

Abstract
Copper is an essential trace element that functions in a diverse array of biochemical processes that include mitochondrial respiration, neurotransmitter biogenesis, connective tissue maturation, and reactive oxygen chemistry. The Ctr1 protein is a high-affinity Cu+ importer that is structurally and functionally conserved in yeast, plants, fruit flies, and humans and that, in all of these organisms, is localized to the plasma membrane and intracellular vesicles. Although intestinal epithelial cell-specific deletion of Ctr1 in mice demonstrated a critical role for Ctr1 in dietary copper absorption, some controversy exists over the localization of Ctr1 in intestinal epithelial cells in vivo. In this work, we assess the localization of Ctr1 in intestinal epithelial cells through two independent mechanisms. Using immunohistochemistry, we demonstrate that Ctr1 localizes to the apical membrane in intestinal epithelial cells of the mouse, rat, and pig. Moreover, biotinylation of intestinal luminal proteins from mice fed a control or a copper-deficient diet showed elevated levels of both total and apical membrane Ctr1 protein in response to transient dietary copper limitation. Experiments in cultured HEK293T cells demonstrated that alterations in the levels of the glycosylated form of Ctr1 in response to copper availability were a time-dependent, copper-specific posttranslational response. Taken together, these results demonstrate apical localization of Ctr1 in intestinal epithelia across three mammalian species and suggest that increased Ctr1 apical localization in response to dietary copper limitation may represent an adaptive response to homeostatically modulate Ctr1 availability at the site of intestinal copper absorption.
Keywords: Copper, Intestine, Mammal, Metals, Transport Metals
Introduction
Copper is an essential trace element that supports catalysis by a wide range of enzymatic activities that include lysyl oxidase, copper/zinc-superoxide dismutase, dopamine β-monooxygenase, and others that carry out key physiological functions (1–5). Mammalian copper deficiency leads to defects in growth, development, and cognition, and recent reports indicate that many patients undergoing bariatric surgery or with other conditions that lead to a copper deficiency may experience severe myeloneuropathy (6, 7).
Consequently, it is important to identify those components that carry out copper uptake, distribution, and utilization and to understand how these components are regulated in their abundance, activity, or subcellular localization. Many proteins have been identified that play key functions in mammalian copper homeostasis, yet we know relatively little about their detailed mechanisms of action in cultured cells or in distinct tissues or cell types in vivo (3, 4, 8).
Previous studies indicate that dietary copper absorption occurs largely in the small intestine (9). Moreover, mammals preconditioned on a copper-deficient diet demonstrate strongly enhanced uptake of copper in a subsequent exposure compared with control subjects, suggesting a regulatory mechanism for controlling dietary copper uptake in response to copper status (10–12). Although the mechanisms underlying this regulation are not understood, Ctr1 is a high-affinity copper(I) transporter that is conserved in overall structure and function from yeast to humans (3, 13, 14). In both yeast and cultured mammalian cells, Ctr1 has been shown to traffic from the plasma membrane to intracellular compartments both constitutively and in response to elevated copper levels, prompting speculation that intestinal Ctr1 may play a significant role in intestinal copper absorption and its regulation (15–17). Mice with an intestinal epithelial cell (IEC)3-specific Ctr1 knock-out (Ctr1int/int) show severe copper deficiency in peripheral tissues and die ∼3 weeks after birth (18). The ability to rescue Ctr1int/int mice with a single intraperitoneal copper injection to bypass intestinal absorption strongly suggests that intestinal epithelial Ctr1 plays a critical role in dietary copper absorption (18).
Previous reports suggest that Ctr1 resides on the apical membrane and in intracellular vesicular compartments of rodent IEC (Refs. 11, 18, and 19 and reviewed in Ref. 8). However, a recent report by Zimnicka et al. (20) suggests that Ctr1 resides exclusively on the basolateral membrane of Caco-2 cells, a cell culture model for polarized epithelial cells, and on the basolateral membrane of mouse IEC. As copper acquisition is critical for normal growth, development, cognition, and neurological function (1–5) and Ctr1 plays a vital role in dietary copper absorption through the intestine (18), it is important to clearly establish the location of Ctr1 in IEC in vivo. In this study, we demonstrate apical localization of Ctr1 in mouse, rat, and pig intestine using immunohistochemistry analysis. Moreover, intestinal luminal cell surface biotinylation and cell culture experiments demonstrate elevated levels of Ctr1 at the apical surface in response to a copper limitation and suggest that changes in the abundance of apical Ctr1, at a posttranslational level, may contribute to the regulation of dietary copper uptake in the intestine as a function of copper status.
MATERIALS AND METHODS
Antibodies
Generation of the anti-Ctr1 antibody was described and characterized previously (18). The anti-Ctr1 amino-terminal antibody was a gift from Dr. Jack H. Kaplan, University of Illinois (Chicago, IL). The anti-hephaestin antibody (HEPH11-A) was purchased from Alpha Diagnostics (San Antonio, TX). The anti-sodium/potassium/chloride co-transporter antibody (NKCC; T4) was obtained from the Developmental Studies Hybridoma Bank at the Department of Biology, University of Iowa (Iowa City, IA). The anti-superoxide dismutase antibody (SOD1; SOD-100) was purchased from Assay Designs (Ann Arbor, MI). The anti-copper chaperone for superoxide dismutase antibody (CCS; FL274) was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The anti-Cox IV subunit IV antibody was purchased from MitoScience Inc. (Eugene, OR). The anti-sodium/glucose co-transporter antibody (SGLT1; ab14686) was purchased from Abcam (Cambridge, MA). The anti-actin antibody (A5060) was purchased from Sigma. The anti-Na+/K+-ATPase α1 subunit antibody was purchased from Millipore (Temecula, CA). The anti-β-tubulin antibody (9F3) was purchased from Cell Signaling Technology (Danvers, MA). Alexa Fluor 488 anti-rabbit IgG and Alexa Fluor 568 anti-mouse IgG were obtained from Invitrogen.
