

Abstract
DNA photolyases use two noncovalently bound chromophores to catalyze photoreactivation, the blue light-dependent repair of DNA that has been damaged by ultraviolet light. FAD is the catalytic chromophore for all photolyases and is essential for photoreactivation. The identity of the second chromophore is often 7,8-didemethyl-8-hydroxy-5-deazariboflavin (FO). Under standard light conditions, the second chromophore is considered nonessential for photoreactivation because DNA photolyase bound to only FAD is sufficient to catalyze the repair of UV-damaged DNA. phr1 is a photoreactivation-deficient strain of Chlamydomonas. In this work, the PHR1 gene of Chlamydomonas was cloned through molecular mapping and shown to encode a protein similar to known FO synthases. Additional results revealed that the phr1 strain was deficient in an FO-like molecule and that this deficiency, as well as the phr1 photoreactivation deficiency, could be rescued by transformation with DNA constructs containing the PHR1 gene. Furthermore, expression of a PHR1 cDNA in Escherichia coli produced a protein that generated a molecule with characteristics similar to FO. Together, these results indicate that the Chlamydomonas PHR1 gene encodes an FO synthase and that optimal photoreactivation in Chlamydomonas requires FO, a molecule known to serve as a second chromophore for DNA photolyases.
Keywords: DNA Damage; DNA Enzymes; DNA Repair; Flavin; Genetics; 7,8-Didemethyl-8-hydroxy-5-deazariboflavin; Cyclobutane Pyrimidine Dimers; Photoreactivation; Ultraviolet Light
Introduction
Cyclobutane pyrimidine dimers (CPDs)2 and 6-(1,2)-dihydro-2-oxo-4-pyrimidinyl-5-methyl-2,4-(1H,3H)-pyrimidinedione photoproducts are the primary forms of DNA damage induced by UV light. These types of DNA damage are hazardous to cells by potentially stalling DNA replication and introducing mutation (1–3). Photoreactivation is a blue light-dependent DNA repair process catalyzed by enzymes known as DNA photolyases. There are different classes of DNA photolyases with distinct repair specificities. For instance, there are DNA photolyases responsible for repairing CPDs and ones specific for repairing (6-4) photoproducts. During photoreactivation, DNA photolyase binds to the UV-induced lesion specific for its class and uses blue light as a cosubstrate to return the DNA to its undamaged state (4).
Each DNA photolyase binds two chromophores noncovalently. The first, FAD, is common to all photolyases. It is the catalytic chromophore, and as such, it is essential for photoreactivation (5). The identity of the second chromophore is variable, but 7,8-didemethyl-8-hydroxy-5-deazariboflavin (FO) serves this role for a number of photolyases (6–10). During photoreactivation, the second chromophore acts as a photoantenna, becoming excited by blue light energy and transferring this energy to FADH−. The excited FADH− reversibly transfers an electron to the damaged DNA allowing the DNA to return to its original undamaged form. Although the antenna chromophore increases the repair rate of DNA photolyase 10–100-fold under limited-light conditions, it is generally considered nonessential for photoreactivation under standard light conditions (4, 11, 12) because DNA photolyase bound to FADH− alone is sufficient to catalyze the repair of UV-induced DNA damage (5, 13–15).
FO is generated from 5-amino-6-ribitylamino-2,4(1H,3H)-pyrimidinedione, an intermediate in riboflavin biosynthesis, and 4-hydroxyphenylpyruvate, a precursor to tyrosine. The reaction is catalyzed by an enzyme known as FO synthase (EC 2.5.1.77), a member of the radical S-adenosylmethionine (AdoMet) superfamily of proteins (16). Members of this family catalyze many diverse reactions, but all use a unique [4Fe-4S] cluster to bind AdoMet as a ligand. The AdoMet is split to form a 5′-deoxyadenosyl radical that abstracts a hydrogen from the substrate initiating the reaction mechanism (17, 18). FO synthases have been partially characterized from mycobacteria and Methanocaldococcus jannaschii (16, 19). BLASTP searches have identified FO synthase homologs in Archaea, some classes of Bacteria, and in the Chlorophyta of the eukaryotes (10, 16, 19). FO is a biosynthetic precursor for the hydride carrier coenzyme F420. Accordingly, FO synthase homologs have been identified in all organisms known to produce F420, including the methanogens and the actinomycetes. However, there are organisms, including the cyanobacteria, that do not produce F420 but still generate FO as an apparent end product (16).
