCuI and NMe2LCuI are the same, but more negative than that for PyLCuI, however the coordination geometries of the copper(II) complexes of ImL (∼ PyL) and NMe2L are different (vide supra). Thus, we conclude that the et properties/kinetics of the O2-interaction somehow leads to the change in mechanism observed. Solvent release is initiated by O2-interaction with copper(I) (i.e., we suggest a CuI-O2 transient forms) prior to et (Scheme 8) giving the observed DLCuII-O2− product. To summarize in other words, the reaction mechanism is governed by the order that et from copper(I) to O2 occurs; for PyL, et occurs before solvent is released, perhaps outer-sphere, but not so for ImL or NMe2L. For ImL and NMe2L, a dissociative interchange mechanism (vide supra) may be an appropriate description in which bond breaking (CuI-solv → CuI + solv) dominates over bond formation and et (CuI + O2 → CuII-O2−).
Table 4.
Comparison of CO and O2 Bimolecular Rates and Binding Constants at 298 K for Select Ligand-Copper(I)-Complexes, Hemocyanins (Hc), and Selected Hemes.
| Compound or Protein | KCO (M-1) | KO2 (M-1) | kCO (M-1 s-1) | kO2 (M-1 s-1) | k-CO (s-1) | k-O2 (s-1) |
|---|---|---|---|---|---|---|
| NMe2LCuI (THF) | 4.9 × 103 | 100 | 2.5 × 109 | 2.3 × 1011 | 5.0 × 105 | 2.3 × 109 |
| ImLCuI (THF) | 2.4 × 103 | 17.4 | 2.8 × 109 | 3.4 × 1010 | 1.1 × 106 | 1.9 × 109 |
| PyLCuI (THF) | 1.2 × 104 | 15.4 | 1.9 × 109 | 1.3 × 109 | 1.6 × 105 | 1.3 × 108 |
| PyLCuI (CH3CN) | 220 | 0.38 | 5.9 × 107 | 5.8 × 107 | 2.7 × 105 | 1.5 × 108 |
| Myoglobin (human) | 2.6 × 107 | (0.74–117) × 104 | 7.6 × 105 | (1.4–25) × 107 | 0.022 | 22 |
| Hemoglobin (human) | 4.6 × 108 | (2.9–48) × 105 | 4.6 × 106 | (2.9 –22) × 107 | 0.009 | 13.1 |
| Limulus Hc (arthropod) | (2.7 – 11.2) × 103 | (2.6 – 5.4) × 105 | (2 – 4.3) × 105 | (1.3 – 1.9) × 106 | 38 – 75 | 2.4 – 7.5 |
| Busycon Hc (mollusk) | 220 × 103 | 1.8 × 105 | 7.7 × 105 | (1.1 – 2.2) × 106 | 3 – 4 | 6.5 – 11.5 |
For all ligand-copper-complexes studied, KCO is much larger than KO2 primarily as a result of the lower CO dissociation rate constant (k−CO < k−O2). Breakage of the Cu-O2 bond is more favorable than Cu-CO bond breakage even though the former involves an et step. The high k−O2 values are a result of the highly favorable ΔS‡ value attributed to a late transition state (which leads to O2-release) and the structural rearrangement that occurs upon redox changes.
