

Abstract
A fragment of the prion protein, PrP(89–143, P101L), bearing a mutation implicated in familial prion disease, forms fibrils that have been shown to induce prion disease when injected intracerebrally into transgenic mice expressing full-length PrP containing the P101L mutation. In this study, we utilize amide hydrogen exchange measurements to probe the organization of the peptide in its fibrillar form. We determined the extent of hydrogen exchange first by tandem proteolysis, liquid chromatography, and mass spectrometry (HXMS) and then by exchange-quenched NMR. Although single amide resolution is afforded by NMR measurements, HXMS is well suited to the study of natural prions because it does not require labeling with NMR active isotopes. Thus, natural prions obtained from infected animals can be compared with model systems such as PrP(89–143, P101L) studied here. In our study, we find two segments of sequence that display a high level of protection from exchange, residues 102–109 and 117–136. In addition, there is a region that displays exchange behavior consistent with the presence of a conformationally heterogeneous turn. We discuss our data with respect to several structural models proposed for infectious PrP aggregates and highlight HXMS as one of the few techniques well suited to studying natural prions.
Keywords: Amyloid, NMR, Prions, Protein Folding, Protein Structure, Hydrogen Exchange
Introduction
Prion diseases lead to neurodegenerative processes that result in spongiform degeneration and astrocytic gliosis in the central nervous system and are known to exist in genetic, sporadic and transmissible forms (1). The agent that is implicated in initiating disease is the protein PrP,4 which can exist in two isoforms as follows: the benign native form PrP cellular and the infectious and neurotoxic form PrPSc (PrP scrapie) (2). Although non-protein factors, including nucleic acids and lipids, have been shown to facilitate the conversion from PrP cellular to PrPSc and modulate prion infectivity, it is clear that the PrP protein is required for prion passage (3–11). PrP is found in all mammalian and avian genomes and, after maturation, consists of approximately 200 residues with one disulfide bridge, two glycosylation sites, and is attached to the membrane via a C-terminal glycosylphosphatidylinositol anchor. Solution structures of PrP cellular, without post-translational modifications, from several organisms have been solved, showing it to be largely α-helical with an unstructured N terminus of approximately 100 residues (12–17). Experiments have shown that PrP binds copper and may be involved in copper homeostasis (18–20); however, the precise biochemical function of PrP remains unclear. For a detailed discussion, see the work by Aguzzi et al. (21). In contrast to the mostly α-helical, soluble PrP cellular, PrPSc is β-rich, with a protease-resistant core and is present as an aggregate (22). When PrPSc is proteolytically cleaved at its N terminus, it readily associates to form fibrils or rods that retain high levels of infectivity (23). Both electron microscopy and molecular mechanics calculations have been used to generate model structures for infectious PrP (24–27); however, due to their propensity to aggregate and the intrinsic insoluble nature, it has been difficult to obtain any high resolution structural information on infectious PrP with which a single model can be validated.
Determining the rates at which amide hydrogens exchange with solvent hydrogens provides a useful probe for hydrogen bonding in proteins. Exchange requires contact between the amide and solvent and catalysis by an acid or base (base dominates at neutral pH). Amides involved in hydrogen bonds thus cannot exchange without first fluctuating to an “open” state. Simple kinetic theories show that at neutral pH, the rates of exchange are determined by the equilibrium constant for opening and the intrinsic rate of exchange of the amide (determined by the identity of the amino acid and its sequence context) (28). Intrinsic exchange rates have been tabulated (29), so the observed exchange rates can be interpreted in terms of opening equilibria. At pH 7 and 25 °C, the intrinsic fully exposed amide hydrogen exchange rate is on the order of 10 s−1. Recently, numerous groups have employed hydrogen exchange-based experiments to study amyloid fibrils formed from a number of different proteins, including PrP(89–230); note that throughout this study we use the mouse PrP numbering scheme (30–40). Interestingly, in each case it was shown that only subsets of residues in the protein were protected from exchange with solvent, with rather different patterns of protected residues for different proteins. The remaining residues exhibited rapid exchange behavior characteristic of exposed amide groups.
One of the earliest animal models of prion disease was generated by expressing human PrP with a leucine substitution at P101L in transgenic mice at elevated levels; this mutation is implicated in Gerstmann-Sträussler-Scheinker disease (GSS), a familial form of prion disease. These mice developed sporadic GSS that could be serially passed to mice bearing the sensitizing mutation (41, 42). Later work reconstituted prion infectivity from a synthetic protein identified as a 55-residue PrP fragment, PrP(89–143, P101L), that readily associated into amyloid fibrils. Intracerebral injection of the aggregated fibrillar peptide initiated GSS in mice that express full-length PrP bearing the P101L mutation, whereas the nonaggregated peptide did not initiate disease (43). Furthermore, the peptide-initiated disease can be serially transmitted in mice bearing the same mutation (44).
Here, we describe amide hydrogen exchange behavior in the fibrillar form of PrP(89–143, P101L) obtained first at medium sequence resolution with tandem proteolysis, liquid chromatography, and mass spectrometry (HXMS) (45) and then at single amide resolution with NMR spectroscopy. We are able to observe regions of the peptide that are highly protected from exchange and others that are not. The strongly protected regions identify the residues in the hydrogen- bonded core of the amyloid fibrils of PrP(89–143, P101L).