Cell Culture, Animals, and Tissue Preparations
The Ctr1 knock-out (Ctr1−/−, E8) and WT E3 mouse embryonic fibroblasts and their culture conditions were described previously (21). At ∼60% confluency, cells were rinsed with ice-cold PBS (pH 7.4), scraped in the same buffer to harvest, and centrifuged at 200 × g for 5 min at 4 °C. The cells were lysed in cell lysis buffer (PBS (pH 7.4), 1% (v/v) Triton X-100, 0.1% (w/v) SDS, 1 mm EDTA) containing a protease inhibitor mixture (Complete, EDTA-free; Roche) at 4 °C for >1 h with gentle agitation, followed by centrifugation at 16,000 × g for 20 min at 4 °C to remove insoluble material. Protein concentrations were measured using a Bio-Rad DC protein assay kit (500-0002, Bio-Rad). The indicated amount of protein extract was fractionated by SDS-gel electrophoresis on 4–20% gradient gels (Bio-Rad) and immunoblotted. The antibodies used are described in the figure legends. Human embryonic kidney cells (HEK293T) were cultured in DMEM supplemented with 10% (v/v) fetal bovine serum. The cells were pretreated with 100 mg/ml cycloheximide for 20 min; treated with CuSO4, bathocuproine disulfonate (BCS), ferric ammonium sulfate, or ZnCl2; and incubated as described in the figure legends. Total protein was extracted and subjected to immunoblotting as described above.
The intestinal Ctr1 knock-out mice (Ctr1int/int) and control mice (Ctr1flox/flox and Ctr1flox/+) were generated as described previously (18). For the copper-deficient diet treatment, WT mice (C57BL/6J, Jackson Laboratory, Bar Harbor, ME) 4 weeks of age received the control diet (CA.170481) or the copper-deficient diet (TD.80388) (Harlan Teklad, Madison, WI) for 5 days and were killed, and the tissues were processed. For the immunohistochemistry analysis, the mice were killed and perfused with PBS followed by 4% (w/v) paraformaldehyde/PBS. An ∼1-cm long section of small intestine, positioned ∼0.5–1 cm from the stomach, was dissected; cut longitudinally to open the tract; and fixed with the same fixative solution overnight at 4 °C with gentle shaking. Rat duodenal samples were prepared at the University of Minnesota (Duluth, MN) using a similar protocol. Tissue was harvested from 7-week-old male Sprague-Dawley rats fed a copper-adequate, semipurified diet (TD.08584). A 5-cm portion of the pig jejunum, located 60 cm from the terminus of the stomach, was dissected; cut longitudinally; rinsed with ice-cold PBS; and fixed in 4% (w/v) paraformaldehyde/PBS at 4 °C overnight. For preparation of copper-deficient hearts, WT postnatal day 2 pups were treated with a control or copper-deficient diet (as described above) by feeding their dams. The pups were weaned at 3 weeks of age. Immediately after weaning, mice were maintained with the same diet. At 5 weeks of age, the mice were killed, their hearts were dissected, and the total protein was extracted and analyzed by immunoblotting as described above.
Immunohistochemistry and Confocal Immunofluorescence Microscopy
Fixed animal tissues were immersed in 70% (v/v) ethanol at 4 °C overnight, embedded in paraffin, and sectioned at a thickness of 8 μm. The deparaffinized sections were heated at 95 °C in Tris-EDTA buffer (10 mm Tris base, 1 mm EDTA, 0.05% (v/v) Tween 20 (pH 9.0)) for 20 min to expose the antigen. For immunohistochemistry analysis, samples were subjected to 3% (v/v) hydrogen peroxide in PBS for 15 min; blocked with 1% (w/v) BSA, 5% (v/v) normal goat serum in PBS for 30 min; and incubated with primary antibody (as described in the figure legends) for 1 h at room temperature. Secondary antibody incubation and detection were performed following the manufacturer's recommendations (Super Sensitive IHC Detection Systems, BioGenex, San Ramon, CA). After immunohistochemical staining, tissues were counterstained with hematoxylin. For confocal immunofluorescence microscopy, samples were blocked with 1% (w/v) BSA, 5% (v/v) normal goat serum in PBS for 30 min and incubated with primary antibody (as described in the figure legends) for 1 h at room temperature. Sections were incubated with a mixture of Alexa Fluor 488 anti-rabbit IgG and Alexa Fluor 568 anti-mouse IgG (1:250 dilution in 1% (w/v) BSA in PBS) overnight at 4 °C, treated with DAPI, washed with PBS, mounted, and visualized with a Leica SP5 confocal microscope.
Biotinylation Experiments
The entire small intestine from PBS-perfused mice was immediately dissected, cut longitudinally, and rinsed with ice-cold PBS containing a protease inhibitor mixture (Complete, EDTA-free). All buffers used in subsequent procedures contained the same protease inhibitor mixture. Biotinylation was performed based on the manufacturer's specifications (Sulfo-NHS-SS-Biotin, 21331, Thermo Scientific, Rockford, IL). Small intestine samples were incubated in 10 ml of PBS containing Sulfo-NHS-SS-Biotin at a final concentration of 0.48 mg/ml for 30 min at room temperature with gentle agitation. Samples were washed three times with ice-cold PBS, and IEC were isolated by incubation of the intestines in PBS containing 1.5 mm EDTA for 1 h at 4 °C with gentle agitation, as described previously (18). The IEC were collected by centrifugation at 400 × g for 5 min at 4 °C, followed by washing three times with ice-cold PBS. Total protein was extracted by incubation of IEC with cell lysis buffer at 4 °C for 1 h with gentle agitation, followed by centrifugation at 16,000 × g for 20 min at 4 °C to remove insoluble material.
