

Abstract
Caspase-6 (Casp6) is activated early in Alzheimer disease and involved in axonal degeneration, but the regulation of Casp6 activity has not been explored. Several alternatively spliced forms of caspases act as inhibitors of caspase activation. The CASP6 gene generates an alternatively spliced transcript known as CASP6β in addition to the CASP6α that encodes pro-Casp6a. Here, we show that the CASP6β transcript and the pro-Casp6b protein are present in many cell lines, in primary human neurons, and in human brains. Unlike most other alternatively spliced caspase transcripts, pro-Casp6b contains a catalytic site. However, purified pro-Casp6b did not have caspase activity, nor did it inhibit already activated Casp6a. Pro-Casp6b prevented the proteolytic activation of pro-Casp6a in vitro and in cells. Pro-Casp6b interacts directly with pro-Casp6a. This work shows that pro-Casp6b is an inhibitor of pro-Casp6a activation. These results imply that pro-Casp6b could negatively regulate pro-Casp6a activation in neurons and prevent Casp6a-mediated axonal degeneration.
Keywords: Alzheimer Disease, Caspase, Neuroblastoma, Neurodegeneration, Neuron, RNA Splicing
Introduction
Caspases are well known cysteinyl proteases that regulate tissue homeostasis and whose deregulation can often lead to disease (1). One mode of regulation of caspases is through alternatively spliced isoforms that act as dominant negative inhibitors of caspase activation. All caspases are constituted of a pro-domain, a large and a small subunit often separated by a linker domain. Caspases are activated through proteolytic processing and reassembly of the large and small subunits into a tetrameric active enzyme. The catalytic site is contained within the large subunit. Most of the alternatively spliced dominant negative forms have lost their catalytic sites and are proposed to associate with the subunits of the active form to inhibit activation of the caspase. The RCasp1ϵ lacking part of the pro-peptide and the p20 subunit interacts with the RCasp1a-p20 subunit and increases cellular survival (2). The Casp3s isoform, which lacks the catalytic site and p12 subunit of Casp3, prevents staurosporin-activated Casp3-dependent poly(ADP-ribose) polymerase cleavage and apoptosis (3). Casp8L lacks the catalytic site and part of the C terminus of Casp8 and interferes with the interaction of Casp8 with the Fas-associated death domain, which is necessary for Casp8-mediated cell death (4, 5). Casp9s lacks the large subunit of normal Casp9 and interacts with Apaf-1 to block recruitment and activation of Casp9 to the apoptosome (6). However, a few alternatively spliced forms retain the catalytic site and can prevent activation of caspases by interfering with regulatory proteins. For example, the Casp2s isoform prevents activation of Casp2 through interaction with the Ich-1S (caspase-2S)-binding protein (7, 8).
The CASP6β transcript is an alternatively spliced form of the CASP6 nascent transcript. The CASP6β mRNA lacks amino acids 15–103, encoding a portion of the pro-domain and the Casp6 p20 subunit but retains the catalytic site (QACRG) and the p10 subunit (9). It is not known if the alternatively spliced CASP6β product, pro-Casp6b, has any catalytic activity or acts as a dominant negative inhibitor of Casp6a. A dominant negative alternative form of Casp6a would be important because increasing evidence suggests that Casp6a has essential physiological functions and can be activated in pathological situations. Casp6a activity precedes Casp3 and Casp7 activity in isolated intestinal epithelial cells submitted to detachment (anoikis) (10). Casp6a is involved in lymphocyte differentiation and proliferation (11, 12). Although debated, Casp6a is thought to be activated during the dissolution of organelles in the lens of the eye (13–16). Casp6a is activated and induces a protracted type of cell death in primary cultures of human neurons in the absence of Casp3 and Casp7 activity (17, 18). Active caspase-1 (RCasp1) activates Casp6a in vitro and in human neurons in culture (19). Casp6a activity is abundant in the neuropil threads, neuritic plaques, and neurofibrillary tangles of sporadic and familial forms of Alzheimer disease brains and in the neurites and nuclei of apoptotic neurons in human fetal and adult ischemic brains (20, 21). Furthermore, active Casp6a is present in some aging brains and correlates negatively with the global cognitive score of aged individuals (22). Casp6a cleaves several proteins of the cytoskeleton and synapses in human neurons and in Alzheimer disease (20, 22, 23). However, in contrast to Casp3 and Casp7, activation of Casp6a in mammalian cells does not induce cell death (24). More recently, Casp6a activity has been shown to be responsible for axonal pruning and degeneration in mouse neurons (25). Abnormal Casp6a cleavage of the DJ-1 and Huntingtin proteins may also be implicated in Parkinson and Huntington diseases, respectively (26, 27). Together, these results indicate that active Casp6a may be predominantly responsible for neurodegeneration rather than cell death.
The regulation of Casp6 activity is not well known. Casp6a is self-activated in vitro and in cells, and this is regulated by the pro-domain (24). Unlike the other two effector caspases, Casp3 and Casp7, Casp6 is not inhibited by inhibitor of apoptosis proteins (IAPs)2 (28). Estrogen induces an inhibitor of the active form of Casp6a in human primary neurons (29). Here, we investigate if the protein product of CASP6β, pro-Casp6b, generates Casp6 activity or acts as an inhibitor toward pro-Casp6a.
EXPERIMENTAL PROCEDURES
DNA Constructs and siRNA
Control siRNA (sc-3007) was obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA), and ON-TARGETplus SMARTpool human CASP6 siRNA (L-004406-00) was purchased from Dharmacon (Lafayette, CO). Custom designed ON-TARGETplus siRNA sequence to the exon 1/exon 5 junction of CASP6β was synthesized by Dharmacon to recognize the target sequence 5′-CACCCGGCAGUGUCAACUGUUUU-3′.
Reagents and Antibodies
Purified recombinant active Casp1 (RCasp1), Ac-VEID-AFC, and Ac-YVAD-AFC were purchased from BioMol (Philadelphia, PA). Anti-Casp6a polyclonal antiserum from Upstate (anti-Casp6a(16–32)) and NeoMarkers Ab-4 (Fremont, CA) recognize the N-terminal region of human Casp6, a region that is lacking in pro-Casp6b. The anti-p10Casp6 (Pharmingen) monoclonal antibody (anti-Casp6(271–285)) recognizes the p10 subunit and full length of pro-Casp6a or pro-Casp6b. The anti-p20Casp6 10630 neoepitope antiserum to the cleaved p20 subunit of active Casp6a (amino acid 179 of human pro-Casp6a) was made in our laboratory (20). The monoclonal anti-β-actin antibody (Sigma) was raised against the N-terminal 16 amino acids of the protein. The anti-His antibody is a monoclonal antibody from Novagen (VWR, Mississauga, Canada).
Cell Lines and Primary Human Neuronal Culture
The HCT116, MCF7, MC-IXC, and BE(2)M17 cell lines were obtained from ATCC (Manassas, VA). The HEK293T cell line was obtained from colleagues at the Lady Davis Institute. Primary human neurons were cultured from fetal brains obtained under ethical approval from McGill University's institutional review board as described previously (30).