EXPERIMENTAL PROCEDURES
Amyloid Fibril Formation
Amyloid fibrils of PrP(89–143, P101L) were formed by dissolving 10 mg of peptide into 500 μl of 20 mm sodium acetate, 100 mm NaCl, pH 5, and adding 500 μl of acetonitrile. Samples were taken at several time points, and EM images were collected. Small fibrils were seen even a short time after preparing the sample. At longer times primarily longer fibrils with more clearly defined edges were seen. Fibrils used in hydrogen exchange experiments were collected after 3 weeks of incubation in the acetonitrile solution. These fibrillization conditions are the same as used in previous bioassay experiments (43).
Electron Microscopy
Samples were loaded onto 1000 mesh copper grids, coated with Formvar, and glow discharged prior to use. The samples were negatively stained with 3% aqueous uranyl acetate, and images were collected on a FEI Tecnai 12 operating at 120 kV.
Hydrogen Exchange Mass Spectrometry
PrP(89–143, P101L)-peptide used in MS analysis was synthesized using standard Fmoc (N-(9-fluorenyl)methoxycarbonyl) chemistry and subsequent cleavage with trifluoroacetic acid. PrP(89–143, P101L) fibrils were collected by centrifugation at 1300 × g in an Eppendorf tube and washed twice with 10 mm sodium phosphate, pH 7.5. Deuterium exchange was initiated by resuspending the fibrils in 100% D2O buffered with 10 mm sodium phosphate at a pH* of 7.5 (pH* is the reading of a pH meter, uncorrected for isotope and solvent effects) to a final peptide concentration of 50 mg/ml. 5-μl aliquots of the peptide suspension were removed after 1, 6, and 21 h and 1, 3, and 6 weeks. 5 μl of the peptide slurry was removed before exposure to deuterium to serve as the 0-h time point. Exchange samples were prepared for MS analysis by dilution with 55 μl of ice-cold 10 mm sodium phosphate buffer and rapid addition of 90 μl of quench buffer (6.8 m guanidine hydrochloride (GdnHCl), 16.6% glycerol, and 0.8% formic acid), yielding a final pH* of 2.1, followed by 5 min of incubation over thawing ice to fully dissolve the fibrils. Each solution was then divided into 50-μl aliquots in individual autosampler vials and rapidly frozen over powdered dry ice. Samples were stored under powdered dry ice in −80 °C freezers.
The extent of deuteration was determined by quickly thawing the frozen samples over melting ice and passing the solution through two immobilized protease columns (pepsin and fungal protease XIII from Sigma, 100 μl/min, 0.05% TFA). Peptides were separated by reverse-phase high pressure liquid chromatography (HPLC) (Vydac C-18 300A, 50 μl/min with a 6.4–38.4% acetonitrile gradient over 30 min, 4 °C). The effluent was directly injected into a mass spectrometer (ThermoFinnigan LCQ, capillary temperature of 200 °C). Initial identification of proteolytic fragments was determined by tandem MS followed by data analysis using the SEQUEST and DXMS software programs (ThermoFinnigan, San Jose, CA, and Sierra Analytics, Modesto, CA, respectively) (46, 47). Peptide deuteration levels were extracted from the centroid of individual mass envelopes according to the method of Zhang and Smith (48) after accounting for end effects and proline residues (29). Peptide deuteration levels were corrected for back exchange during proteolysis and chromatography based on their retention time in the chromatography step. Back exchange at the beginning of chromatography was found to be 15 and 20% at the end according to fully deuterated reference protein samples.
Cloning, Expression, and Purification
A synthetic gene for PrP(89–143, P101L) with an N-terminal six residue histidine tag and tobacco etch virus cleavage site was designed based on Escherichia coli optimum codon usage and synthesized by GenScript (Piscataway, NJ) in a pUC19 plasmid. This gene was subcloned into the pIVEX 2.4c vector between the NdeI and BamHI sites.
E. coli cells (BL21 (DE3)) containing a plasmid, pACYC, that constitutively expresses LacI were transformed using the hexamine cobalt chloride method (49) and grown on MDG (50) agar plates (1.5%) containing ampicillin and kanamycin at 37 °C for 12 h. A single colony was then picked and inoculated into 2 ml of MDAG media (100 μg/ml ampicillin, 50 μg/ml kanamycin) (50) and allowed to grow at 37 °C for 6 h. To obtain 15N-labeled protein, 1 ml of this starter culture was used to inoculate 1 liter of N-5052 media (50) contained in a 2.8-liter Fernbach flask. Cells were allowed to grow for 48 h at 37 °C with shaking at 240 rpm. Cells expressing 13C,15N-labeled protein were obtained according to the method of Marley et al. (51) with the substitution of MDAG media for LB.
The cells were recovered by centrifugation at 5,000 rpm for 10 min and resuspended in ∼40 ml of 50 mm KPi, pH 8.0, and lysed by sonication at 4 °C. The resulting suspension was centrifuged for 30 min at 30,000 rpm. The supernatant was discarded, and the inclusion body was resolubilized by applying three rounds of resuspension in 6 m GdnHCl, 200 mm NaPi, pH 8.0, 300 mm NaCl, 5 mm β-mercaptoethanol followed by centrifugation for 30 min at 30,000 rpm. The supernatants were pooled together and loaded onto nickel-nitrilotriacetic acid resin (6 ml). The column was then washed with a linear gradient from 6 to 0 m GdnHCl, 50 mm NaPi, pH 8.0, 300 mm NaCl, and 15 mm imidazole. The protein was eluted from the column with 50 mm NaPi, pH 8.0, 300 mm NaCl, and 500 mm imidazole and then dialyzed extensively against 50 mm Tris, pH 8.0, 1 mm DTT, 1 mm EDTA at 4 °C. One milligram of tobacco etch virus protease was added to the dialysis bag, and the cleavage reaction was monitored by HPLC. After complete proteolysis to remove the His tag, the solution was dialyzed against 50 mm NaPi, pH 8.0, 300 mm NaCl, and 15 mm imidazole and then passed over nickel-nitrilotriacetic acid resin to remove both the His tag fragment and tobacco etch virus and desalted using a C18 Sep-Pak cartridge (Waters). The solution was frozen and lyophilized. Dry peptide was stored in a desiccator. On average, 15 mg of purified 15N-labeled PrP(89–143, P101L) was obtained per 1 liter of growth. 0.5 mg of purified 13C,15N-labeled PrP(89–143, P101L) was obtained from 0.5 liter of M9 media.