Affinity purification of biotinylated proteins was performed using the Pierce monomeric avidin kit (20227, Thermo Scientific). Columns (bed volume, ∼0.5 ml) of monomeric avidin-agarose (20228, Thermo Scientific) were prepared at room temperature by equilibration with 2 ml of cell lysis buffer, 2 ml of biotin blocking/elution buffer containing 1% (v/v) Triton X-100 and 0.1% (w/v) SDS, 3 ml of regeneration buffer supplied with 1% (v/v) of Triton X-100 and 0.1% (w/v) SDS, and then 3 ml of cell lysis buffer. Protein extracts of 0.5-ml volume containing 1 mg of protein (adjusted in cell lysis buffer) were applied to the columns. Columns were washed with 3 ml of cell lysis buffer, and bound protein was eluted with 3 ml of biotin blocking/elution buffer containing 1% (v/v) Triton X-100 and 0.1% (w/v) SDS. Elution fractions were concentrated using a centrifugal filter concentrator (Centricon-10, Millipore Corp., Billerica, MA).
RESULTS
Re-evaluation of Anti-Ctr1 Polyclonal Antibody Specificity
Given the importance of Ctr1 in intestinal copper absorption and the reports of apical (11, 18, 19) or basolateral (20) localization in IEC, experiments were carried out to further ascertain Ctr1 subcellular localization in IEC. Fig. 1A shows a model for Ctr1 topology based on previous protease shaving experiments, epitope-specific immunoreactivity, and cryo-EM studies (22, 23). An ∼56-amino acid amino-terminal domain is proposed to be extracellular (for plasma membrane-localized Ctr1) and luminal (for vesicular Ctr1), with an ∼53-residue cytoplasmic loop, three transmembrane domains, and an ∼15-amino acid cytosolic tail. A polyclonal antibody was raised in rabbits against an 18-amino acid peptide derived from the sequence of the cytosolic loop of human Ctr1 (Fig. 1A) and affinity-purified as described previously (18). Because this antibody had been stored at −80 °C for several years, we recharacterized its specificity for endogenous Ctr1 by immunoblotting Triton X-100-solubilized protein extracts from WT mouse embryonic fibroblasts (E3) and Ctr1−/− mouse embryonic fibroblasts from littermates (E8) (21). As shown in Fig. 1B, anti-Ctr1 antibody detects major 35–37-kDa polypeptide species and minor lower molecular weight polypeptides in E3 cell extracts. Although these polypeptide species were not observed in the Ctr1−/− (E8) cells, prolonged exposure of the immunoblot showed poorly reactive bands at ∼17, 26, 36, and 74 kDa (Fig. 1B). As we show the entire length of the electrophoresis distance on this immunoblot, no additional species were detected, and approximately equivalent loading of the SDS-polyacrylamide gel was apparent from detection of copper/zinc-superoxide dismutase (SOD1) in each extract. The 25- and 35–37-kDa species have previously been demonstrated to represent the primary translation product and mature O- and N-linked glycosylated Ctr1 species, respectively (24, 25).
FIGURE 1.

Re-evaluation of anti-Ctr1 polyclonal antibody specificity. A, primary and predicted secondary structure of mouse Ctr1. The arrowhead indicates the location of the Ctr1 extracellular lysine residue. The box indicates the antigen peptide sequence within the Ctr1 cytosolic loop against which anti-Ctr1 antibody was generated. B, immunoblotting with anti-Ctr1 antibody. Fifty micrograms of total protein extract from mouse embryonic fibroblasts of WT and Ctr1 knock-out (Ctr1−/−) mice were assayed. The upper panel shows immunoblot results with anti-Ctr1 antibody (at a 1:1,000 dilution). The arrowheads labeled u and g indicate the unglycosylated and glycosylated monomer of Ctr1, respectively. The arrowhead labeled t indicates the amino-terminal truncation form of Ctr1. The lower panel shows immunoblot results with anti-SOD1 antibody (1:1,000 dilution) as a loading control.
Ctr1 Localizes to the Apical Membrane in Mouse, Rat, and Pig Enterocytes
Given the specificity of the anti-Ctr1 antibody shown in Fig. 1B, we evaluated IEC Ctr1 localization across these three mammalian species from freshly dissected and fixed samples. First, immunohistochemical analysis was carried out on jejunal sections from 14-day-old control mice (Ctr1flox/flox) and IEC-specific Ctr1 knock-out mice (Ctr1int/int), generated as described previously (18). As shown in Fig. 2A at lower magnification and Fig. 2B at higher magnification, the Ctr1 antibody immunoreactivity occurred predominantly at the cell surface, indicative of apical membrane localization, with low levels of immunoreactivity appearing intracellularly. Although antibody against hephaestin, a basolateral multicopper oxidase, clearly confirmed a basolateral location for this protein (Fig. 2C), no Ctr1 immunoreactivity was detected on the basolateral surface of mouse IEC (Fig. 2, A and B). As a further test of the specificity of the anti-Ctr1 antibody in immunohistochemical experiments, jejunal samples from Ctr1int/int littermates were probed with anti-Ctr1 antibody. As shown at low magnification in Fig. 2D and higher magnification in Fig. 2E, very little anti-Ctr1 antibody immunoreactivity was observed at any location in the Ctr1int/int IEC. Although, as expected, hephaestin localized to the basolateral membrane of Ctr1int/int IEC (Fig. 2F), the immunoreactive signal was weaker relative to control IEC, likely due to the enhanced turnover of hephaestin under conditions of copper deficiency (26). An alternative method of immunohistochemical analysis using double-label confocal immunofluorescence microscopy also showed the predominant localization of Ctr1 on the apical surface (Fig. 2G, green), with no overlap with the basolateral membrane Na+/K+-ATPase α1 subunit (Fig. 2G, red) (27). Taken together, these experiments strongly support the predominant localization of Ctr1 to the apical surface of mouse IEC, with some intracellular staining that may represent vesicles.