RT-PCR Analysis
Total RNA was isolated from HCT116, HEK293T, MCF7, MC-IXC, and BE(2)M17 cell lines and from primary human neurons with TRIzol reagent (Invitrogen) to determine endogenous levels of CASP6α and CASP6β. RNA was extracted 24 h post-transfection from MCF7 cells transfected with CASP6α- and CASP6β-expressing constructs and run as markers. The first strand cDNA synthesis reaction (20 μl) was performed with 1 μg of total RNA, 1 mm dNTP, 0.5 μl of RNAsin (Roche Applied Science), 5 μm oligo(dT) (Amersham Biosciences), and 40 units of avian myeloblastosis virus reverse transcriptase (Roche Applied Science) for 10 min at 25 °C and 60 min at 42 °C and followed by 10 min at 70 °C. The cDNA was used as template to amplify CASP6α and CASP6β, using the primers 5′-CGC GGA TCC ACC ATG AGC TCG GCC TCG-3′ (primer 1 in Fig. 1A) and 5′-CGG AGG CTG CAG CCA CCT CAG TTA TG-3′ (primer 2 in Fig. 1A). The PCRs (25 μl) were performed with 0.025 units/μl FideliTaq DNA polymerase (U.S. Biochemical Corp.), 0.2 mm each dNTP, 1 μm each primer, and 2 μl of cDNA. The reactions were incubated for 5 min at 95 °C, followed by 25 cycles of 30 s at 95 °C, 1 min at 55 °C, and 1 min at 68 °C, followed by 5 min at 68 °C. To amplify the p20p10 CASP6α product, PCR was conducted using forward primer 5′-CGG GGT ACC ATG GCC TTC TAT AAA AGA GAA ATG-3′ and primer 2. The β-actin amplification was performed using forward primer 5′-CTG GAA CGG TGA AGG TGACA-3′ and reverse primer 5′-AAG GGA CTT CCT GTA ACA ATG CA-3′. The products were analyzed on 1.5–2.5% agarose gel electrophoresis and visualized by ethidium bromide staining.
FIGURE 1.

Expression of CASP6α and CASP6β mRNA and proteins. A, left, schematic diagram of the exons and introns in the CASP6 gene and the resulting CASP6α and CASP6β transcripts. Numbering starts at the first nucleotide of the first exon. The arrows represent primers used for the RT-PCR. Right, schematic diagram showing the main domains of pro-Casp6a and pro-Casp6b and the position of the epitopes for the antibodies that were used in the study. Pro, pro-domain. B, ethidium bromide-stained agarose gel of CASP6α and CASP6β PCR products with primer pairs indicated in A and with β-actin primers. The marker is a 100-bp DNA ladder. Densitometry of CASP6 transcripts was analyzed by ImageQuant software and expressed as a ratio of CASP6α/CASP6β. C, ethidium bromide-stained agarose gel of CASP6α and CASP6β PCR products from CASP6 siRNA-transfected MCF7 cells and control CASP6α- and CASP6β-transfected MCF7 cells. D, ethidium bromide-stained agarose gel showing the NcoI restriction digestion of purified PCR-amplified CASP6β and of non-purified PCR amplified CASP6α and CASP6β. E, Western blot analysis with anti-p10Casp6 antibody on total protein extracts from cell lines and human primary neurons. F, Western blot analysis with anti-p10Casp6 antibody on total protein extracts from MCF7 cells transfected with CASP6 siRNA. G, Western blot analysis with Neomarkers or Upstate α-Casp6a antisera of total protein extracts from MCF7 and HEK293T cells. H, Western blot analysis with anti-p10Casp6 or β-actin antibodies of total protein extracts from human temporal cortex, frontal cortex, or cerebellum.
CASP6 Silencing
MCF7 cells were transfected with 40 nm scrambled or CASP6 siRNA (Dharmacon ThermoScientific) and 5 μl of Lipofectamine 2000 reagent (Invitrogen). Cells were given a second treatment of 10 nm siRNA at 48 h and harvested at 72 h for total RNA and protein.
Enzymatic Digestion
To confirm the identity of the PCR-amplified CASP6α and CASP6β bands, amplicons were digested with 50 units of NcoI enzyme (New England BioLabs, Pickering, Canada), in a 25-μl reaction containing NEB buffer 3 for 2 h at 37 °C. Digested products were separated on a 2% agarose gel and visualized by ethidium bromide staining.
Protein Extractions
Total proteins were extracted from cell cultures in radioimmunoprecipitation assay buffer (1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 100 mm Tris, pH 8.0) supplemented with protease inhibitors (0.1 μg/ml TLCK, 0.5 μg/ml leupeptin, 38 mg/ml AEBSF, and 0.1 μg/ml pepstatin A). Frozen tissues of nine non-cognitively impaired brains were obtained from Dr. Catherine Bergeron (University of Toronto). Tissues were homogenized mechanically with a Polytron homogenizer in 5× radioimmunoprecipitation assay buffer, containing 5% Nonidet P-40, 2.5% sodium deoxycholate, 0.5% SDS, 500 mm Tris, pH 8.0) supplemented with protease inhibitors (0.1 μg/ml TLCK, 0.5 μg/ml leupeptin, 38 mg/ml AEBSF, and 0.1 μg/ml pepstatin A). Temporal, frontal, and cerebellar cortices were analyzed. Protein extracts lysed in radioimmunoprecipitation assay buffer were cleared by centrifugation at 13,000 × g for 20 min. For Casp6 activity assays, transfected HCT116 cells were lysed in CHAPS buffer (50 mm HEPES, 0.1% CHAPS, 0.1 mm EDTA, 1 mm DTT) containing the same set of protease inhibitors as above. Protein extracts were quantified with the BCA protein assay (ThermoScientific, Rockford, IL).
Cloning of CASP6β
The pro-Casp6b cDNA was obtained by targeted PCRs from the pET23b Casp6a-His-tagged expressing vector (kind gift from Dr. Guy Salvesen, Burnham Institute, La Jolla, CA). The oligonucleotides 5′-CTA ACC AGT AAG GCA ACC CC-3′ and 5′-CAG TTG ACA CTG CCG GGT GCC CCC TGC GG-3′ amplified the 5′-end, generating a 0.23-kb fragment, and 5′-CAC CCG GCA GTG TCA ACT GTT AGC CAC GCA G-3′ and 5′-CCG GAA TTC GCA GCC GGA TCT CAG TGG-3′ amplified the 3′-end, generating a 0.61-kb fragment. The two fragments were ligated and served as a template for a second PCR using the oligonucleotides 5′-CGC GGA TCC ATG AGC TCG GAA TCG-3′ and 5′-CCG GAA TTC GCA GCC GGA TCT CAG TGG-3′. The PCR product was digested with BamHI and XhoI, and cloned into the pET23b (Novagen, Madison, WI) prokaryotic expression vector. The pro-Casp6a was subcloned into pCep4β eukaryotic and pIVEX prokaryotic vectors via the XhoI/NotI and SpeI/XhoI restriction sites, respectively.