As emphasized, the copper(II)-η1-superoxo species generated from PyLCuI, ImLCuI, and NMe2LCuI, are spectroscopically very similar. In fact, spectroscopically (and therefore structurally) similar Cu/O2 = 2:1 adducts also form, dicopper(II)-μ-1,2-(end-on)-peroxo species [{(DL)CuII}2(O22−)]2+ analogous to the well characterized case for PyL (Scheme 11).8,61,62,65 However, the conditions (i.e. temperature) under which the [{(DL)CuII}2(O22−)]2+ complexes are observed, and/or their relative stability, varies significantly. Thus, reaction dynamics, i.e., kinetics and thermodynamics, governing each reaction step likely vary significantly, as observed in the present study on the Cu/O2 = 1:1 adducts. For example, the peroxo species of ImL (Figure 7A) can only be observed at −128 °C in 2-methyl tetrahydrofuran (MeTHF).65 In THF at −80 °C, a Cu-O2 intermediate is not observed.65 Quite differently, formation of PyLCuII-O2− can be detected on the bench-top in MeTHF at −128 °C before complete conversion to the peroxo species (Figure 7B), which is stable for weeks at −80°C in THF.61 Interestingly, for NMe2LCuI + O2, a new as yet uncharacterized species is detected in MeTHF at −128 °C in addition to the peroxo adduct (Figure 7C).76 The new UV-vis feature(s) may suggest a different type or additional intermediate forms on the pathway to produce [{(NMe2L)CuII}2(O22−)]2+; further studies are required.
Scheme 11.
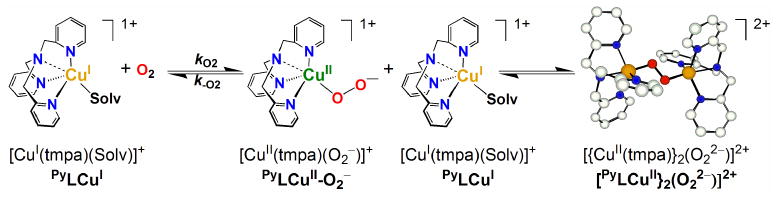
Figure 7.

UV-visible spectra in MeTHF at −128 °C representing O2 + DLCuI (dark blue) to yield: A, [{(ImL)CuII}2(O22−)]2+ (purple); B, PyLCuII-O2− (green) and [{(PyL)CuII}2(O22−)]2+ (purple); C, [{(NMe2L)CuII}2(O22−)]2+ (purple) and a new copper-dioxygen adduct of NMe2L (brown).
Comparison to natural copper and heme systems
The kinetics and thermodynamics of CO and O2 binding to the ligand-copper(I)-complexes (DLCuI) reported here are in line with studies carried out on natural and synthetic heme systems (Table 4). As said, carbon monoxide coordinates to the cuprous center (DLCuI) with a higher affinity than that of O2 (KCO > KO2), allowing for extension of the “flash-and-trap” experiment as inspired by extensive studies on a variety of heme systems. Furthermore, like our synthetic species, KCO is much higher than KO2 for the porphyrinate systems as a result of the lower CO dissociation rate (k−CO < k−O2); Cu-O2 bond breakage is more favorable than breakage of the Cu-CO bond. The bimolecular rate constants for the copper complexes (kCO and kO2) are much higher than typically exhibited by heme systems and the equilibrium binding constants are much lower. Therefore, the trends observed here are in line with conclusions drawn concerning gaseous small molecule binding at the active site of the heme-copper heterobimetallic enzyme cytochrome c oxidase (CcO) (vide supra; see Introduction). In CcO, where one can imagine some competition for gases by both the heme and copper, CO coordinates to the CuIB site before thermally transferring to the FeII-hemea3 site (Figure 1). We have previously constructed a model binucleating iron and copper containing ligand system as well as 1:1 heme/copper component systems that demonstrate this CO migration phenomenon, and most recently have additionally examined NO transfer from iron-to-copper in the latter 1:1 component system.22,25
The interaction of CO and O2 with hemocyanins (Hc) has also been extensively studied and the characteristics are unlike hemes. For Hc, lower equilibrium CO binding constants in comparison to O2 (KO2 > KCO) has been attributed to the higher rate of CO dissociation in comparison to that for O2 (k−CO > k−O2) see Table 4. This behavior most likely occurs because both copper(II) centers are involved in O2-binding and release, whereas only one copper center binds CO; more structural rearrangements must occur in the release of O2. Conversely, the high O2 association rates (kO2) are a result of the 2:1 Cu/O2 binding nature of the binuclear copper protein, a characteristic that is not exhibited upon CO binding (1:1 Cu/CO binding), vide supra.