FIGURE 2.

Localization of Ctr1 in the jejuna of control and Ctr1int/int mice in vivo. Jejuna from control mice (Ctr1flox/flox or Ctr1flox/+) and Ctr1int/int P14 mice were subjected to immunohistochemistry (A–F) and confocal immunofluorescence microscopy (G) analysis. A and B, control, anti-Ctr1 (1:200). C, control, anti-hephaestin (1:100). D and E, Ctr1int/int, anti-Ctr1 (1:200). F, Ctr1int/int, anti-hephaestin (1:100). The arrowheads indicate regions of immunopositive signals. G, confocal microscopy of Ctr1 in the jejuna of WT mice; green, anti-Ctr1 (1:200); red, anti-Na+/K+-ATPase α1 subunit (1:200); blue, DAPI. A and D, scale bars = 50 μm; B, C, and E–G, scale bars = 20 μm.
Given the precise conservation of the epitope used to generate this anti-Ctr1 antibody across many mammalian species, we evaluated intestinal sections from both rats and pigs for Ctr1 localization using immunohistochemistry. As shown in Fig. 3 (A and B) for rat duodenum and in Fig. 3 (C and D) for pig jejunum, immunoreactivity with the anti-Ctr1 antibody occurred predominantly on the apical surface of the IEC. Although there is also some immunoreactivity in intracellular compartments of unknown identity (Fig. 3, B and D) for both rat and pig tissue, these experiments revealed no evidence of Ctr1 in a basolateral location in IEC from duodenal or jejunal samples.
FIGURE 3.

Localization of Ctr1 in rat duodenum and pig jejunum. Tissues were probed with anti-Ctr1 antibody (1:200) and analyzed by immunohistochemistry. A and B, rat duodenum. C and D, pig jejunum. The arrowheads indicate some regions of immunopositive staining. A and C, scale bars = 50 μm; B and D, scale bars = 20 μm.
Elevated Levels of Ctr1 at the IEC Apical Surface in Response to Copper Limitation
Previous cell culture studies of yeast and mammalian Ctr1 demonstrated that cell surface Ctr1 is stimulated to internalize in response to copper concentrations near the Km for copper(I) uptake (15–17). Moreover, using an antibody that is distinct from that used in this and previous studies (18), Ctr1 immunoreactivity on the surface of mouse IEC was shown to be greater after perinatal copper deficiency than immunoreactivity in age-matched mice raised on a diet adequate in copper (19). As an independent means to assess Ctr1 localization in mouse small intestine, cell surface biotinylation experiments were carried out on freshly dissected small intestines from 5-week-old mice fed a control or copper-deficient diet for 5 days. The primary covalent binding site for Sulfo-NHS-SS-Biotin is the ϵ-amino group of lysine, of which one is present in the predicted extracellular domain of Ctr1 (Fig. 1A). A section of the small intestine was longitudinally sliced to expose the lumen, the fragment was incubated with biotin, and IEC were purified away from the substratum, as described previously (18). Proteins were solubilized, and the biotinylated proteins were purified by avidin affinity chromatography. Both total and affinity-purified proteins were fractionated by SDS-PAGE and analyzed by immunoblotting. As a control for the specificity of this reaction, when the biotinylation experiments were carried out on intestines dissected from 2-week-old control mice (Ctr1flox/+ or Ctr1flox/flox) or Ctr1int/int mice, Ctr1 was readily detected in the control mouse, but there was no detectable Ctr1 in total protein extract or in eluates from avidin-purified extracts from the Ctr1int/int mouse (Fig. 4A). Very low levels of the cytosolic protein GAPDH were detected in the eluate fractions, indicating minimal lysis of IEC during the biotinylation and sample preparation process.
FIGURE 4.
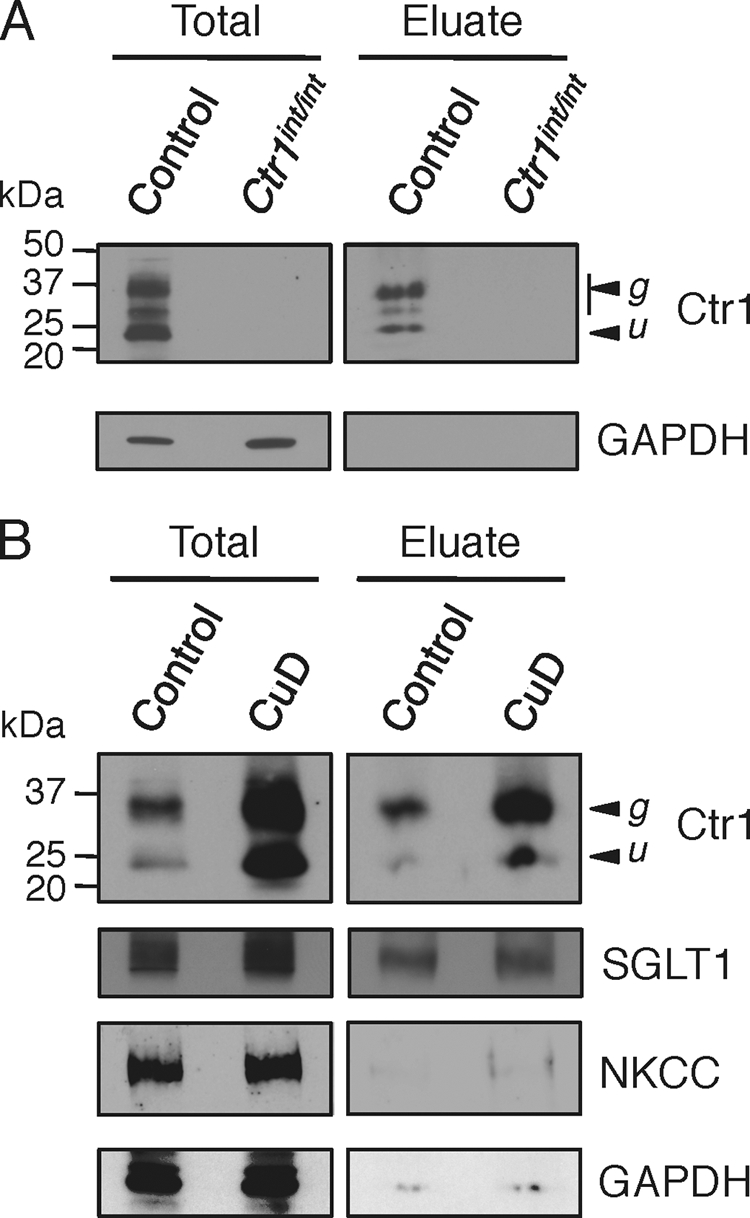
Apical surface biotinylation assays. A, small intestines from control mice (Ctr1flox/flox) and Ctr1int/int mice were subjected to apical surface biotinylation. Twenty micrograms of total protein (Total) and avidin column-purified fraction (Eluate) from control and Ctr1int/int mice were subjected to immunoblotting with anti-Ctr1 (1:1,000) and anti-GAPDH (1:1,000) antibodies. B, small intestines from 5-week-old mice fed a control or copper-deficient diet (CuD) for 5 days were used for apical surface biotinylation and analyzed as in A. Ctr1 (1:1,000), anti-SGLT1 (1:1,000), anti-NKCC (1:1,000), and anti-GAPDH (1:1,000) antibodies are shown. The glycosylated (g) and unglycosylated (u) forms of Ctr1 are indicated with arrowheads.