Recombinant Protein Expression and Purification
Catalytic mutant pro-Casp6aC163A (20) was expressed in BL21 (DE3) Escherichia coli (Novagen) and purified as described previously (31). His-tagged pro-Casp6b was expressed in the BL21 (DE3) E. coli strain. Overnight starter culture was diluted 50× in 2 liters of 2× YT medium supplemented with 100 μg/ml ampicillin and grown at 37 °C until an A600 of 0.6 was reached. Protein expression was induced with 500 μm isopropyl 1-thio-β-d-galactopyranoside (Fisher) for 4 h at 28 °C. Cells were collected by centrifugation at 6000 × g for 15 min and lysed by sonication in resuspension buffer (50 mm Tris-HCl, pH 8, 300 mm NaCl) with 1 mg/ml lysozyme (Sigma). The lysate was cleared by centrifugation at 26,000 × g for 1 h at 4 °C. The supernatant was then loaded on a 3-ml nickel-Sepharose-6 Fast Flow column (GE Healthcare) pre-equilibrated in buffer (50 mm Tris-HCl, pH 8, 300 mm NaCl, 10 mm imidazole). Bound proteins were washed with a 10× column volume of wash buffer (50 mm Tris-HCl, pH 8, 300 mm NaCl, 20 mm imidazole). The proteins were eluted with 50 mm Tris-HCl, pH 8, 300 mm NaCl, 500 mm imidazole elution buffer over 14 1-ml fractions. Proteins from each fraction were separated on a 15% polyacrylamide gel and immunoblotted with anti-p10Casp6 antibody, or the gel was directly stained with 2.5 g/liter Coomassie Brilliant Blue R250 in 45% methanol and 10% acetic acid. Fractions predominantly containing pro-Casp6b were pooled, dialyzed against caspase buffer (50 mm Tris-HCl, pH 8, 100 mm NaCl) in 10,000 molecular weight cut-off dialysis cassettes (Thermo Scientific, Rockland, IL) for 8 h at 4 °C, and concentrated on a 15,000 molecular weight cut-off Amicon Ultra column. Pro-Casp6b was detected as described above.
Effect of Pro-Casp6b on RCasp6a and RCasp1 Activity
Recombinant Casp6 activity was detected by measuring the release of AFC from Ac-VEID-AFC (Sigma) at 37 °C using a Bio-Rad (Hercules, CA) Fluoromark fluorometer at an excitation wavelength of 390 nm and an emission wavelength of 538 nm. Measurements were read every 2 min for 1 h, and the number of released mol of AFC was calculated from a standard curve of 0–2500 pmol of free AFC. The reactions were performed in a 50-μl total volume with Stennicke's buffer (50 mm HEPES, pH 7.4, 100 mm NaCl, 0.1% CHAPS, 10 mm DTT, 1 mm EDTA, 10% sucrose) and the following conditions. 1) For measurement of Casp6 activity, 69 μm Ac-VEID-AFC, 46 nm RCasp6a, and the indicated concentrations of pro-Casp6b were used with the final total protein concentration adjusted to 200 ng/μl with BSA. 2) For measurement of RCasp1 activity, 5 μm Ac-YVAD-AFC, 56 nm pro-Casp6aC163A, 56 nm pro-Casp6b, and 2.2 nm RCasp1 were used as indicated. For the latter, pro-Casp6aC163A was preincubated with pro-Casp6b for 1 h at 37 °C prior to the addition of RCasp1 and the substrate. The reaction mix was then boiled in Laemmli buffer, and 50 μl was loaded on a 15% SDS-polyacrylamide gel and analyzed by Western blotting with anti-p10Casp6 antibodies and anti-p20Casp6 antiserum.
In Vitro Translation (IVT) of Pro-Casp6a
The pro-Casp6a cDNA was subcloned into the pIVEX vector, and translated in E. coli with the Rapid Translation System (Roche Applied Science).
Effect of Pro-Casp6b on Caspase-mediated Activation and Processing of IVT Pro-Casp6a
For measurement of RCasp6a and RCasp1 activity, pro-Casp6b and IVT pro-Casp6a were mixed with Stennicke's buffer containing 5 μm Ac-VEID-AFC or Ac-YVAD-AFC, and 2.2 nm RCasp1 was added immediately prior to the start of the activity assay. The activity assays were performed as described above followed by Western blot analysis using anti-p10 and anti-p20 Casp6 antibodies.
Effect of Pro-Casp6b on the Activation of p20p10Casp6a in Vivo
Human Neurons
The expression of pro-Casp6b from the pCep4β construct was verified by transfecting SK-N-SH cells (ATTC, Manassas, VA) with 3 μg of DNA per 35-mm well using ExGen500 (Fermentas, Burlington, Canada) according to the manufacturer's instructions. Proteins were extracted and Western blotted against the anti-p10Casp6 antibody. Primary human neurons prepared as described previously (30) were transfected with the Helios Gene Gun system (Bio-Rad) at a shooting pressure of 100 p.s.i. according to the manufacturer's protocol. Transfection cartridges were prepared with a 1:3 ratio of pCep4β or pCep4β-pro-Casp6b DNA to pCep4β-EGFP, 4.2 mg of gold microcarrier beads in 0.1 ml of 1 m calcium chloride, and 0.1 ml of 0.05 m spermidine, as described previously (32). 48 h post-transfection, the primary human neurons were serum-deprived for 18 h. Cells were stained with Hoechst 33342, and condensed chromatin EGFP-positive neurons were counted as a percentage of all EGFP-positive neurons to give a percentage of cell death. Neurons were counted blindly.
HCT116 Cells
HCT116 cells were plated overnight in a 6-well plate at a density of 350,000 cells/well in McCoy's medium (Invitrogen) supplemented with 10% fetal bovine serum (Fisher). Transfections were performed on the following day using a DNA/polyethyleneimine (Polysciences Inc., Warrington, PA) ratio of 1:5. The transfection complex was prepared in 100 μl of serum-free medium. 6 h following transfection, complete medium was replaced, and cells were incubated for an additional 18 h to allow for expression. Cells were harvested in CHAPS buffer containing protease inhibitors. Caspase activity assays were performed using 10 μg of total protein lysate and 10 μm Ac-VEID-AFC. The caspase assay reaction mix was analyzed by Western blotting using anti-p10Casp6, anti-p20Casp6, and anti-β-actin antibodies.
Effect of Proteosomal Inhibition on Pro-Casp6b Protein Stability
HCT116 cells were transfected as described above with 4 μg of total cDNA and 20 μg of polyethyleneimine. Epoxomicin was added at a final concentration of 0.1 μm immediately following transfection for 24 h.
Co-immunoprecipitation
Recombinant proteins were incubated for 4 h at 4 °C with protein A-Sepharose beads and Upstate α-Casp6a antibody at a dilution of 1:100 in 50 mm Tris, pH 8.0, 100 mm NaCl. The beads were preblocked for 1 h at 4 °C with 1% BSA solution. Beads were vigorously washed three times with the buffer and then resuspended in Laemmli SDS buffer. Samples were boiled and loaded on SDS-polyacrylamide gels and immunoblotted with the α-p10Casp6 antibody.
Statistical Evaluations
Statistical evaluations of the data were performed by analysis of variance and post hoc analysis as indicated in the legend of each figure.