NMR Experiments (Assignments)
All data were collected on a Bruker Avance DRX-500 or an Avance DRX-800 spectrometer equipped with a cryogenic probe, with samples kept at 25 °C. A single 15N,13C-labeled sample of PrP(89–143, P101L) consisting of 0.5 mg of protein dissolved in 500 μl of DMSO (5% H2O, 0.03% TFA) was used for backbone assignments. Assignment of the 1H,15N HSQC spectrum was accomplished via inter-residue connection of the Cα, Cβ, and N chemical shifts using the HNCA, CBCA(CO)NH, and HNCACB experiments (52–54). Data were processed with NMRPipe (55). For the three-dimensional experiments, data were linear predicted to 128 pts in the 15N dimension. Analysis and integration of data were performed with NMRView or CARA (56, 57).
Hydrogen Exchange NMR Spectroscopy
Fibrillization of 15N-labeled PrP(89–143, P101L) was performed as outlined above. Approximately 0.5 mg of fibrillized 15N-labeled PrP(89–143, P101L) was aliquoted into a separate Eppendorf tube for each exchange time point. Fibrils were resuspended in 10 mm KPi, pD 7.5, and incubated at room temperature for varying periods of time. At the end of each time point, the fibrils were pelleted by centrifugation and washed twice with D2O at 4 °C. After removing the supernatant, the fibrils were frozen over dry ice and lyophilized. Lyophilized fibrils were dissolved in DMSO containing 5% D2O and 0.03% TFA-D, pH* 5. A series of 1H,15N HSQCs was collected over 8 h to correct for exchange with solvent under the quenching NMR sample conditions (32). The dead time from the addition of quenching/denaturing solvent to the fibrils to the acquisition of the first time point of the two-dimensional experiment was ∼10 min (allowing for mixing to aid fibril dissolution and optimizing parameters for the NMR experiment). Each 1H,15N HSQC signal was the average of four transients, with 128 complex points acquired in the t1 dimension, for a total acquisition time of 20 min per two-dimensional data set.
The intrinsic exchange rate in DMSO was determined by first integrating the volumes for each peak in each 1H,15N HSQC spectrum acquired over the 8 h. These individual volumes versus time were then plotted and fitted to single exponentials. The calculated rates from the exponential fits were then used to account for exchange during the dead time of the experiment. The intrinsic exchange rates are shown in supplemental Table S1. A 1–1-echo one-dimensional 1H NMR spectrum was acquired for each sample, and the methyl region of the spectrum was integrated to normalize sample concentrations, which did not deviate by more than 3% between samples. Hydrogen exchange incubation times were 0, 1, 6, and 21 h and 1 and 6 weeks.
RESULTS
Hydrogen Exchange Mass Spectrometry of PrP(89–143, P101L) Fibrils
Synthetic PrP(89–143, P101L) fibrils that had been grown in H2O were incubated in D2O (buffered with sodium phosphate at pD 7.5) and aliquots removed after incubation for 1, 6, and 21 h and 1, 3, and 6 weeks. To determine the deuterium content of the fibrils using electrospray ionization mass spectrometry, the fibrils were redissolved by the addition of an aqueous buffer containing guanidine hydrochloride at low pH to quench hydrogen exchange, limiting back exchange during the mass analysis. To identify sites retaining deuterium in the primary sequence, exchange-quenched peptide solutions were fragmented using two protease columns containing immobilized pepsin and fungal protease XIII. Both proteases retain activity under quenching conditions. The peptide fragments were then separated by reverse-phase HPLC with the eluent running directly into the mass spectrometer. The ability to resolve slowly exchanging sites depends on the degree of fragmentation obtained, which in optimal circumstances can discriminate single amides. Peptides were identified in a reference experiment in which nondeuterated PrP(89–143, P101L) was proteolyzed in the same manner, and eluting fragments were then analyzed by tandem MS (58). Peptides in the deuterated samples were then identified by matching both mass and retention time.
Fig. 1 shows the extent of amide exchange of PrP(89–143, P101L) in fibrils after incubation in D2O for 6 weeks observed using mass spectrometry. 34 “primary” peptide fragments were obtained with good signal to noise ratios in repeated experiments at each time point studied. Exchange in 23 additional “secondary” peptide segments could be analyzed via deuterium level subtraction of N-terminally aligned peptides of different length; N-terminal alignment of peptides is required to correct for end effects. Although PrP(89–143, P101L) was not proteolyzed sufficiently before mass spectrometry to afford single site resolution, a consensus exchange behavior can be approximated by calculating an average of the exchange behavior in which the contribution from each observed peptide is weighted according to the information content of the peptide. The longer the peptide, the less information for localization of exchange it contains. The averaging is performed according to Equation 1,
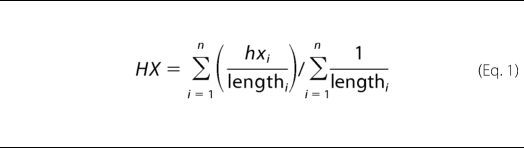 |
where HX is the averaged percent protection; hxi is the percent protection of each peptide i; n represents the number of peptides in which a given residue is observed, and lengthi is the length of each peptide i. Although the data do not afford single amide resolution, it is clear that there is a strongly protected region between residues 119 and 130, increased protection at the C terminus, and two regions from 99 to 107 and 133 to 139 that show significantly lower protection from exchange with solvent.