As shown from biotinylation experiments of whole small intestines illustrated in Fig. 4B, the steady-state levels of both the Ctr1 primary translation product and the mature glycosylated form were elevated in IEC from WT mice fed a copper-deficient diet compared with those fed the control diet. No changes in the levels of the sodium/glucose co-transporter SGLT1 (28), an apical protein, and the sodium/potassium/chloride co-transporter NKCC (29), a basolateral protein, were detected in response to a copper-limited diet (Fig. 4B). Moreover, the steady-state levels of the copper chaperone CCS and the mitochondrial cytochrome oxidase subunit Cox IV did not change (data not shown), suggesting that these cells did not experience a strong copper deficiency over the course of the 5 days of a copper-limited diet. After purification of IEC apical surface proteins by avidin affinity chromatography, followed by SDS-PAGE and immunoblotting, Ctr1 was detected in mice fed a control diet, and the levels of apical Ctr1 were enhanced ∼2-fold in mice fed the copper-deficient diet. This increase in Ctr1 protein levels in response to copper deficiency was specific, as the levels of another apical protein, SGLT1, did not change in either total or eluate fractions (Fig. 4B). Consistent with the established basolateral localization of NKCC and the cytosolic protein GAPDH, only a small amount of these proteins was detected in the eluate after avidin chromatography (Fig. 4B). Control immunoblots of extracts that were avidin-purified without prior biotinylation demonstrated that biotinylation is essential for avidin affinity purification of Ctr1 (data not shown). Taken together, these biotinylation experiments independently demonstrated the localization of Ctr1 to the apical membrane of mouse IEC and indicate that dietary copper deficiency results in elevated levels of total Ctr1 protein and elevated levels of Ctr1 at the apical membrane.
Posttranslational Control of Ctr1 in Response to Copper
A previous report showed no change in Ctr1 mRNA levels in purified rat IEC as a function of dietary copper status (12), suggesting the possibility of posttranscriptional regulation of steady-state Ctr1 protein levels in mouse IEC. To explore this possibility, HEK293T cells were treated with copper or the copper(I) chelator BCS in the presence of the translational inhibitor cycloheximide, and the steady-state levels of Ctr1 protein were analyzed by immunoblotting over time. Fig. 5A shows that the steady-state levels of the glycosylated form of Ctr1 were decreased in response to exposure of HEK293T cells to 100 mm copper in a time-dependent manner, whereas no notable decrease was observed under copper-deficient conditions imposed by the copper(I) chelator BCS. The addition of leupeptin, a broad-spectrum protease inhibitor, partially abrogated the decrease in Ctr1 levels in response to copper (Fig. 5A, asterisks), suggesting that the decrease in Ctr1 protein levels was due, in large part, to proteolysis that was directly or indirectly stimulated by copper. The degradation of Ctr1 that was stimulated by copper was dose dependent, as we observed a clear decrease in Ctr1 protein levels, even with the addition of 1 mm copper to the medium (Fig. 5B). Furthermore, this decrease in Ctr1 protein was specific for copper, as exposure to neither iron nor zinc at 10-fold molar excess over copper resulted in substantial reductions in Ctr1 protein levels (Fig. 5C). Although exposure of HEK293T cells to copper resulted in clear reductions in the steady-state levels of the full-length glycosylated form of Ctr1, steady-state levels of a previously characterized Ctr1 species that was truncated 29–33 residues from the amino terminus (25, 30) were not significantly altered in response to any metal tested (Fig. 5). Immunoblotting with an antibody raised against the Ctr1 amino terminus (30) showed cross-reactivity with the 35–37-kDa glycosylated form of Ctr1 and not the ∼17-kDa amino-terminally truncated form (Fig. 5D), supporting the assignment of these polypeptide species.
FIGURE 5.