RESULTS
CASP6β mRNA and Its Protein Product, Pro-Casp6b, Are Expressed in Various Cell Lines, Primary Human Neurons, and Adult Human Brains
The CASP6 gene has been shown to yield two alternatively spliced mRNAs called MCH2α (CASP6α) and MCH2β (CASP6β) (9) (Fig. 1A). Pro-Casp6a, translated from the CASP6α mRNA, generates the proenzyme form that is proteolytically processed into a fully active caspase, whereas the pro-Casp6b results from the translation of the shorter CASP6β mRNA. To assess if the CASP6β mRNA is generated with the CASP6α mRNA, we performed RT-PCR on total RNA extracted from various cell types with CASP6-specific primers that would amplify both cDNAs (indicated by the arrows in Fig. 1A). The expected 564-bp PCR product of CASP6α was detected in human colon carcinoma HCT116, embryonic kidney 293 (HEK293T), breast carcinoma MCF7, and neuroblastoma MC-IXC and BE(2)M17 cells (Fig. 1B). In addition, a second PCR-amplified DNA band of 325 bp corresponding to the CASP6β transcript was also detected. The 564 and 325 bp bands were strongly amplified from CASP6α- and CASP6β-transfected MCF7 cells. The ratio of CASP6α over CASP6β transcripts was consistently greater than 1 in HCT116, HEK293T, MCF7, and MC-IXC and less than 1 in the BE(2)M17 cell line. In cultures of human primary neurons, CASP6β mRNA levels slightly exceeded those of CASP6α. CASP6 siRNA strongly reduced the levels of PCR-amplified CASP6α but had only slight effects on the levels of CASP6β (Fig. 1C). The lack of down-regulation of CASP6β by siRNA was surprising because the four pooled CASP6 siRNAs target sequences in exons 5, 6, and 7 are shared by CASP6α and CASP6β. To confirm the identity of the two CASP6 transcripts, we subjected purified CASP6β or unpurified PCR-amplified CASP6α and CASP6β to enzymatic digestion with NcoI (Fig. 1D). Digestion of the 325 bp band generated the expected 219-bp product indicative of a unique NcoI site cleavage at 93 bp. Digestion of both transcripts generated a digested product of CASP6α at 360 bp and the same 219 bp CASP6β band. An attempt to generate a custom-made siRNA to the exon 1/exon 5 junction of CASP6β also failed to down-regulate CASP6β (results not shown). We concluded that the siRNA may not effectively down-regulate CASP6β because of a low turnover of the CASP6β mRNA or its interaction with RNA-binding proteins (33).
The pro-Casp6b protein resulting from CASP6β translation lacks part of the pro-domain and part of the large p20 subunit of pro-Casp6a (Fig. 1A). Therefore, the expression of pro-Casp6a can be differentiated from that of pro-Casp6b with the α-Casp6a antiserum, but both pro-Casp6a and pro-Casp6b should be recognized with the α-p10Casp6 antibody (Fig. 1A). Western blot analysis of total proteins showed the presence of two α-p10Casp6 immunoreactive proteins in most cell lines and primary human neurons (Fig. 1E). The 34- and 23-kDa proteins corresponded to the expected size for pro-Casp6a and pro-Casp6b, respectively. The level of pro-Casp6a relative to pro-Casp6b protein was consistent with the observed levels of transcripts in Fig. 1B. CASP6 siRNA reduced the levels of pro-Casp6a significantly but only minimally reduced the levels of pro-Casp6b (Fig. 1F), consistent with the observed effect of CASP6 siRNA on the mRNA levels. To confirm that the 23-kDa protein detected with α-p10Casp6 antibody was indeed pro-Casp6b, we also performed Western blot analyses with the Neomarker and Upstate α-Casp6a antisera. Although immunoblot analysis revealed the pro-Casp6a and the p20 subunit of recombinant Casp6, they did not detect any protein at 23 kDa, consistent with the lack of this epitope in pro-Casp6b (Fig. 1G). Western blot analysis of proteins extracted from human temporal cortex, frontal cortex, and cerebellar tissues with α-p10Casp6 did not reveal a 32-kDa protein expected for pro-Casp6a but showed a 23-kDa protein consistent with pro-Casp6b (Fig. 1H). The 23-kDa immunoreactive protein in human brains migrated exactly as that observed in MCF7 cells (not shown). This protein varied in intensity in different cases but was consistently present in the three areas of the brain investigated. The identity of an additional lower molecular weight protein was not pursued, but it could represent a post-translationally modified form of pro-Casp6b or a nonspecific cross-reacting protein. Together, these results confirmed that the CASP6β transcript, as shown by Northern blot analysis previously (9), and the pro-Casp6b protein were expressed in various cell lines, primary human neurons, and normal human brain tissue.
Purified Bacterially Expressed Recombinant Pro-Casp6b Does Not Possess Catalytic Activity and Does Not Inhibit the Active Form of Casp6a
To assess the function of pro-Casp6b, C-terminally His-tagged bacterially expressed pro-Casp6b was purified. With the addition of the His tag to pro-Casp6b, the protein had the expected mass of 27 kDa. We isolated the isopropyl 1-thio-β-d-galactopyranoside-induced 27-kDa pro-Casp6b protein to over 90% purity (not shown). The pro-Casp6b protein was immunoreactive with α-p10Casp6 (Fig. 2A). The α-p10Casp6 antibody also recognized the catalytic mutant pro-Casp6aC163A, pro-Casp6a, and the p10 subunit of active Casp6a. As expected, the α-Casp6a antiserum (Fig. 1C) recognized the pro-Casp6a and the pro-Casp6aC163A but not pro-Casp6b. The neoepitope α-p20Casp6 antiserum recognized only the p20 subunit in active Casp6a, indicating that the Asp179 site was not cleaved in bacterially expressed pro-Casp6b. In contrast, bacterially expressed pro-Casp6a was always self-proteolytically cleaved into its p20 and p10 subunits. Nevertheless, pro-Casp6b contains the catalytic QACRG site of caspases. To determine if pro-Casp6b might retain some catalytic activity, pure pro-Casp6b was incubated at 37 °C for 1 h (Fig. 2B). This did not result in any proteolytic processing, indicating that pro-Casp6b did not have self-processing activity in vitro. Furthermore, even at very high concentrations, pro-Casp6b did not proteolytically process Ac-VEID-AFC substrate (Fig. 2C). To determine if pro-Casp6b inhibited active RCasp6a, we assessed the VEIDase activity of active Casp6a in the absence or presence of increasing amounts of purified pro-Casp6b (Fig. 2D). In this experiment, the RCasp6a was incubated with different concentrations of the substrate Ac-VEID-AFC and pro-Casp6b. We did not observe any variation in the specific activity of 46 nm active RCasp6a with up to 100 nm pro-Casp6b. These results indicated that pro-Casp6b was not catalytically active and could not directly inhibit the already activated RCasp6a.
FIGURE 2.

Purification of pro-Casp6b from E. coli lysate. A, Western blots of pro-Casp6a, active RCasp6a, inactive pro-Casp6aC163A, and pro-Casp6b with anti-p10Casp6 (top), Upstate anti-pro-Casp6a (middle), and neoepitope anti-p20Casp6 (bottom). B, Western blot with anti-p10Casp6 of purified pro-Casp6b incubated for 1 h at 37 °C. C, relative VEIDase activity with increasing amounts of pro-Casp6b relative to active RCasp6a. D, specific VEIDase Casp6 activity (Spec. Act.) for purified recombinant active Casp6a in the absence or presence of pro-Casp6b at the indicated concentrations. Error bars, S.E.
Pro-Casp6b Inhibits RCasp1-mediated Processing of Pro-Casp6a in Vitro
To determine if pro-Casp6b can inhibit the activation of pro-Casp6a, we activated pro-Casp6a by adding active RCasp1 (19). The addition of active RCasp1 to IVT pro-Casp6a resulted in processing of pro-Casp6a at Asp23 and Asp179, thereby producing the p20p10Casp6a and the p20 subunit detected with the anti-p20 neoepitope antiserum, respectively (Fig. 3A). Consequently, because p20p10Casp6a and p20 subunits were generated, the full-length pro-Casp6a decreased (Fig. 3A). Pro-Casp6b completely inhibited the processing of IVT pro-Casp6a at Asp23 and Asp179 as evident by the lack of p20p10Casp6a and p20 subunits. In contrast, pro-Casp6a levels were maintained in the presence of pro-Casp6b, indicating a complete inhibition of pro-Casp6a processing. These results showed that pro-Casp6b had robust inhibitory activity.