As stated in the Introduction, an experiment such as the flash-and-trap experiment cannot be utilized for the binuclear copper proteins because KO2 > KCO. Laser flash photolysis has been used by Hirota et al. to examine O2 binding by tyrosinase (Tyr) from Streptomyces antibioticus.44 Following O2-photoejection from oxy-Tyr to form deoxy-Tyr, reformation of the starting dicopper(II)-μ-η2:η2-peroxo species was observed while an initial 1:1 Cu/O2 adduct was not detected. In a more recent study, Hirota, Bubacco, and coworkers reported thermodynamic data for oxygenation of Hc to form the dicopper(II)-(side-on)-peroxo species.45 Interestingly, the values were comparable to the superoxo model compounds measured here, suggesting that formation of the 1:1 adduct is the rate-determining step and that oxyHc is easily produced through a nearly simultaneous two et step. This experimental finding fits with Solomon and coworkers' prior computational studies on the O2-binding process that occurs in Hc.92
Summary and Conclusions
In the present report, we have detailed the kinetics, thermodynamics, and coordination dynamics of 1:1 CuI/CO and CuI/O2 binding within tetradentate ligand-copper(I)-complexes with analogous coordination frameworks (Chart 2). The major findings are:
In the ligand-copper(I) carbonyl complexes (DLCuI), the variable N-donor arm of the synthetic ligand is dangling; all species possess the same four-coordinate core structure in solution where the cuprous ion binds to the bispicolylamine (PY1) tridentate chelate along with the CO molecule.
The fast kinetics of CO binding are measurably influenced by the exact nature of that uncoordinated donor arm. With change in ligand where KCO increases, kCO and k−CO decrease; k−CO decreases more substantially because the dangling donor ligand partially interacts (but to different extents) before it fully coordinates with the Cu(I) ion as CO dissociates.
In previous studies where the exact same PY1 ligand framework was present but with more electron releasing 4-pyridyl substituents, KCO increased, as well as kCO, very different from what was found here. In other words, ligand electronic variations previously led to the standard expected results, but here, (small) changes in the identity of D (the fourth donor, 3rd ligand arm, Chart 1) led to a change in the relationship of KCO to kCO.
Using nanosecond laser flash photolysis to initiate gas (CO, O2) reactions with copper(I) complexes, the unique finding of an extremely large rate constant for O2 binding, as previously described for PyLCuI, is shown to be a general phenomenon amongst D-PY1 ligand copper(I) complexes. Further, as seen before, kO2 and kCO values are quite similar to each other.
Changing one donor arm N-ligand from pyridyl to imidazolyl or to dimethylamino results in a change in the mechanism of O2-binding to the respective copper(I) complexes, associative for PyLCuI but dissociative for ImLCuI and NMe2LCuI. We attribute this to aspects of the timing of et from copper(I) to O2 as this molecule coordinates, in relation to the dissociation of the bound solvent molecule THF. Based on our other experiments, the change in mechanism is not simply related to a change in the DLCuII/I structures or the order that O2/THF coordinate.
The work discussed here establishes transient absorbance laser flash photolysis as an invaluable tool for determining fast small molecule (CO and O2) binding kinetics and thermodynamics to synthetic copper complexes such as DLCuI. Further, the ability to combine this utility with variable low-temperature control has enabled us to fully characterize 1:1 binding of O2 to ImLCuI and NMe2LCuI, which was previously unobtainable with other relatively fast physical methods such as low-temperature stopped flow kinetics/spectroscopy.