Copper-dependent changes in steady-state levels of the mature, glycosylated form of Ctr1. A, HEK293T cells were incubated with cycloheximide and copper or the copper(I) chelator BCS for the indicated times. Total protein extracts of 100 μg were subjected to immunoblotting with anti-Ctr1 (1:1,000) and anti-actin (1:5,000) antibodies. B, HEK293T cells were incubated with BCS or copper at the concentrations indicated in the presence of cycloheximide for 8 h and subjected to immunoblotting as described in A. C, HEK293T cells were incubated with cycloheximide and BCS, copper, ferric ammonium sulfate (Fe), or ZnCl2 (Zn) for 8 h, and protein extracts were subjected to immunoblotting as described in A. D, two independently prepared protein extracts (lanes 1 and 2) from HEK293T cells treated with cycloheximide and BCS for 8 h as described in B and C were immunoblotted with antibodies that recognize either the cytosolic loop domain (Ctr1 loop; 1:1,000) or amino-terminal domain (Ctr1 N-term; 1:500) of Ctr1. Actin (1:5,000) levels were assayed as a loading control. The arrowhead labeled g indicates the glycosylated monomer of Ctr1, and the arrowhead labeled t indicates the amino-terminal truncation form of Ctr1. The asterisk indicates a polypeptide species that cross-reacts with the Ctr1 N-terminal antibody.
Copper Deficiency Increases Heart Ctr1 Protein Levels
Due to the energy requirements for cardiac contractility provided by mitochondrial cytochrome oxidase, the oxidative stress protection provided by copper/zinc-superoxide dismutase, and other factors, cardiac tissue exhibits a strong requirement for copper. Mice bearing a cardiac-specific Ctr1 deletion exhibit strong copper deficiency, reductions in copper-dependent enzymes, and a lethal cardiac hypertrophy (31). Given this high demand for copper yet the propensity for copper to generate reactive oxygen species, it would be advantageous for cardiac tissue to tightly regulate the copper homeostasis machinery. We examined the abundance of Ctr1 in cardiac tissue from mice fed a control or copper-deficient diet for 5 weeks starting at postnatal day 2. As shown in Fig. 6, the hearts from mice fed a copper-deficient diet exhibited dramatically elevated levels of the glycosylated form of Ctr1 compared with mice fed a control diet (two mice from each dietary group were assessed). These results suggest that not only the intestinal epithelium but also other tissues are responsive to dietary copper to regulate the abundance of the mature glycosylated form of the Ctr1 copper importer.
FIGURE 6.
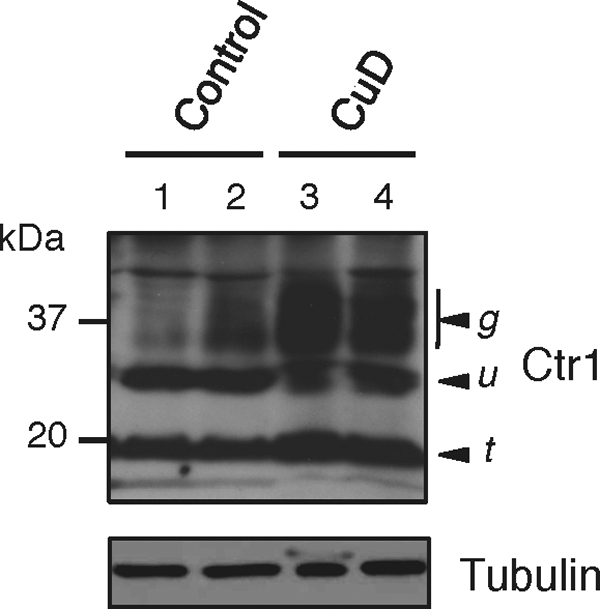
Cardiac Ctr1 protein levels respond to dietary copper status. Hearts were dissected from two independent mice fed either a control diet (lanes 1 and 2) or a copper-deficient diet (CuD; lanes 3 and 4), and total protein extract was analyzed by immunoblotting. Arrowheads labeled u, g, and t indicate the unglycosylated, glycosylated, and amino-terminally truncated forms of Ctr1, respectively. Tubulin (1:5,000) levels were assayed as a loading control.
DISCUSSION
Previous studies demonstrated that IEC Ctr1 is critical for normal copper acquisition, growth, cardiac function, and viability in mice (18). Although that study and two additional investigations (11, 19) presented data indicating that Ctr1 resides predominantly on the apical membrane of IEC, a recent study (Ref. 20 and reviewed in Ref. 32) suggests that Ctr1 functions in intestinal copper import at the basolateral membrane of polarized Caco-2 cells and in mouse intestine. Recognizing the importance of firmly establishing Ctr1 localization in the intestine, and the variability in antibodies, experimental conditions, and cell culture models of the intestine (33–35), we evaluated Ctr1 localization in mouse intestine and in that of rat and pig. We re-evaluated the specificity of our anti-peptide Ctr1 polyclonal antibody and used immunohistochemistry and biotinylation of intestinal luminal cell surface proteins to determine the subcellular location of Ctr1. The data presented here strongly support predominant apical membrane localization of IEC Ctr1 in mice, rats, and pigs, with some Ctr1 localizing to intracellular vesicles that may correspond to endosomes.
In contrast to a recent report (Ref. 20 and reviewed in Ref. 32), the results presented here based on light microscopy, confocal microscopy, and biotinylation analyses of mouse intestinal sections confirm and extend previous reports (11, 18, 19) that showed no evidence for the localization of Ctr1 to the basolateral membrane of IEC. Our results are also consistent with the gut epithelial cell apical localization of the structurally and functionally related copper transporter, Ctr1B, from Drosophila melanogaster (36). Taken together, these data strongly suggest that metazoan Ctr1 proteins import copper(I) from the diet across the apical membrane of IEC. It is unclear why the results from this and other studies (11, 18, 19) are in opposition to previous results using polarized Caco-2 cells and a mouse intestine sample (Ref. 20 and reviewed in Ref. 32). One possibility may be the wide variability in different laboratory Caco-2 cell lines that leads to differences in gene expression, tight junction integrity, and other variables that could lead to changes in Ctr1 trafficking (33–35). Although Caco-2 cells are often used as a model for polarized IEC, they are not authentic intestinal tissue. Another variable may the differences in the Ctr1 epitope used for antibody generation or differences in the specificity of the antibody generated.