FIGURE 3.

Effect of pro-Casp6b on RCasp1-mediated processing of IVT pro-Casp6a. A, Western blot with anti-p10Casp6 or the neoepitope anti-p20Casp6 antiserum showing the effect of pro-Casp6b on RCasp1-mediated processing of the catalytically competent pro-Casp6a. B, VEIDase activity of IVT pro-Casp6a treated with or without active RCasp1 in the absence or presence of pro-Casp6b in three independent experiments. The results from the third experiment show that with freeze-thaw cycles and prolonged storage time in freezer, the purified caspases and pro-Casp6b are unstable. C, RCasp1 activity in the experiments shown in A on Ac-YVAD-AFC substrate. Error bars, S.E.
To assess pro-Casp6a activation in these conditions, we measured IVT Casp6a VEIDase activity in three independent experiments. The molar amount of purified pro-Casp6b was assessed by spectrophotometry, whereas IVT pro-Casp6a in the E. coli lysate was assessed relative to pro-Casp6b by immunoblotting with anti-p10Casp6 antibody. Variations in specific activities were observed in each experiment and depended on the number of freeze-thaw cycles of RCasp1, pro-Casp6b, and IVT pro-Casp6a. Pro-Casp6b and active RCasp1 had no VEIDase activity but IVT pro-Casp6a displayed a small amount of activity, consistent with a certain amount of self-activation during synthesis in E. coli lysates. The addition of RCasp1 to IVT pro-Casp6a resulted in increased VEIDase activity, consistent with the RCasp1-mediated increased processing of pro-Casp6a into its active subunits (Fig. 3B). The addition of an equimolar amount of pro-Casp6b to IVT pro-Casp6a inhibited RCasp1-mediated activation of pro-Casp6a by 50–90% in three independent experiments. Adding twice the amount of pro-Casp6b resulted in 90–100% inhibition. Pro-Casp6b had no effect on RCasp1 YVADase activity in the absence or presence of IVT pro-Casp6a (Fig. 3C).
These results showed that pro-Casp6b inhibited RCasp1-mediated activation of pro-Casp6a. The results excluded the possibility that pro-Casp6b directly inhibited RCasp1 activity. From this, we deduced only two other possibilities. Pro-Casp6b could interact with pro-Casp6a and block the access of RCasp1 to the processing sites of pro-Casp6a. Alternatively, pro-Casp6b could inhibit the self-processing activity of p20p10Casp6a generated by RCasp1 cleavage at the Asp23 site of the pro-domain (24).
Effect of Pro-Casp6b on RCasp1-Mediated Processing of Pro-Casp6aC163A in Vitro
To determine if pro-Casp6b inhibits RCasp1-mediated p20p10Casp6a self-activation, we repeated the above experiments with catalytically incompetent pro-Casp6aC163A. Active RCasp1 was incubated with the catalytically inactive pro-Casp6aC163A in the absence and presence of an equal amount of pro-Casp6b (Fig. 4A). RCasp1 only minimally processed pro-Casp6aC163A at Asp23, resulting in a small amount of p20p10Casp6aC163A. Pro-Casp6b did not inhibit RCasp1-mediated cleavage of pro-Casp6aC163A at Asp23. We excluded the possibility that RCasp1 was inhibited by pro-Casp6b or pro-Casp6aC163A by measuring RCasp1 YVADase activity in each of the samples shown in the Western blot (Fig. 4B). Neither pro-Casp6aC163A nor pro-Casp6b had any YVADase activity, and RCasp1 YVADase activity was unaffected in the presence of pro-Casp6aC163A, pro-Casp6b, or both. From these results, we concluded that pro-Casp6b is inhibiting p20p10Casp6a self-processing. Furthermore, RCasp1 did not process the pro-Casp6b (Fig. 4A), excluding the possibility that a truncated Casp6b p20 subunit associated with p20 subunits of Casp6a to act in a dominant negative manner.
FIGURE 4.

Effect of pro-Casp6b on RCasp1-mediated processing of pro-Casp6aC163A. A, Western blot with anti-p10Casp6 showing the effect of pro-Casp6b on RCasp1-mediated processing of the catalytically inactive pro-Casp6aC163A mutant. B, RCasp1 activity in the experiments shown in A on Ac-YVAD-AFC substrate. Error bars, S.E.
Surprisingly, when pro-Casp6b was added to pro-Casp6aC163A, a protein fragment migrated at 26 kDa, slightly below the pro-Casp6b, and a very faint amount of p10 subunit appeared despite the absence of RCasp1. Both of these fragments were detected with the anti-p10Casp6 antibody. Because the pro-Casp6aC163A is catalytically inactive, these results indicated that under some circumstances, the pro-Casp6b can have minor catalytic activity at Asp193 of pro-Casp6a to generate the p10 subunit and additionally cleaved within the p20 subunit to yield a 26-kDa fragment containing the p10 subunit. Processing at this site was partially inhibited when active RCasp1 was added to pro-Casp6aC163A and pro-Casp6b, suggesting that RCasp1-mediated cleavage at the Asp23 site decreased the accessibility of this additional processing site to pro-Casp6b. These results showed that the pro-Casp6b must inhibit p20p10Casp6a self-activation rather than inhibit RCasp1-mediated activation.
Pro-Casp6b Inhibits Pro-Casp6a Activation in Mammalian Cells
To test if pro-Casp6b inhibits pro-Casp6a activation in cells, we used a model of serum-deprived human primary neurons where pro-Casp6a is activated via RCasp1 (19). Unfortunately, the low efficiency of transfection of these primary neurons and the lack of a specific pro-Casp6b antibody does not allow biochemical analysis of the expression of the exogenously expressed pro-Casp6b. However, we confirmed expression of pro-Casp6b from the pCep4β eukaryotic construct in the SK-N-SH cell line (Fig. 5A). In mock- and vector-transfected cells, there were low levels of 23-kDa pro-Casp6b. This protein increased significantly in CASP6β cDNA-transfected cells. In human neurons, we found that serum-deprived non-transfected or pCep4βEGFP-transfected neurons underwent 50% cell death with serum deprivation (Fig. 5B). CASP6β cDNA-transfected neurons showed significantly less cell death. These results suggested that pro-Casp6b prevented cell death in primary human neurons.
FIGURE 5.