Overall the results further support the general finding that subtle changes in coordination environment, as occurs over time through evolution in nature or through controlled ligand design in synthetic systems, can dictate the observed chemistry in terms of reaction kinetics, structure and reactivity, and thus function. Many mononuclear and binuclear copper proteins exist that have an active site ligation of three imidazolyl donors per copper center, yet have very different functions, such as dioxygen transport in hemocyanin (Hc), dioxygen activation and phenol o-hydroxylation in tyrosinase (Tyr), electron-transfer (et) at the CuH site in peptidylglycine α-hydroxylating monooxygenase (PHM) and/or dopamine β-monooxygenase, or O2-binding (and/or et) at CuB in cytochrome c oxidases (CcO), see Introduction. Therefore, by gaining a deeper understanding of how the coordination environment and/or subtle changes in the surroundings influence the reactivity and/or binding properties of the copper center may help in understanding protein active site ligand dynamics. Such information may also provide insight into secondary coordination sphere contributions that account for N3CuI active site functional differences.
Supplementary Material
Acknowledgments
We are grateful to the National Institutes of Health (K.D.K., NIH GM28962; G.J.M., NSF CHE0911558) for support of this research. We also acknowledge Drs. Amy A. Narducci Sarjeant and Maxime A. Siegler for relevant X-ray structural determination.
Footnotes
Supporting Information Available. X-ray data files (CIF), UV-visible data including spectrophotometric CO titrations, Van't Hoff plots, ΔA spectra (O2 and CO binding), linear [CO] and [O2] dependence, and Eyring plots are all available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kim E, Chufan EE, Kamaraj K, Karlin KD. chem Rev. 2004;104:1077–1133. doi: 10.1021/cr0206162. [DOI] [PubMed] [Google Scholar]
- 2.Wasser IM, de Vries S, Moenne-Loccoz P, Schroder I, Karlin KD. Chem Rev. 2002;102:1201–1234. doi: 10.1021/cr0006627. [DOI] [PubMed] [Google Scholar]
- 3.Lucas HR, Karlin KD. Met Ions Life Sci. 2009;6:295–362. doi: 10.1039/BK9781847559159-00295. [DOI] [PubMed] [Google Scholar]
- 4.Lewis EA, Tolman WB. Chem Rev. 2004;104:1047–1076. doi: 10.1021/cr020633r. [DOI] [PubMed] [Google Scholar]
- 5.Mirica LM, Ottenwaelder X, Stack TDP. Chem Rev. 2004;104:1013–1045. doi: 10.1021/cr020632z. [DOI] [PubMed] [Google Scholar]
- 6.Solomon EI, Szilagyi RK, George SD, Basumallick L. Chem Rev. 2004;104:419–458. doi: 10.1021/cr0206317. [DOI] [PubMed] [Google Scholar]
- 7.Woertink JS, Smeets PJ, Groothaert MH, Vance MA, Sels BF, Schoonheydt RA, Solomon EI. Proc Nat Acad Sci. 2009;106:18908–18913. doi: 10.1073/pnas.0910461106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang CX, Kaderli S, Costas M, Kim E, Neuhold YM, Karlin KD, Zuberbuhler AD. Inorg Chem. 2003;42:1807–1824. doi: 10.1021/ic0205684. [DOI] [PubMed] [Google Scholar]