Previous studies demonstrated that exposure of mammals to a low-copper diet results in enhanced copper absorption upon subsequent exposure to copper (10–12). However, the molecular mechanisms underlying this response are not understood. It has been reported that after copper deficiency, choroid plexus Ctr1 steady-state protein levels are increased (37), and enhanced apical density of Ctr1 in the IEC and the choroid plexus was observed by others using rats and another strain of mouse with a different Ctr1 antibody (19). Here, we have shown that mice exposed to a copper-limited diet had notably elevated steady-state levels of the full-length glycosylated Ctr1 protein, both in IEC and in cardiac tissue, compared with mice fed a control diet. The biotinylation assays of intestinal luminal proteins demonstrated that mice fed a copper-deficient diet display a corresponding increase in the levels of Ctr1 on the apical membrane of the IEC. Previous studies in rat (12) demonstrated that mRNA levels of intestinal epithelial Ctr1 do not change in response to a copper-deficient diet, suggesting that the increase in Ctr1 protein levels observed here may be due to posttranscriptional regulation. The degradation of the human Ctr1 35-kDa glycosylated monomer and 60–70-kDa dimer by elevated copper was previously observed in HEK293 cells overexpressing Myc-tagged Ctr1 (38). Our results obtained from HEK293T cells with endogenous Ctr1 suggested that the mature, glycosylated form of Ctr1 was degraded in response to exposure to elevated copper levels and was stabilized by copper limitation. That the enhanced Ctr1 turnover in vivo in response to elevated copper may be due to proteolytic activity is suggested by partial inhibition of the reduction in Ctr1 in the presence of leupeptin, a broad-spectrum protease inhibitor. Despite evidence for the degradation of mature, glycosylated Ctr1, levels of the amino-terminal truncation form of Ctr1 protein did not show notable changes in response to copper deficiency under these conditions. It has been reported that the Ctr1 amino-terminal truncation form has ∼50% of copper-transporting activity when it is overexpressed in HEK293T cells (25). However, the precise localization, regulation, turnover, and physiological function of the truncated form of Ctr1 in vivo are not well understood. Although these experiments were carried out by evaluating endogenous Ctr1 in cultured HEK293T cells, these results may explain the increase in the steady-state Ctr1 protein levels in IEC from mice exposed to a transient copper-deficient diet. Although our results and previous work suggest the presence of regulatory responses that modulate the abundance of the Ctr1 copper(I) transporter at the apical surface in response to dietary copper availability, further studies are required to fully elucidate the mechanisms underlying changes in Ctr1 protein levels in vivo. It is also important to note that although previous studies reported changes in Ctr1 localization in cell culture as a function of exogenous copper (16, 24, 39), this was not observed in all cell lines. This variation could represent cell type-specific differences in the protein-trafficking machinery or in the regulation of this machinery and further underscores the importance of ultimately evaluating protein localization and physiological regulatory mechanisms for Ctr1 in tissues.
It is interesting that despite strong changes in Ctr1 abundance in parallel with a transient 5-day exposure to a dietary copper deficiency, we did not detect a concomitant increase in the CCS copper chaperone or a decrease in the Cox IV subunit of mitochondrial cytochrome oxidase. Perhaps an early response to copper deficiency is the elevation of Ctr1 steady-state levels and apical Ctr1 to deliver more copper to the interior of the cells, thereby maintaining the levels of CCS and Cox IV. If copper deficiency persists over a longer time or is of a severity at which elevated apical Ctr1 cannot import sufficient copper, then the IEC may adapt by elevating the levels of CCS and reducing the levels of Cox IV. Additional regulatory changes (e.g. phosphorylation/dephosphorylation) may also ensue, such as have been observed for the copper-responsive trafficking of ATP7A (40), or there may be alterations in ATP7A abundance in liver and intestine in response to copper deficiency in peripheral organs (31).
Acknowledgments
We thank Scott McNaughton for excellent technical assistance, members of the Thiele laboratory for critical reading of the manuscript, Jack H. Kaplan and Edward Maryon for anti-Ctr1 amino-terminal antibody, and the Duke University Light Microscopy Core Facility for technical assistance and advice.
This work was supported, in whole or in part, by National Institute of Health Grants DK074192-09 (to D. J. T.) and HD039708-08 (to J. R. P.). This work was also supported by American Heart Association Postdoctoral Fellowship 09POST2251047 (to B.-E. K.).
- IEC
- intestinal epithelial cell(s)
- E
- embryonic day
- BCS
- bathocuproine disulfonate.