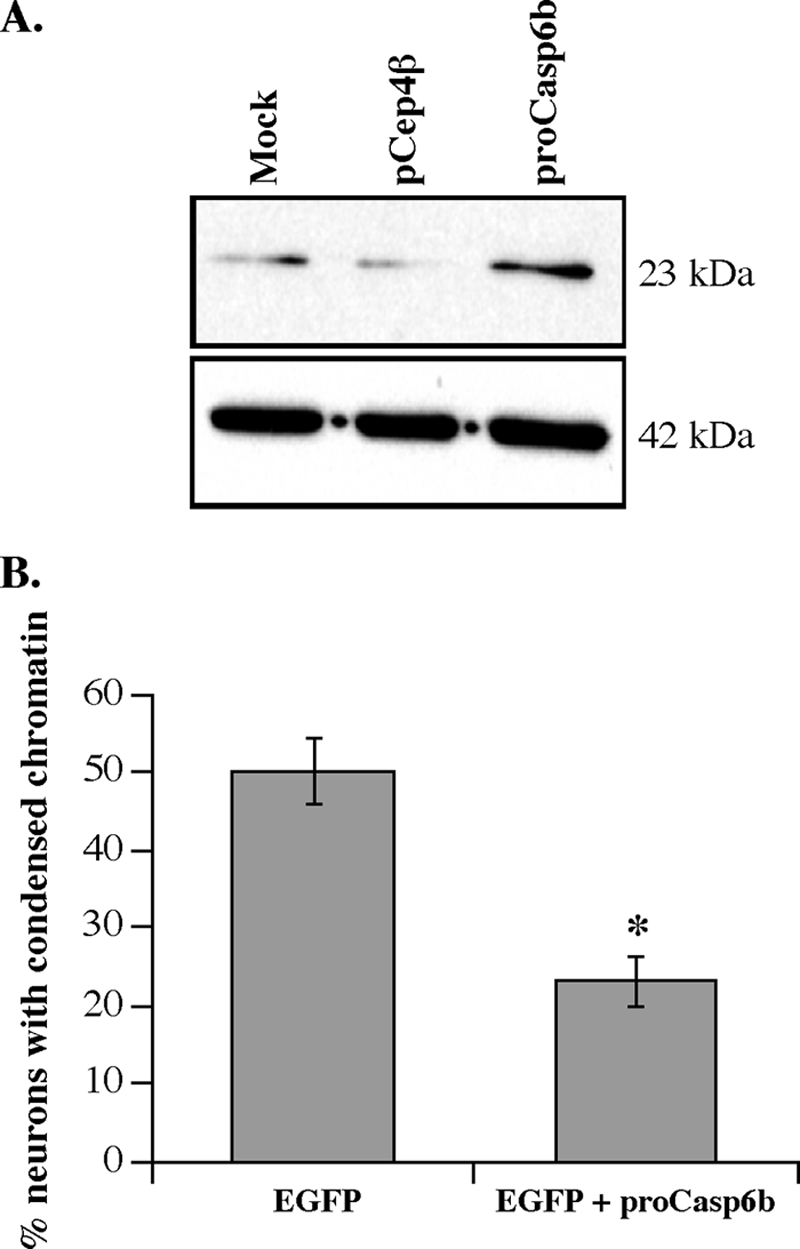
Pro-Casp6b inhibits Casp6a-mediated cell death in primary cultures of human neurons. A, Western blot of protein extracts of mock, pCep4β, or pCep4β-pro-Casp6b cDNA-transfected cells with anti-p10Casp6 and β-actin antibodies. B, neuronal cell death of neurons transfected with pCep4β/EGFP alone or pCep4β/EGFP and pCep4β/pro-Casp6b cDNA (ratio 1:3) and serum-deprived. The base line of cell death in non-transfected and non-serum-deprived neurons was 5%. Non-transfected neurons had ∼50% cell death due to serum deprivation, indicating that EGFP is not additionally toxic to these neurons. *, p < 0.0005 by analysis of variance and Scheffé's post hoc analysis. Error bars, S.E.
To confirm that pro-Casp6b inhibited pro-Casp6a activation in cells, we used an alternate cellular model that allows verification of the expression and activity of Casp6a. HCT116 cells were transfected with increasing concentrations of CASP6β cDNAs, and the cellular extracts were tested for VEIDase activity 24 h after the transfection (Fig. 6A). As observed in vitro, the expression of pro-Casp6b, detected by Western blotting with either the anti-p10Casp6 or anti-His antibodies, did not generate VEIDase activity. Similar to previous observations in HEK293 cells (24), the p20p10Casp6α cDNA-transfected HCT116 cells exhibited significant VEIDase activity compared with control cDNA-transfected cells (Fig. 6A). Co-transfection of pro-Casp6b cDNA with the p20p10Casp6a cDNA resulted in a 4-fold inhibition of the VEIDase activity generated by p20p10Casp6a (Fig. 6B).
FIGURE 6.

Pro-Casp6b inhibits p20p10Casp6a-mediated processing and activation in HCT116 cells. A, VEIDase activity and Western blot analysis with anti-p10Casp6 and anti-His antibodies of protein extracts from HCT116 cells transfected with vector alone (pCep4β), pCep4βEGFP, pCep4βpro-Casp6b, or pCep4βp20p10Casp6a. B, VEIDase activity and Western blot analysis with anti-p10Casp6 and anti-p20 neoepitope antiserum of protein extracts from HCT116 cells transfected with vector alone (pCep4β)-, pCep4βp20p10-, pCep4βp20p10/pCep4βpro-Casp6b (1:3 cDNA ratio)-, or pCep4βp20p10/pCep4βpro-Casp6b (1:9 cDNA ratio)-transfected HCT116 cells. C, ethidium bromide-stained agarose gel of RT-PCR products from HCT116-transfected cells. D, Western blot analysis with anti-p10Casp6 antibody (top), anti-neoepitope p20 antiserum (middle), and anti-β-actin antibodies of proteins extracted from transfected HCT116 cells treated in the absence or presence of epoxomicin (EPOX). Error bars, S.E.
However, the co-expression of pro-Casp6b with p20p10Casp6a also considerably decreased the abundance of the p20p10Casp6a and pro-Casp6b proteins (Fig. 6B). To investigate if co-transfections altered the mRNA levels of p20p10Casp6a and pro-Casp6b, we performed RT-PCR on RNA extracted from HCT116-transfected cells. Two different primer pairs had to be specifically designed for pro-Casp6b and p20p10Casp6a cDNAs because the p20p10Casp6a lacks the pro-domain sequence that is shared by pro-Casp6b and pro-Casp6a mRNA. The p20p10Casp6a primers amplified the expected 564-bp CASP6α DNA from pCep4β vector but not in non-transfected cells, indicating an increase in endogenously expressed CASP6α mRNA during transfection. CASP6α mRNA was slightly increased in cells transfected with the p20p10Casp6a cDNA (Fig. 6C). The pro-Casp6b primers amplified the expected 325-bp DNA from pro-Casp6b-transfected cells. Co-transfection of pro-Casp6b and p20p10Casp6a cDNAs did not alter the levels of pro-Casp6b mRNA. Reduced mRNA levels were observed in vector and non-transfected cells. However, in p20p10Casp6a-transfected cells, there was an increased level of pro-Casp6b PCR product. These results indicated that pro-Casp6b and p20p10Casp6a expression affect endogenously expressed pro-Casp6a or pro-Casp6b mRNA levels. More work will be required for a thorough understanding of this phenomenon. However, the detection of CASP6α and CASP6β mRNAs in the p20p10Casp6a and pro-Casp6b co-transfected cells indicated that the reduction of protein levels observed in Fig. 5B was not the result of attenuated mRNA. These results suggest that the decreased levels of p20p10Casp6a and pro-Casp6b proteins in co-transfected cells may be due to either increased protein degradation or decreased translation.
We have previously demonstrated that the p20 and p10 subunits of Casp6a are degraded by the proteasome (34). To determine if proteasomal degradation is responsible for the reduction of p20p10Casp6a and pro-Casp6b protein levels in the co-transfected HCT116 cells, we compared protein extracts from transfected cells treated with or without the irreversible proteasomal inhibitor, epoxomicin (Fig. 6D). The p10 subunit became detectable in the epoxomicin-treated p20p10Casp6a-transfected HCT116 cells, and the p20 subunit increased in both the p20p10Casp6a and co-transfected cells. However, the levels in co-transfected cells were still lower than in the p20p10Casp6a-transfected cells. Furthermore, the p20p10Casp6a protein did not increase in epoxomicin-treated cells. These results indicated that the p20p10Casp6a was still converted into the p20 subunit when co-expressed with pro-Casp6b and that there was proteasomal degradation of p20 and p10 subunits in these cells. However, the lower levels of p20p10 in co-transfected cells also could indicate that the co-transfection inhibited the translation of these proteins or increased degradation of p20p10Casp6a and its proteolytic subunits through a proteasome-independent pathway.