- 9.Himes RA, Karlin KD. Curr Opin Chem Biol. 2009;13:119–131. doi: 10.1016/j.cbpa.2009.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maiti D, Lee DH, Gaoutchenova K, Wurtele C, Holthausen MC, Sarjeant AAN, Sundermeyer J, Schindler S, Karlin KD. Angew Chem Int Ed. 2008;47:82–85. doi: 10.1002/anie.200704389. [DOI] [PubMed] [Google Scholar]
- 11.Würtele C, Gaoutchenova E, Harms K, Holthausen MC, Sundermeyer J, Schindler S. Angew Chem Int Ed. 2006;45:3867–3869. doi: 10.1002/anie.200600351. [DOI] [PubMed] [Google Scholar]
- 12.Fujisawa K, Tanaka M, Morooka Y, Kitajima N. J Am Chem Soc. 1994;116:12079–12080. [Google Scholar]
- 13.Aboelella NW, Kryatov SV, Gherman BF, Brennessel WW, Young VG, Sarangi R, Rybak-Akimova EV, Hodgson KO, Hedman B, Solomon EI, Cramer CJ, Tolman WB. J Am Chem Soc. 2004;126:16896–16911. doi: 10.1021/ja045678j. For comparison, kO2 ∼ 104 M-1 s-1 for βDkCuI. [DOI] [PubMed] [Google Scholar]
- 14.Weitzer M, Schindler S, Brehm G, Schneider S, Hormann E, Jung B, Kaderli S, Zuberbuhler AD. Inorg Chem. 2003;42:1800–1806. doi: 10.1021/ic025941m. [DOI] [PubMed] [Google Scholar]
- 15.Fry HC, Scaltrito DV, Karlin KD, Meyer GJ. J Am Chem Soc. 2003;125:11866–11871. doi: 10.1021/ja034911v. [DOI] [PubMed] [Google Scholar]
- 16.Itoh S, Fukuzumi S. Acc Chem Res. 2007;40:592–600. doi: 10.1021/ar6000395. [DOI] [PubMed] [Google Scholar]
- 17.Itoh S. Curr Opin Chem Biol. 2006;10:115–122. doi: 10.1016/j.cbpa.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 18.Prigge ST, Eipper BA, Mains RE, Amzel LM. Science. 2004;304:864–867. doi: 10.1126/science.1094583. [DOI] [PubMed] [Google Scholar]
- 19.Humphreys KJ, Mirica LM, Wang Y, Klinman JP. J Am Chem Soc. 2009;131:4657–4663. doi: 10.1021/ja807963e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kitagawa T, Ogura T. Prog Inorg Chem. 1997;45:431–479. [Google Scholar]
- 21.Larsen RW, Mikšovská J. Coord Chem Rev. 2007;251:1101–1127. [Google Scholar]
- 22.Fry HC, Cohen AD, Toscano JP, Meyer GJ, Karlin KD. J Am Chem Soc. 2005;127:6225–6230. doi: 10.1021/ja043199e. [DOI] [PubMed] [Google Scholar]
- 23.Fry HC, Hoertz PG, Wasser IM, Karlin KD, Meyer GJ. J Am Chem Soc. 2004;126:16712–16713. doi: 10.1021/ja045195f. [DOI] [PubMed] [Google Scholar]
- 24.Fry HC, Lucas HR, Zakharov LN, Rheingold AL, Meyer GJ, Karlin KD. Inorg Chim Acta. 2008;361:1100–1115. [Google Scholar]
- 25.Lucas HR, Meyer GJ, Karlin KD. J Am Chem Soc. 2009;131:13924–5. doi: 10.1021/ja906172c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vos MH. Biochim Biophys Acta. 2008;1777:15–31. doi: 10.1016/j.bbabio.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 27.Dyer RB, Peterson KA, Stoutland PO, Woodruff WH. J Am Chem Soc. 1991;113:6276–6277. [Google Scholar]
- 28.Spiro TG, Wasbotten IH. J Inorg Biochem. 2005;99:34–44. doi: 10.1016/j.jinorgbio.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 29.Rousseau DL, Han S. Method Enzymol. 2002;354:351–368. doi: 10.1016/s0076-6879(02)54028-3. [DOI] [PubMed] [Google Scholar]