FIGURE 1.

Exchange behavior of PrP(89–143, P101L) fibrils observed by mass spectrometry. A, shows the mass envelope of the 1+ charge state of the proteolytic fragment of PrP(89–143, P101L) corresponding to residues 133–139. B, mass envelope of the same peptide obtained from PrP fibrils that had been incubated in D2O for 1 week. C, exchange behavior of proteolytic fragments of PrP(89–143, P101L) after 6 weeks of incubation in D2O. The extent of exchange is depicted as a percent protection of exchangeable backbone amides. Each primary peptide is numbered. The identity of the two primary peptides used to obtain each secondary peptide is indicated to the left of the secondary peptides. D, consensus exchange behavior calculated according to Equation 1.
Recombinant Expression and Fibrillization of PrP(89–143, P101L)
Amide hydrogen exchange at individual sites can be detected with NMR, but it requires isotopically labeled protein. To obtain uniformly and isotopically labeled PrP(89–143, P101L) for heteronuclear NMR experiments, we developed an E. coli expression system in which purification of the peptide was expedited by the presence of an N-terminal His6 tag. The supplemental Fig. S1 shows the results from a typical purification process and shows an electron micrograph of PrP(89–143, P101L) fibrils produced from recombinant peptide. Fibrils were grown with the same procedure used to fibrillize synthetic peptide that initiated prion disease in sensitized transgenic mice (43).
Sequence-specific Detection of Hydrogen Exchange in PrP(89–143, P101L) Fibrils
As in the mass spectrometry exchange experiments, detecting the extent of hydrogen exchange in amyloid fibrils with NMR requires the denaturation of fibrils under exchange quenching conditions prior to data collection. Acidic dimethyl sulfoxide (DMSO) is an excellent quench solvent to dissolve amyloid fibrils (31, 32, 34, 59, 60). The minimum rate for amide hydrogen exchange in DMSO occurs at pH* 5 and is typically on the order of 10−5 s−1 (61). All non-proline residues have been assigned for the 55-mer with the exception of the N-terminal glycine that exchanges too quickly to be observed in the NMR experiment and four ambiguous glycine residues that occur in glycine-glycine pairs in the sequence. These residues have very similar exchange behavior; therefore, it is not important to individually assign them.
Dissolution of the fibrils and optimization of the NMR experiment lead to a “dead time” of approximately 10 min for each sample before NMR data collection. Because the DMSO contains a small amount of D2O, multiple 1H,15N HSQC spectra were collected over a period of 6–8 h after dissolving for each exchange time point to determine the intrinsic rate of exchange in DMSO for each amide. This allows one to discriminate between the exchange that occurs during the incubation of the fibrils in deuterated buffer and the exchange that occurs during the dead time for handling the sample. The intrinsic rate of amide exchange under quenching NMR conditions was slow for all residues as confirmed by exchange measurements on fibrils that had not been exposed to deuterated buffers. The measured rate of exchange for all residues was on the order of 10−5 s−1 (data not shown), which corresponds to a half-life of hours, so that little exchange occurs during the measurement time of the experiment.
Given the small number of time points in the experimental data set and the limited range of time sampled, the slow exchange rates of the backbone cannot be quantified with certainty. Therefore, we present the exchange data using a percent intensity (IP) given by Equation 2,
where It is the integrated intensity (corrected for back exchange and normalized by concentration) for a given amide cross-peak at time t, and I0 is the integrated intensity (corrected for back exchange and normalized by concentration) for the 0-h time point. The calculated IP values for each residue for each experimental time point are shown in Table 1. Four types of exchange behavior were observed and are summarized in Fig. 2. The amide hydrogens of residues 90 and 143, the terminal amides, exchange completely before the first time point. Residues 95–101 and 137–141 exchange more slowly, with a half-life of approximately a week, but are completely replaced after 6 weeks. Residues 110–116 exchange slowly as well, with similar extents of exchange after a week as residues 95–101, but retain IP ∼50 even after 6 weeks, which implies a multiexponential behavior in the rates. Finally, residues 102–109 and 117–135 remain highly protected even after 6 weeks (IP >90).
TABLE 1.
Percent intensity (see Equation 2)
A listing of measured IP (Equation 2) values for each residue of PrP(89–143, P101L) amyloid fibrils from the hydrogen exchange experiment. The exchange behavior of the four ambiguous glycine residues (marked by an asterisk) is reported as an average, the exchange behavior of each was similar.

FIGURE 2.

Exchange behavior of PrP(89–143, P101L) fibrils observed with NMR. A, shows the assigned 1H,15N HSQC spectrum of PrP(89–143, P101L) under the conditions of a typical 0-h time point, dissolving fibrils that had not been exposed to deuterium in DMSO containing 5% D2O and 0.03% TFA-D, pH* 5. The four ambiguous glycine residues are denoted with an asterisk. B, summary of the exchange behavior of PrP(89–143, P101L) fibrils.