Co-immunoprecipation of Pro-Casp6a and Pro-Casp6b
To investigate the underlying mechanism of pro-Casp6b inhibition of pro-Casp6a activation, we performed an immunoprecipitation with preimmune serum and α-Casp6a from a mixture of purified recombinant pro-Casp6a and pro-Casp6b and Western blotted with α-p10Casp6. The Western blot showed that α-Casp6a antiserum co-immunoprecipitated pro-Casp6b with pro-Casp6a and Casp6a lacking the pro-domain (Fig. 7). Similarly, α-Casp6a antiserum co-immunoprecipitated the catalytically inactive pro-Casp6C163A and pro-Casp6b. In contrast, neither pro-Casp6a nor pro-Casp6b immunoprecipitated with the preimmune serum.
FIGURE 7.

Co-immunoprecipitation of purified recombinant pro-Casp6a and pro-Casp6b. Western blot analysis with α-p10Casp6 of a mixture of purified recombinant pro-Casp6a and pro-Casp6b immunoprecipitated with α-Casp6 or preimmune antisera.
DISCUSSION
Understanding the regulation of Casp6a activity could provide a viable treatment to inhibit and possibly reverse neuronal degeneration in several pathological conditions. Indeed, Casp6a is highly activated in mild cognitive impairment and at all stages of sporadic Alzheimer disease and in familial Alzheimer disease (20–22). Furthermore, Casp6 activity may be involved in the clinical manifestation of Huntington and Parkinson diseases (26, 27). In addition, the role of Casp6 in neurodegeneration rather than cell death has recently been well documented (20, 24, 25). Therefore, finding a role for pro-Casp6b as a potential inhibitor of pro-Casp6a activation provides a novel therapeutic target against neurodegeneration.
We investigated the pro-Casp6b protein product of the alternatively spliced mRNA of the CASP6 gene because several alternatively spliced isoforms of caspases regulate their cognate caspase activity. We confirmed that the pro-Casp6b is expressed as mRNA and protein in several cell lines. The existence of the pro-Casp6b mRNA has been previously demonstrated in Jurkat cells (9); our results further indicated that pro-Casp6b expression is ubiquitous in different cell types, including human colon carcinoma, breast carcinoma, neuroblastoma cells, primary human neurons, and human adult normal brains.
Pro-Casp6b did not have significant catalytic activity despite retaining its catalytic site. We cloned and purified the recombinant pro-Casp6b and investigated if it demonstrated either self-processing or VEIDase activity as does bacterially expressed and purified recombinant pro-Casp6a. We did not find any evidence of pro-Casp6b self-processing by Western blotting, and the pro-Casp6b did not display any processing activity on the Ac-VEID-AFC substrate that is well cleaved by Casp6a. However, we did observe the production of a 26-kDa protein when pro-Casp6b was added to pro-Casp6aC163A. The processing occurred at an unexpected site within the p20 subunit domain. Because this processing occurred with the catalytically inactive pro-Casp6aC163A, the activity must arise from the pro-Casp6b catalytic site. Which of the two proteins is cleaved is not clear at this time, but this result hints at the ability of pro-Casp6b to carry minor catalytic activity in some situations.
Pro-Casp6b cannot inhibit Casp6a once it is activated. In vitro, the addition of pro-Casp6b to recombinant purified active Casp6a did not alter its VEIDase activity. However, the pro-Casp6b inhibited the self-activation of pro-Casp6a. This was deduced from the fact that pro-Casp6b inhibited RCasp1-mediated activation of IVT pro-Casp6a but had no effect on the RCasp1-mediated processing of the catalytically incompetent pro-Casp6aC163A protein. This type of inhibition is consistent with the mechanism by which other alternatively spliced forms of caspases inhibit their cognate caspases (2, 5, 8, 9).
Pro-Casp6b also inhibited Casp6a activation in transfected human neurons and in HCT116 cells. We confirmed inhibition in serum-deprived human neurons, a condition known to induce RCasp1-mediated activation of Casp6a (19). However, because of the inherent difficulty of transfecting human neurons with viral vectors or liposome-based products, we were limited to performing single cell analyses on pro-Casp6b Genegun-transfected neurons. Therefore, to confirm the ability of pro-Casp6b to inhibit pro-Casp6a activation in cells, we took advantage of a recently developed system where the pro-domain lacking p20p10Casp6a induced self-processing and VEIDase activity when transfected in mammalian cells (24). In HCT116 cells, pro-Casp6b did not demonstrate any VEIDase catalytic activity, but p20p10Casp6a did. In cells co-transfected with pro-Casp6b and p20p10Casp6a cDNAs, the p20p10Casp6a-mediated VEIDase activity was significantly inhibited.
We could exclude two potential mechanisms to explain pro-Casp6b inhibitory activity. First, pro-Casp6b did not inhibit RCasp1 activity (Fig. 8A). Second, because pro-Casp6b was not processed by either RCasp1 or activated Casp6a, we can exclude the possibility that the truncated p20 subunit of pro-Casp6b might have formed a complex with Casp6a subunits and resulted in the dominant negative inhibition of the active enzyme (Fig. 8B). Our results showed that pro-Casp6b inhibited the self-processing of pro-Casp6a after the removal of the pro-domain (Fig. 8C). A likely mechanism by which pro-Casp6b inhibited RCasp1-activated Casp6a self-processing is via an association between the p20p10Casp6a and pro-Casp6b. Both the proenzyme and the active form of Casp6a exist naturally as symmetrical dimers, which form through an interaction of the p10 subunits. Because the p10 region of pro-Casp6b is intact, it is possible that it interacts with pro-Casp6a to form an asymmetrical dimer. Indeed, a direct interaction of pro-Casp6a and pro-Casp6b was observed by co-immunoprecipitation. This interaction possibly disrupts the catalytic activity of Casp6a.
FIGURE 8.

Schematic diagram of three possible mechanisms for pro-Casp6b inhibition of pro-Casp6a activation. A, mechanism by which pro-Casp6b inhibits the active Casp1. Pro, pro-domain; L, linker; LSCasp6b, pro-Casp6b large subunit. B, dominant negative inhibition of pro-Casp6a by pro-Casp6b because the large subunit of pro-Casp6b could replace one p20 subunit of Casp6a in the tetrameric enzyme. C, pro-Casp6b inhibition of pro-Casp6a self-processing at Asp179 and Asp193.
This model predicts that pro-Casp6b should inhibit pro-Casp6a activation at equimolar concentrations. Accordingly, we observed that concentrations of pro-Casp6b equimolar to those of pro-Casp6a resulted in 50–90% inhibition of RCasp1 activation of pro-Casp6a in vitro. In cell lines and in human primary neurons, the levels of pro-Casp6a and pro-Casp6b were mostly equivalent, suggesting that transient activation of pro-Casp6a could be kept in check by pro-Casp6b. In the adult human brain tissue, the levels of pro-Casp6b largely exceeded those of pro-Casp6a, suggesting that pro-Casp6b could act as a mechanism to prevent Casp6a activation in vivo. This may be particularly important given the role of active Casp6 in neurodegeneration (25).