- 30.Ionascu D, Gruia F, Ye X, Yu AC, Rosca F, Beck C, Demidov A, Olson JS, Champion PM. J Am Chem Soc. 2005;127:16921–16934. doi: 10.1021/ja054249y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kapetanaki SM, Field SJ, Hughes RJL, Watmough NJ, Liebl U, Vos MH. Biochim Biophys Acta. 2008;1777:919–924. doi: 10.1016/j.bbabio.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 32.Gibson QH, Greenwood C. Biochem J. 1963;86:541–&. doi: 10.1042/bj0860541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greenwood C, Gibson QH. J Biol Chem. 1967;242:1782–&. [PubMed] [Google Scholar]
- 34.Einarsdottir O, Szundi I. Biochim Biophys Acta. 2004;1655:263–73. doi: 10.1016/j.bbabio.2003.07.011. [DOI] [PubMed] [Google Scholar]
- 35.Szundi I, Rose MJ, Sen I, Eroy-Reveles AA, Mascharak PK, Einarsdottir O. Photochem Photobiol. 2006;82:1377–84. doi: 10.1562/2006-07-25-rc-984. [DOI] [PubMed] [Google Scholar]
- 36.Soldatova AV, Ibrahim M, Olson JS, Czernuszewicz RS, Spiro TG. J Am Chem Soc. 132:4614–25. doi: 10.1021/ja906233m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Birukou I, Schweers RL, Olson JS. J Biol Chem. 2010;285:8840–8854. doi: 10.1074/jbc.M109.053934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohta K, Muramoto K, Shinzawa-Itoh K, Yamashita E, Yoshikawa S, Tsukihara T. Acta Crystallogr F. 2010;66:251–253. doi: 10.1107/S1744309109055109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Babcock GT, Floris R, Nilsson T, Pressler M, Varotsis C, Vollenbroek E. Inorg Chim Acta. 1996;243:345–353. [Google Scholar]
- 40.Oliveberg M, Malmstrom BG. Biochemistry. 1992;31:3560–3563. doi: 10.1021/bi00129a002. [DOI] [PubMed] [Google Scholar]
- 41.Muramoto K, Ohta K, Shinzawa-Itoh K, Kanda K, Taniguchi M, Nabekura H, Yamashita E, Tsukihara T, Yoshikawa S. Proc Nat Acad Sci. 2010;107:7740–5. doi: 10.1073/pnas.0910410107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Magnus KA, Hazes B, Tonthat H, Bonaventura C, Bonaventura J, Hol WGJ. Proteins. 1994;19:302–309. doi: 10.1002/prot.340190405. [DOI] [PubMed] [Google Scholar]
- 43.Solomon EI, Tuczek F, Root DE, Brown CA. Chem Rev. 1994;94:827–856. [Google Scholar]
- 44.Hirota S, Kawahara T, Lonardi E, de Waal E, Funasaki N, Canters GW. J Am Chem Soc. 2005;127:17966–17967. doi: 10.1021/ja0541128. [DOI] [PubMed] [Google Scholar]
- 45.Hirota S, Kawahara T, Beltramini M, Di Muro P, Magliozzo RS, Peisach J, Powers LS, Tanaka N, Nagao S, Bubacco L. J Biol Chem. 2008;283:31941–31948. doi: 10.1074/jbc.M803433200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Floyd JS, Haralampus-Grynaviski N, Ye T, Zheng B, Simon JD, Edington MD. J Phys Chem B. 2001;105:1478–1483. [Google Scholar]
- 47.Guëll M, Siegbahn PEM. J Biol Inorg Chem. 2007;12:1251–1264. doi: 10.1007/s00775-007-0293-z. [DOI] [PubMed] [Google Scholar]