DISCUSSION
Previous solid-state NMR studies of selectively and isotopically labeled PrP(89–143, P101L) fibrils from our laboratory have demonstrated that residues 112–124 adopt an extended β-sheet conformation with evidence for conformational heterogeneity between residues 112 and 114. Furthermore, multiple quantum NMR experiments indicated that PrP(89–143, P101L) does not adopt an in-register parallel β-sheet (62). To extend our knowledge of the conformation of fibrillar PrP(89–143, P101L), we measured the extent of backbone hydrogen exchange at various time points up to 6 weeks of exchange. In the most favorable cases, following hydrogen exchange with tandem proteolysis and LC-MS (HXMS) can lead to single amide resolution. Moreover, HXMS is ideally suited to study prions as it does not require isotopically labeled samples; thus, it is one of the few techniques suited to studying the conformation of PrPSc obtained directly from infected animals. The exchange behavior of natural PrPSc could then be compared with that of different constructs of recombinant or synthetic PrP, which are much more tractable to analysis.
In our MS study of exchange in fibrillar PrP(89–143, P101L), the limited number of peptide fragments observed with sufficient signal to noise for analysis precluded single amide resolution of exchange behavior. However, the data from mass spectrometry clearly demonstrate that there are regions of the peptide that are strongly protected from exchange with solvent, whereas other regions are much less protected from exchange. This is expected as the 55-residue peptide is too long to exist as a single, stably hydrogen-bonded, extended strand within the fibril as the fibril diameter is known to be ∼8 nm. The exchange profile reveals a highly protected β-strand region with residues 119–130, flanked by two segments that are within a less protected region, encompassing residues 99–107 and 133–139. Fragmentation was poor near the N terminus preventing analysis of that region.
To generate a higher resolution map of protection, more detailed exchange data were required. After assigning the resonances of PrP(89–143, P101L), we collected amide exchange profiles by following exchange with NMR. Many of the residues show the slow exchange behavior expected from amides involved in stable hydrogen bonds. Two regions, encompassing residues 102–109 and 117–135, remained protected from exchange after 6 weeks and likely constitute the β-sheet core of the fibrils. The high degree of protection is not surprising in that some residues in β2-microglobulin amyloid have been shown to have exchange half-lives of weeks (39) and some residues in Aβ amyloid have been shown to have half-lives of months (33, 34). Another set of residues, close to the N terminus and at the C terminus, were completely exchanged after 6 weeks of incubation in D2O. Although residues 95–101 and 137–141 have moderate protection factors that are typically afforded by secondary structure conformations in soluble proteins, they exchange more rapidly than the β-sheet core residues of other reported amyloid fibrils. The lower protection in these two regions is consistent with the mass spectrometry measurements.
Interestingly, residues 110–116 show exchange behavior characteristic of multiple exponentials. This group of residues undergoes exchange to ∼50% after 1 week. However, after 6 weeks, no further exchange has occurred. This behavior can be explained by the presence of two (or more) conformations for these residues present in the sample with different exchange rates. One component has a half-life of approximately 1 week, and the second component has a much longer half-life. The mass spectrometry data in this region are also consistent with intermediate protection, but we lack sufficient sequence resolution to observe a bimodal deuterium distribution for this region of the peptide that would confirm the presence of two populations of conformers, one more accessible to exchange with solvent than the other. Furthermore, the residues 110–116 encompass the region of the peptide that displayed conformational heterogeneity in past solid-state NMR experiments (62). It is possible that different conformers could give rise to prion strains observed in vivo (63).
Currently, there are several different models for the conformation of PrP in prions (64). One is a left-handed β-helix proposed by Govaerts et al. (24) from two-dimensional electron crystallography data on proteolyzed native PrPSc. Another is a “spiral” model identified in a molecular dynamics study of PrP fibrillization that was also shown to be consistent with the electron crystallography data (27). Neither the β-helix nor the spiral model requires the fibril to be formed by an in-register parallel β-sheet as seen in Aβ fibrils (34), consistent with previous observations from solid-state NMR (62). A solid-state NMR study on fibrils of another prion variant, human PrP(Y145Stop), has demonstrated that the N terminus of the protein, up to residue 112, remains highly mobile in the fibril and that residues 112–115, 118–122, and 130–139 form β-strands (65); the corresponding regions in the mouse protein are residues 111–114, 117–121, and 129–138. Another recent structural model has been obtained from HXMS experiments conducted on prion fibrils of PrP(89–230), in that work protection from exchange is only observed in the C-terminal region of the protein, approximately residues 168–220 (3, 40). This in disagreement with the previously discussed structural models and also lies outside of the region of interest studied in this work (residues 89–143). As the Y145Stop variant has been identified in human prion disease, the prion determining region of PrP cannot be wholly contained to the C terminus of the protein (66, 67). Moreover, it is well documented that different strains of prions can assume drastically different conformations. For example, the highly protected core of Sup35 yeast prions consists of ∼40 residues in one strain and ∼70 residues in a different strain (68). Finally, the protease-resistant PrP present in patients with GSS contains a significant amount of 7–8-kDa PrP fragments. These fragments possess ragged N termini beginning around residue 80 and also ragged C termini ending around residue 150, encompassing the region of interest in this study (69, 70). Furthermore, it has been shown that the synthetic prions used in the HXMS study by Lu et al. (40, 71) are more susceptible to degradation by proteinase K than “classical” PrPSc and are similar to a subpopulation of PrPSc, recently identified in patients, rather than the classical PrPSc. In fact, further HXMS experiments from the Surewicz laboratory demonstrate that PrPSc-seeded recombinant PrP aggregates are significantly more protected from exchange than the spontaneously aggregated proteins studied by Lu et al. (40, 71). Also, similarly to PrP(89–143, P101L) fibrils, the PrPSc-seeded aggregates contain a protected region between residues 116 and 132 (72). For the reasons outlined above, we argue that the precise location of the fibrillar core in prion aggregates varies significantly based on both the prion strain, the particular PrP construct employed, and the conditions under which the peptide is aggregated.