The role of pro-Casp6b as an inhibitor of pro-Casp6a activation could be very important physiologically. Given the recent finding that active Casp6a induces neurodegeneration and not cell death (25), regulation of pro-Casp6b expression could prevent Casp6a-mediated damage to neurons. Not much is known about the regulation of pro-Casp6a activity. The IAPs do not inhibit Casp6a despite being strong inhibitors of the other two effector caspases, Casp3 and Casp7 (28). Estrogen induces an inhibitor of active Casp6a in human primary neurons (29). Our present findings thus provide a novel type of regulation of Casp6a activity.
Acknowledgments
We thank Catherine Bergeron for the frozen brain tissues and Jennifer Hammond for culturing the human neurons. We are grateful to the Birth Defects Research Laboratory (University of Washington, Seattle, WA) for providing conceptual tissue for research.
This work was supported, in whole or in part, by National Institutes of Health Grant MS/MH40965-01. This work was also supported by Canadian Institutes of Health Research (CIHR) Grants MOP81146 and CCI-85682, the CIHR-National Science Foundation of China Joint Health Initiative, the James McGill Professorship, and the Fonds de la Recherche en Santé (to A. C. L.).
- IAP
- inhibitor of apoptosis protein
- AEBSF
- 4-(2-aminoethyl)-benzenesulfonyl fluoride
- IVT
- in vitro translation
- TLCK
- Nα-p-tosyl-l-lysine chloromethyl ketone.
REFERENCES
- 1.Kumar S. (2007) Cell Death Differ. 14, 32–43 [DOI] [PubMed] [Google Scholar]
- 2.Alnemri E. S., Fernandes-Alnemri T., Litwack G. (1995) J. Biol. Chem. 270, 4312–4317 [DOI] [PubMed] [Google Scholar]
- 3.Huang Y., Shin N. H., Sun Y., Wang K. K. (2001) Biochem. Biophys. Res. Commun. 283, 762–769 [DOI] [PubMed] [Google Scholar]
- 4.Mohr A., Zwacka R. M., Jarmy G., Büneker C., Schrezenmeier H., Döhner K., Beltinger C., Wiesneth M., Debatin K. M., Stahnke K. (2005) Oncogene 24, 2421–2429 [DOI] [PubMed] [Google Scholar]
- 5.Himeji D., Horiuchi T., Tsukamoto H., Hayashi K., Watanabe T., Harada M. (2002) Blood 99, 4070–4078 [DOI] [PubMed] [Google Scholar]
- 6.Seol D. W., Billiar T. R. (1999) J. Biol. Chem. 274, 2072–2076 [DOI] [PubMed] [Google Scholar]
- 7.Wang L., Miura M., Bergeron L., Zhu H., Yuan J. (1994) Cell 78, 739–750 [DOI] [PubMed] [Google Scholar]
- 8.Ito A., Uehara T., Nomura Y. (2000) FEBS Lett. 470, 360–364 [DOI] [PubMed] [Google Scholar]
- 9.Fernandes-Alnemri T., Litwack G., Alnemri E. S. (1995) Cancer Res. 55, 2737–2742 [PubMed] [Google Scholar]
- 10.Grossmann J., Mohr S., Lapentina E. G., Fiocchi C., Levine A. D. (1998) Am. J. Physiol. 274, G1117–G1124 [DOI] [PubMed] [Google Scholar]
- 11.Olson N. E., Graves J. D., Shu G. L., Ryan E. J., Clark E. A. (2003) J. Immunol. 170, 6065–6072 [DOI] [PubMed] [Google Scholar]
- 12.Watanabe C., Shu G. L., Zheng T. S., Flavell R. A., Clark E. A. (2008) J. Immunol. 181, 6810–6819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morozov V., Wawrousek E. F. (2006) Development 133, 813–821 [DOI] [PubMed] [Google Scholar]
- 14.Zandy A. J., Lakhani S., Zheng T., Flavell R. A., Bassnett S. (2005) J. Biol. Chem. 280, 30263–30272 [DOI] [PubMed] [Google Scholar]
- 15.Zandy A. J., Bassnett S. (2007) Invest. Ophthalmol. Vis. Sci. 48, 293–302 [DOI] [PubMed] [Google Scholar]
- 16.Foley J. D., Rosenbaum H., Griep A. E. (2004) J. Biol. Chem. 279, 32142–32150 [DOI] [PubMed] [Google Scholar]
- 17.LeBlanc A., Liu H., Goodyer C., Bergeron C., Hammond J. (1999) J. Biol. Chem. 274, 23426–23436 [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y., Goodyer C., LeBlanc A. (2000) J. Neurosci. 20, 8384–8389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo H., Pétrin D., Zhang Y., Bergeron C., Goodyer C. G., LeBlanc A. C. (2006) Cell Death Differ. 13, 285–292 [DOI] [PubMed] [Google Scholar]
- 20.Guo H., Albrecht S., Bourdeau M., Petzke T., Bergeron C., LeBlanc A. C. (2004) Am. J. Pathol. 165, 523–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Albrecht S., Bogdanovic N., Ghetti B., Winblad B., LeBlanc A. C. (2009) J. Neuropathol. Exp. Neurol. 68, 1282–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Albrecht S., Bourdeau M., Bennett D., Mufson E. J., Bhattacharjee M., LeBlanc A. C. (2007) Am. J. Pathol. 170, 1200–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klaiman G., Petzke T. L., Hammond J., LeBlanc A. C. (2008) Mol. Cell Proteomics 7, 1541–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klaiman G., Champagne N., LeBlanc A. C. (2009) Biochim. Biophys. Acta 1793, 592–601 [DOI] [PubMed] [Google Scholar]
- 25.Nikolaev A., McLaughlin T., O'Leary D. D., Tessier-Lavigne M. (2009) Nature 457, 981–989 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Giaime E., Sunyach C., Druon C., Scarzello S., Robert G., Grosso S., Auberger P., Goldberg M. S., Shen J., Heutink P., Pouysségur J., Pagès G., Checler F., Alves da Costa C. (2010) Cell Death Differ. 17, 158–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Graham R. K., Deng Y., Slow E. J., Haigh B., Bissada N., Lu G., Pearson J., Shehadeh J., Bertram L., Murphy Z., Warby S. C., Doty C. N., Roy S., Wellington C. L., Leavitt B. R., Raymond L. A., Nicholson D. W., Hayden M. R. (2006) Cell 125, 1179–1191 [DOI] [PubMed] [Google Scholar]
- 28.Vaux D. L., Silke J. (2005) Nat. Rev. Mol. Cell Biol. 6, 287–297 [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y., Tounekti O., Akerman B., Goodyer C. G., LeBlanc A. (2001) J. Neurosci. 21, RC176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.LeBlanc A. (1995) J. Neurosci. 15, 7837–7846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Denault J., Salvesen G. (2002) in Current Protocols in Protein Science, Unit 21.13.21–21.13.15, John Wiley & Sons, Inc., New York [Google Scholar]
- 32.Roucou X., Giannopoulos P. N., Zhang Y., Jodoin J., Goodyer C. G., LeBlanc A. (2005) Cell Death Differ. 12, 783–795 [DOI] [PubMed] [Google Scholar]
- 33.Dorsett Y., Tuschl T. (2004) Nat. Rev. Drug Discov. 3, 318–329 [DOI] [PubMed] [Google Scholar]
- 34.Tounekti O., Zhang Y., Klaiman G., Goodyer C. G., LeBlanc A. (2004) J. Neurochem. 89, 561–568 [DOI] [PubMed] [Google Scholar]