- 48.Jaron S, Blackburn NJ. Biochemistry. 1999;38:15086–15096. doi: 10.1021/bi991341w. [DOI] [PubMed] [Google Scholar]
- 49.Jaron S, Mains RE, Eipper BA, Blackburn NJ. Biochemistry. 2002;41:13274–13282. doi: 10.1021/bi020404z. [DOI] [PubMed] [Google Scholar]
- 50.Wilmot CM, Hajdu J, McPherson MJ, Knowles PF, Phillips SEV. Science. 1999;286:1724–1728. doi: 10.1126/science.286.5445.1724. [DOI] [PubMed] [Google Scholar]
- 51.Mukherjee A, Smirnov VV, Lanci MP, Brown DE, Shepard EM, Dooley DM, Roth JP. J Am Chem Soc. 2008;130:9459–9473. doi: 10.1021/ja801378f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shepard EM, Okonski KM, Dooley DM. Biochemistry. 2008;47:13907–13920. doi: 10.1021/bi8011516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hirota S, Iwamoto T, Tanizawa K, Adachi O, Yamauchi O. Biochemistry. 1999;38:14256–14263. doi: 10.1021/bi991129s. [DOI] [PubMed] [Google Scholar]
- 54.Alben JO, Moh PP, Fiamingo FG, Altschuld RA. Proc Nat Acad Sci. 1981;78:234–237. doi: 10.1073/pnas.78.1.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hill J, Goswitz VC, Calhoun M, Garciahorsman JA, Lemieux L, Alben JO, Gennis RB. Biochemistry. 1992;31:11435–11440. doi: 10.1021/bi00161a023. [DOI] [PubMed] [Google Scholar]
- 56.Einarsdottir O, Killough PM, Fee JA, Woodruff WH. J Biol Chem. 1989;264:2405–2408. [PubMed] [Google Scholar]
- 57.Zhang HM, Boulanger MJ, Mauk AG, Murphy MEP. J Phys Chem B. 2000;104:10738–10742. [Google Scholar]
- 58.Fager LY, Alben JO. Biochemistry. 1972;11:4786–4792. doi: 10.1021/bi00775a023. [DOI] [PubMed] [Google Scholar]
- 59.Fry HC, Lucas HR, Sarjeant AAN, Karlin KD, Meyer GJ. Inorg Chem. 2008;47:241–256. doi: 10.1021/ic701903h. [DOI] [PubMed] [Google Scholar]
- 60.Lucas HR, Li L, Sarjeant AAN, Vance MA, Solomon EI, Karlin KD. J Am Chem Soc. 2009;131:3230–3245. doi: 10.1021/ja807081d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tyeklár Z, Jacobson RR, Wei N, Murthy NN, Zubieta J, Karlin KD. J Am Chem Soc. 1993;115:2677–2689. [Google Scholar]
- 62.Weitzer M, Schatz M, Hampel F, Heinemann FW, Schindler S. J Chem Soc, Dalton Trans. 2002:686–694. [Google Scholar]
- 63.Wei N, Murthy NN, Chen Q, Zubieta J, Karlin KD. Inorg Chem. 1994;33:1953–1965. [Google Scholar]
- 64.Maiti D, Lucas HR, Sarjeant AAN, Karlin KD. J Am Chem Soc. 2007;129:6998–6999. doi: 10.1021/ja071704c. [DOI] [PubMed] [Google Scholar]
- 65.Lee Y, Park GY, Lucas HR, Vajda PL, Kamaraj K, Vance MA, Milligan AE, Woertink JS, Siegler MA, Sarjeant AAN, Zakharov LN, Rheingold AL, Solomon EI, Karlin KD. Inorg Chem. 2009;48:11297–11309. doi: 10.1021/ic9017695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liang HC, Kim E, Incarvito CD, Rheingold AL, Karlin KD. Inorg Chem. 2002;41:2209–2212. doi: 10.1021/ic010816g. [DOI] [PubMed] [Google Scholar]
- 67.Lim BS, Holm RH. Inorg Chem. 1998;37:4898–4908. doi: 10.1021/ic9801793. [DOI] [PubMed] [Google Scholar]
- 68.See Supporting Information.