In the β-helical model, three 5-residue strands are joined by a single residue that has torsion angles characteristic of a left-handed helix to form a triangular 18-residue “rung,” and these rungs are then stacked in a stably hydrogen-bonded network, including a hydrogen bond at each residue, to form the β-helix. Our exchange data show that the region from 102 to 136 is resistant to exchange with some degree of conformational heterogeneity from 110 to 116. This span of the peptide is essentially long enough to support two rungs of a β-helix, but PrP(89–143, P101L) is rather unlikely to adopt this conformation as in nature the minimal number of “rungs” found experimentally to date is four (seen in the structure of the C-terminal domain of N-acetylglucosamine-1-phosphate uridyltransferase from E. coli, Protein Data Bank code 1FXJ). However, in the fibril, monomers could be packing against each other to extend the helix. More difficult to reconcile than the length issue is the fact that the previous solid-state NMR experiments on PrP(89–143, P101L) did not find any evidence of turn conformations between residues 112 and 118 and between residues 122 and 124 (62). If the core of the fibril is formed by a β-helix, some of these residues should be clearly involved in turns.
The core of the spiral model of PrPSc consists of four short β-strands in which three short strands (E1, E2, E3; residues 115–118, 128–131, 159–163, respectively) form an anti-parallel intramolecular sheet where the extra strand (E4, residues 134–139) adds to the sheet on an adjacent molecule through a parallel E1-E4 interface. E3 is not essential for the formation of the intermolecular interface; however, the positions of the protected residues in PrP(89–143, P101L) fibrils are not consistent with the location of E1, E2, and E4 in the model, although the spiral architecture could explain the protection factors and the observation that PrP(89–143, P101L) fibrils do not form an in-register β-sheet.
In summary, our hydrogen exchange experiments identify two regions of the protein, encompassing residues 102–109 and 117–135, that display strong protection from exchange with solvent even after 6 weeks and are likely to constitute the core of the fibril. Two other regions, encompassing residues 95–101 and 137–141, exchange completely with solvent after 1 week, an exchange time expected for residues involved in hydrogen bonds but not in the amyloid core. Interestingly, the region of the protein between residues 110 and 116 exhibits intermediate exchange behavior that can be explained by the existence of two populations of conformers in the fibril that exchange with solvent at different rates. This interpretation of the intermediate protection factors is corroborated by our previous finding that this region possesses some conformational heterogeneity (62). The core of the PrP(89–43, P101L) amyloid fibril consists of two highly protected β-strands (β1 and β2, residues 102–109 and 117–136, respectively) that are joined by a hydrogen-bonded yet conformationally heterogeneous turn. The overall fibril architecture must be different from that seen in fibrils of Aβ as the PrP fibrils have been shown to not be in register. Assuming slightly different locations of strands E1, E2, and E4 from the spiral model, the core of the fibril could form from an intramolecular sheet between β1 and the first section of β2 where the intermolecular interface is between the second section of β2 and β1 on the adjacent molecule. Finally, we stress that HXMS is one of the few techniques that can assess the conformation of natural prions and will be extremely valuable in comparing synthetic prions to those obtained directly from infected animals.
Supplementary Material
Acknowledgments
We thank Prof. Stan Prusiner and co-workers for discussions about prion behavior and Dr. David King for the synthesis of PrP(89-143, P101L).
This work was supported by the Army Prion Research Program Grant DMAD17-03-1-0476. Development of the HXMS methodology was supported by National Institutes of Health Grants AI076961, AI081982, AI2008031, AI072106, AI068730, GM037684, GM020501, and GM066170 (to V. L. W.), Innovative Technologies for the Molecular Analysis of Cancer Program Grants CA099835 and CA118595 (to V. L. W.), and Discovery Grant UC10591 from the University of California IUCRP Program, BiogenIDEC corporate sponsor (to V. L. W.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1, Fig. S1, and an additional reference.
- PrP
- prion protein
- HXMS
- tandem proteolysis, liquid chromatography and mass spectrometry
- PrPC
- PrP cellular
- PrPSc
- PrP scrapie
- GdnHCl
- guanidine hydrochloride
- HSQC
- heteronuclear single quantum correlation
- GSS
- Gerstmann-Sträussler-Scheinker.