REFERENCES
- 1.Linder M. C. (1991) Biochemistry of Copper, Plenum Press, New York [Google Scholar]
- 2.Prohaska J. R. (2006) in Present Knowledge in Nutrition (Bowman B., Russell R. eds) 9th Ed., pp. 458–470, ISLI Press, Washington, DC [Google Scholar]
- 3.Kim B. E., Nevitt T., Thiele D. J. (2008) Nat. Chem. Biol. 4, 176–185 [DOI] [PubMed] [Google Scholar]
- 4.Turski M. L., Thiele D. J. (2009) J. Biol. Chem. 284, 717–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collins J. F., Prohaska J. R., Knutson M. D. (2010) Nutr. Rev. 68, 133–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumar N. (2006) Mayo Clin. Proc. 81, 1371–1384 [DOI] [PubMed] [Google Scholar]
- 7.Neerman M. F., Kiefhaber K., Barrera R. D. (2007) Lab. Med. 38, 608–609 [Google Scholar]
- 8.van den Berghe P. V., Klomp L. W. (2009) Nutr. Rev. 67, 658–672 [DOI] [PubMed] [Google Scholar]
- 9.Crampton R. F., Matthews D. M., Poisner R. (1965) J. Physiol. 178, 111–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turnlund J. R., Keyes W. R., Anderson H. L., Acord L. L. (1989) Am. J. Clin. Nutr. 49, 870–878 [DOI] [PubMed] [Google Scholar]
- 11.Bauerly K. A., Kelleher S. L., Lönnerdal B. (2004) J. Nutr. Biochem. 15, 155–162 [DOI] [PubMed] [Google Scholar]
- 12.Lee J., Prohaska J. R., Dagenais S. L., Glover T. W., Thiele D. J. (2000) Gene 254, 87–96 [DOI] [PubMed] [Google Scholar]
- 13.Lee J., Penã M. M., Nose Y., Thiele D. J. (2002) J. Biol. Chem. 277, 4380–4387 [DOI] [PubMed] [Google Scholar]
- 14.Petris M. J. (2004) Eur. J. Physiol. 447, 752–755 [DOI] [PubMed] [Google Scholar]
- 15.Ooi C. E., Rabinovich E., Dancis A., Bonifacinol J. S., Klausner R. D. (1996) EMBO J. 15, 3515–3523 [PMC free article] [PubMed] [Google Scholar]
- 16.Guo Y., Smith K., Lee J., Thiele D. J., Petris M. J. (2004) J. Biol. Chem. 279, 17428–17433 [DOI] [PubMed] [Google Scholar]
- 17.Molloy S. A., Kaplan J. H. (2009) J. Biol. Chem. 284, 29704–29713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nose Y., Kim B. E., Thiele D. J. (2006) Cell Metab. 4, 235–244 [DOI] [PubMed] [Google Scholar]
- 19.Kuo Y. M., Gybina A. A., Pyatskowit J. W., Gitschier J., Prohaska J. R. (2006) J. Nutr. 136, 21–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zimnicka A. M., Maryon E. B., Kaplan J. H. (2007) J. Biol. Chem. 282, 26471–26480 [DOI] [PubMed] [Google Scholar]
- 21.Lee J., Petris M. J., Thiele D. J. (2002) J. Biol. Chem. 277, 40253–40259 [DOI] [PubMed] [Google Scholar]
- 22.Puig S., Lee J., Lau M., Thiele D. J. (2002) J. Biol. Chem. 277, 26021–26030 [DOI] [PubMed] [Google Scholar]
- 23.De Feoa C. J., Allera S. G., Siluvaib G. S., Blackburnb N. J., Unger V. M. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 4237–4242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klomp A. E., Tops B. B., Van Denberg I. E., Berger R., Klomp L. W. (2002) Biochem. J. 364, 497–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maryon E. B., Molloy S. A., Kaplan J. H. (2007) J. Biol. Chem. 282, 20376–20387 [DOI] [PubMed] [Google Scholar]
- 26.Nittis T., Gitlin J. D. (2004) J. Biol. Chem. 279, 25696–25702 [DOI] [PubMed] [Google Scholar]
- 27.Amerongen H. M., Mack J. A., Wilson J. M., Neutra M. R. (1989) J. Cell Biol. 109, 2129–2138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turk E., Martín M. G., Wright E. M. (1994) J. Biol. Chem. 269, 15204–15209 [PubMed] [Google Scholar]
- 29.D'Andrea L., Lytle C., Matthews J. B., Hofman P., Forbush B., 3rd, Madara J. L. (1996) J. Biol. Chem. 271, 28969–28976 [DOI] [PubMed] [Google Scholar]
- 30.Maryon E. B., Zhang J., Jellison J. W., Kaplan J. H. (2009) J. Biol. Chem. 284, 28104–28114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim B. E., Turski M. L., Nose Y., Casad M., Rockman H. A., Thiele D. J. (2010) Cell Metab. 11, 353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaplan J. H., Lutsenko S. (2009) J. Biol. Chem. 284, 25461–25465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seithel A., Karlsson J., Hilgendorf C., Björquist A., Ungell A. L. (2006) Eur. J. Pharm. Sci. 28, 291–299 [DOI] [PubMed] [Google Scholar]
- 34.Hughes P., Marshall D., Reid Y., Parkes H., Gelber C. (2007) BioTechniques 43, 575–586 [DOI] [PubMed] [Google Scholar]
- 35.Hayeshi R., Hilgendorf C., Artursson P., Augustijns P., Brodin B., Dehertogh P., Fisher K., Fossati L., Hovenkamp E., Korjamo T., Masungi C., Maubon N., Mols R., Müllertz A., Mönkkönen J., O'Driscoll C., Oppers-Tiemissen H. M., Ragnarsson E. G., Rooseboom M., Ungell A. L. (2008) Eur. J. Pharm. Sci. 35, 383–396 [DOI] [PubMed] [Google Scholar]
- 36.Balamurugan K., Egli D., Hua H., Rajaram R., Seisenbacher G., Georgiev O., Schaffner W. (2007) EMBO J. 26, 1035–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gybina A. A., Prohaska J. R. (2006) Genes Nutr. 1, 51–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petris M. J., Smith K., Lee J., Thiele D. J. (2003) J. Biol. Chem. 278, 9639–9646 [DOI] [PubMed] [Google Scholar]
- 39.Eisses J. F., Chi Y., Kaplan J. H. (2005) J. Biol. Chem. 280, 9635–9639 [DOI] [PubMed] [Google Scholar]
- 40.Lutsenko S., Petris M. J. (2003) J. Membr. Biol. 191, 1–12 [DOI] [PubMed] [Google Scholar]