- 69.Pasquali M, Marini G, Floriani C, Gaetanimanfredotti A, Guastini C. Inorg Chem. 1980;19:2525–2531. [Google Scholar]
- 70.Kitajima N, Fujisawa K, Fujimoto C, Morooka Y, Hashimoto S, Kitagawa T, Toriumi K, Tatsumi K, Nakamura A. J Am Chem Soc. 1992;114:1277–1291. [Google Scholar]
- 71.Karlin KD, Tyeklar Z, Farooq A, Haka MS, Ghosh P, Cruse RW, Gultneh Y, Hayes JC, Toscano PJ, Zubieta J. Inorg Chem. 1992;31:1436–1451. [Google Scholar]
- 72.Ardizzoia GA, Beccalli EM, Lamonica G, Masciocchi N, Moret M. Inorg Chem. 1992;31:2706–2711. [Google Scholar]
- 73.Imai S, Fujisawa K, Kobayashi T, Shirasawa N, Fujii H, Yoshimura T, Kitajima N, Moro-oka Y. Inorg Chem. 1998;37:3066–3070. [Google Scholar]
- 74.Conry RR, Ji GZ, Tipton AA. Inorg Chem. 1999;38:906–913. doi: 10.1021/ic980851w. [DOI] [PubMed] [Google Scholar]
- 75.Reger DL, Collins JE. Organometallics. 1996;15:2029–2032. [Google Scholar]
- 76.Unpublished work.
- 77.Himes RA, Park GY, Barry AN, Blackburn NJ, Karlin KD. J Am Chem Soc. 2007;129:5352–5353. doi: 10.1021/ja0708013. [DOI] [PubMed] [Google Scholar]
- 78.Pasquali M, Floriani C. Copper(I)-Carbon Monoxide Chemistry: Recent Advances and Perspectives. In: Karlin KD, Zubieta J, editors. Copper Coordination Chemistry: Biochemical & Inorganic Perspectives. Adenine Press; New York: 1983. pp. 311–330. [Google Scholar]
- 79.Rondelez Y, Seneque O, Rager MN, Duprat AF, Reinaud O. Chem Eur J. 2000;6:4218–4226. doi: 10.1002/1521-3765(20001117)6:22<4218::aid-chem4218>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 80.Jonas RT, Stack TDP. Inorg Chem. 1998;37:6615–6629. doi: 10.1021/ic9713487. [DOI] [PubMed] [Google Scholar]
- 81.Fujisawa K, Ono T, Ishikawa Y, Amir N, Miyashita Y, Okamoto K, Lehnert N. Inorg Chem. 2006;45:1698–1713. doi: 10.1021/ic051290t. [DOI] [PubMed] [Google Scholar]
- 82.Dias HVR, Lu HL. Inorg Chem. 1995;34:5380–&. [Google Scholar]
- 83.Carrier SM, Ruggiero CE, Houser RP, Tolman WB. Inorg Chem. 1993;32:4889–4899. [Google Scholar]
- 84.Hu ZB, Williams RD, Tran D, Spiro TG, Gorun SM. J Am Chem Soc. 2000;122:3556–3557. [Google Scholar]
- 85.Zhang CX, Liang HC, Kim EI, Shearer J, Helton ME, Kim E, Kaderli S, Incarvito CD, Zuberbuhler AD, Rheingold AL, Karlin KD. J Am Chem Soc. 2003;125:634–635. doi: 10.1021/ja028779v. [DOI] [PubMed] [Google Scholar]
- 86.τ values (α-β/60°) calculated based on the X-ray crystallographic data reported in cited references.
- 87.Addison AW, Rao TN, Reedijk J, Vanrijn J, Verschoor GC. J Chem Soc, Dalton Trans. 1984:1349–1356. [Google Scholar]
- 88.Karlin KD, Hayes JC, Juen S, Hutchinson JP, Zubieta J. Inorg Chem. 1982;21:4106–4108. [Google Scholar]
- 89.Shearer J, Zhang CX, Zakharov LN, Rheingold AL, Karlin KD. J Am Chem Soc. 2005;127:5469–5483. doi: 10.1021/ja045191a. [DOI] [PubMed] [Google Scholar]
- 90.Smirnov VV, Roth JP. J Am Chem Soc. 2006;128:3683–3695. doi: 10.1021/ja056741n. [DOI] [PubMed] [Google Scholar]
- 91.At much higher O2 concentrations, the formation of a new Cu-O2 adduct was observed and will be discussed in a different publication.
- 92.Metz M, Solomon EI. J Am Chem Soc. 2001;123:4938–4950. doi: 10.1021/ja004166b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.