REFERENCES
- 1.DeArmond S. J., Prusiner S. B. (1996) Current Topics in Microbiology and Immunology (Prusiner S. B. ed) Vol. 207, pp. 125–146, Springer, New York: [DOI] [PubMed] [Google Scholar]
- 2.Prusiner S. B. (2004) in Prion Biology and Diseases (Prusiner S. B. ed) pp. 89–141, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 3.Legname G., Baskakov I. V., Nguyen H. O., Riesner D., Cohen F. E., DeArmond S. J., Prusiner S. B. (2004) Science 305, 673–676 [DOI] [PubMed] [Google Scholar]
- 4.Wang F., Wang X., Yuan C. G., Ma J. (2010) Science 327, 1132–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deleault N. R., Harris B. T., Rees J. R., Supattapone S. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 9741–9746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deleault N. R., Geoghegan J. C., Nishina K., Kascsak R., Williamson R. A., Supattapone S. (2005) J. Biol. Chem. 280, 26873–26879 [DOI] [PubMed] [Google Scholar]
- 7.Geoghegan J. C., Valdes P. A., Orem N. R., Deleault N. R., Williamson R. A., Harris B. T., Supattapone S. (2007) J. Biol. Chem. 282, 36341–36353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baron G. S., Magalhães A. C., Prado M. A., Caughey B. (2006) J. Virol. 80, 2106–2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gabizon R., McKinley M. P., Prusiner S. B. (1987) Proc. Natl. Acad. Sci. U.S.A. 84, 4017–4021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Silva J. L., Lima L. M., Foguel D., Cordeiro Y. (2008) Trends Biochem. Sci. 33, 132–140 [DOI] [PubMed] [Google Scholar]
- 11.Lima L. M., Cordeiro Y., Tinoco L. W., Marques A. F., Oliveira C. L., Sampath S., Kodali R., Choi G., Foguel D., Torriani I., Caughey B., Silva J. L. (2006) Biochemistry 45, 9180–9187 [DOI] [PubMed] [Google Scholar]
- 12.Lysek D. A., Schorn C., Nivon L. G., Esteve-Moya V., Christen B., Calzolai L., von Schroetter C., Fiorito F., Herrmann T., Güntert P., Wüthrich K. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 640–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calzolai L., Lysek D. A., Pérez D. R., Güntert P., Wüthrich K. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 651–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gossert A. D., Bonjour S., Lysek D. A., Fiorito F., Wüthrich K. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 646–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.James T. L., Liu H., Ulyanov N. B., Farr-Jones S., Zhang H., Donne D. G., Kaneko K., Groth D., Mehlhorn I., Prusiner S. B., Cohen F. E. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 10086–10091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hornemann S., Schorn C., Wüthrich K. (2004) EMBO Rep. 5, 1159–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Riek R., Hornemann S., Wider G., Billeter M., Glockshuber R., Wüthrich K. (1996) Nature 382, 180–182 [DOI] [PubMed] [Google Scholar]
- 18.Brown D. R. (2003) J. Neurochem. 87, 377–385 [DOI] [PubMed] [Google Scholar]
- 19.Garnett A. P., Viles J. H. (2003) J. Biol. Chem. 278, 6795–6802 [DOI] [PubMed] [Google Scholar]
- 20.Sakudo A., Lee D. C., Yoshimura E., Nagasaka S., Nitta K., Saeki K., Matsumoto Y., Lehmann S., Itohara S., Sakaguchi S., Onodera T. (2004) Biochem. Biophys. Res. Commun. 313, 850–855 [DOI] [PubMed] [Google Scholar]
- 21.Aguzzi A., Baumann F., Bremer J. (2008) Annu. Rev. Neurosci. 31, 439–477 [DOI] [PubMed] [Google Scholar]
- 22.Pan K. M., Baldwin M., Nguyen J., Gasset M., Serban A., Groth D., Mehlhorn I., Huang Z., Fletterick R. J., Cohen F. E., Prusiner S. B. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 10962–10966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prusiner S. B., McKinley M. P., Bowman K. A., Bolton D. C., Bendheim P. E., Groth D. F., Glenner G. G. (1983) Cell 35, 349–358 [DOI] [PubMed] [Google Scholar]
- 24.Govaerts C., Wille H., Prusiner S. B., Cohen F. E. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 8342–8347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wille H., Michelitsch M. D., Guenebaut V., Supattapone S., Serban A., Cohen F. E., Agard D. A., Prusiner S. B. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 3563–3568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DeMarco M. L., Silveira J., Caughey B., Daggett V. (2006) Biochemistry 45, 15573–15582 [DOI] [PubMed] [Google Scholar]
- 27.DeMarco M. L., Daggett V. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 2293–2298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Englander S. W., Mayne L., Bai Y., Sosnick T. R. (1997) Protein Sci. 6, 1101–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bai Y., Milne J. S., Mayne L., Englander S. W. (1993) Proteins Struct. Funct. Genet. 17, 75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Del Mar C., Greenbaum E. A., Mayne L., Englander S. W., Woods V. L., Jr. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 15477–15482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoshino M., Katou H., Hagihara Y., Hasegawa K., Naiki H., Goto Y. (2002) Nat. Struct. Biol. 9, 332–336 [DOI] [PubMed] [Google Scholar]
- 32.Ippel J. H., Olofsson A., Schleucher J., Lundgren E., Wijmenga S. S. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 8648–8653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kheterpal I., Zhou S., Cook K. D., Wetzel R. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 13597–13601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lührs T., Ritter C., Adrian M., Riek-Loher D., Bohrmann B., Döbeli H., Schubert D., Riek R. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 17342–17347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nazabal A., Maddelein M. L., Bonneu M., Saupe S. J., Schmitter J. M. (2005) J. Biol. Chem. 280, 13220–13228 [DOI] [PubMed] [Google Scholar]
- 36.Olofsson A., Ippel J. H., Wijmenga S. S., Lundgren E., Ohman A. (2004) J. Biol. Chem. 279, 5699–5707 [DOI] [PubMed] [Google Scholar]
- 37.Olofsson A., Sauer-Eriksson A. E., Ohman A. (2006) J. Biol. Chem. 281, 477–483 [DOI] [PubMed] [Google Scholar]
- 38.Wang S. S., Tobler S. A., Good T. A., Fernandez E. J. (2003) Biochemistry 42, 9507–9514 [DOI] [PubMed] [Google Scholar]
- 39.Yamaguchi K., Katou H., Hoshino M., Hasegawa K., Naiki H., Goto Y. (2004) J. Mol. Biol. 338, 559–571 [DOI] [PubMed] [Google Scholar]
- 40.Lu X., Wintrode P. L., Surewicz W. K. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 1510–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hsiao K. K., Scott M., Foster D., Groth D. F., DeArmond S. J., Prusiner S. B. (1990) Science 250, 1587–1590 [DOI] [PubMed] [Google Scholar]
- 42.Hsiao K. K., Groth D., Scott M., Yang S. L., Serban H., Rapp D., Foster D., Torchia M., Dearmond S. J., Prusiner S. B. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 9126–9130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaneko K., Ball H. L., Wille H., Zhang H., Groth D., Torchia M., Tremblay P., Safar J., Prusiner S. B., DeArmond S. J., Baldwin M. A., Cohen F. E. (2000) J. Mol. Biol. 295, 997–1007 [DOI] [PubMed] [Google Scholar]
- 44.Tremblay P., Ball H. L., Kaneko K., Groth D., Hegde R. S., Cohen F. E., DeArmond S. J., Prusiner S. B., Safar J. G. (2004) J. Virol. 78, 2088–2099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Woods V. L., Jr., Hamuro Y. (2001) J. Cell. Biochem. Suppl. 37, 89–98 [DOI] [PubMed] [Google Scholar]
- 46.Pantazatos D., Kim J. S., Klock H. E., Stevens R. C., Wilson I. A., Lesley S. A., Woods V. L., Jr. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 751–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Englander J. J., Del Mar C., Li W., Englander S. W., Kim J. S., Stranz D. D., Hamuro Y., Woods V. L., Jr. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 7057–7062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Z., Smith D. L. (1993) Protein Sci. 2, 522–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hanahan D. (1983) J. Mol. Biol. 166, 557–580 [DOI] [PubMed] [Google Scholar]
- 50.Studier F. W. (2005) Protein Expr. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 51.Marley J., Lu M., Bracken C. (2001) J. Biomol. NMR 20, 71–75 [DOI] [PubMed] [Google Scholar]
- 52.Grzesiek S., Bax A. (1992) J. Magn. Res. 96, 432–440 [Google Scholar]
- 53.Grzesiek S., Bax A. (1993) J. Biomol. NMR 3, 185–204 [DOI] [PubMed] [Google Scholar]
- 54.Wittekind M., Mueller L. (1993) J. Magn. Res. B101, 201–205 [Google Scholar]
- 55.Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 56.Johnson B. A., Blevins R. A. (1994) J. Biomol. NMR 4, 603–614 [DOI] [PubMed] [Google Scholar]
- 57.Keller R. (2004) The Computer Aided Resonance Assignment Tutorial, Cantina Verlag, Goldau, Switzerland [Google Scholar]
- 58.Eng J. K., McCormack A. L., Yates J. R. (1994) J. Am. Soc. Mass Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 59.Hirota-Nakaoka N., Hasegawa K., Naiki H., Goto Y. (2003) J. Biochem. 134, 159–164 [DOI] [PubMed] [Google Scholar]
- 60.Ritter C., Maddelein M. L., Siemer A. B., Lührs T., Ernst M., Meier B. H., Saupe S. J., Riek R. (2005) Nature 435, 844–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Y. Z., Paterson Y., Roder H. (1995) Protein Sci. 4, 804–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lim K. H., Nguyen T. N., Damo S. M., Mazur T., Ball H. L., Prusiner S. B., Pines A., Wemmer D. E. (2006) Solid State Nuclear Magnetic Resonance 29, 183–190 [DOI] [PubMed] [Google Scholar]
- 63.Legname G., Nguyen H. O., Baskakov I. V., Cohen F. E., Dearmond S. J., Prusiner S. B. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 2168–2173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cobb N. J., Surewicz W. K. (2009) Biochemistry 48, 2574–2585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Helmus J. J., Surewicz K., Nadaud P. S., Surewicz W. K., Jaroniec C. P. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 6284–6289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ghetti B., Piccardo P., Spillantini M. G., Ichimiya Y., Porro M., Perini F., Kitamoto T., Tateishi J., Seiler C., Frangione B., Bugiani O., Giaccone G., Prelli F., Goedert M., Dlouhy S. R., Tagliavini F. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 744–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kundu B., Maiti N. R., Jones E. M., Surewicz K. A., Vanik D. L., Surewicz W. K. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 12069–12074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Toyama B. H., Kelly M. J., Gross J. D., Weissman J. S. (2007) Nature 449, 233–237 [DOI] [PubMed] [Google Scholar]
- 69.Parchi P., Chen S. G., Brown P., Zou W., Capellari S., Budka H., Hainfellner J., Reyes P. F., Golden G. T., Hauw J. J., Gajdusek D. C., Gambetti P. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 8322–8327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Piccardo P., Liepnieks J. J., William A., Dlouhy S. R., Farlow M. R., Young K., Nochlin D., Bird T. D., Nixon R. R., Ball M. J., DeCarli C., Bugiani O., Tagliavini F., Benson M. D., Ghetti B. (2001) Am. J. Pathol. 158, 2201–2207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bocharova O. V., Breydo L., Salnikov V. V., Gill A. C., Baskakov I. V. (2005) Protein Sci. 14, 1222–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smirnovas V., Kim J. I., Lu X., Atarashi R., Caughey B., Surewicz W. K. (2009) J. Biol. Chem. 284, 24233–24241 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.