The phr1 strain of the unicellular green alga Chlamydomonas reinhardtii was isolated following chemical mutagenesis and shown to have a defect in the photoreactivation of CPDs (20). In an attempt to identify the PHR1 gene, a homology-based method was used to clone a gene of Chlamydomonas that encoded a CPD-specific DNA photolyase. However, the newly cloned gene was not closely linked to the phr1 mutation and was thus named PHR2 (21). Moreover, further work revealed that PHR2p requires PHR1p for full activity, indicating that photoreactivation in Chlamydomonas requires the products of two genes (22). This discovery marked phr1 as an unmatched photoreactivation mutant. In this work, we report the identification and cloning of the PHR1 gene of C. reinhardtii, which encodes the first characterized eukaryotic FO synthase. Furthermore, we show that FO, a known second chromophore for DNA photolyases, is critical for photoreactivation in Chlamydomonas.
EXPERIMENTAL PROCEDURES
Molecular Mapping
phr1 was mated with S1-D2 (cc-2290), a polymorphic field isolate of Chlamydomonas (23). Progeny from this cross that were photoreactivation-deficient, and thus had inherited the phr1 mutation, were used as the mapping population. Genomic DNA was isolated from each member of the mapping population and used as the template for PCRs with primers from the Chlamydomonas molecular mapping kit (24). The mapping kit uses PCR to reveal polymorphisms in the mapping population, providing a means for measuring the cosegregation frequency of molecular markers and the mutation of interest. The phr1 mutation was localized to LG XIX using the Zys1B marker from the molecular mapping kit. The location of the phr1 mutation was further refined using primers for the FLA10 marker (5′-CTGCGCGCCAGCAAGCTCAAGT-3′ and 5′-GGTAACAGCCCGTCTTCCAGGGCC-3′). These primer sequences were obtained from a data base of primer sets for additional markers not included in the mapping kit.
Complementation Assays
phr1 mt(+) was mated with cw15arg7 mt(−) (cc-1618), a cell wall-less and arginine-requiring strain of Chlamydomonas. The resulting phr1cw15 arg7 strain was transformed with 5 μg of bacterial artificial chromosome (BAC) or plasmid DNA containing the PHR1 gene and with 1 μg of cotransforming DNA, pUC19 Arg7.8, using the glass bead method (25). The plasmid pUC19 ARG7.8 was derived from pARG7.8 (26) by ligation of a 7.8-kb SalI-BamHI fragment into pUC19. To test for complementation of the phr1 photoreactivation deficiency, transformants were cultured in TAP medium (27) and spotted on separate TAP plates. One plate served as a no UV growth control, and the other was irradiated with 400 J/m2 UV light and was immediately placed under normal growth conditions. This includes continuous photoreactivating light provided by two cool white fluorescent bulbs at a distance of 30 cm (57 μmol m−2 s−1). Under these conditions, wild type and complemented strains survive and form a dense green spot on the test plate after 5–6 days, whereas strains with the phr1 mutation do not.
Measuring FO Levels in Chlamydomonas Cells
Cell pellets from Chlamydomonas cultures were resuspended in 50% methanol, boiled for 5 min, and vortexed briefly. Cell debris was pelleted and the supernatant reserved. The pellet was extracted a second time with 50% methanol, and the two supernatants were combined. Chlorophyll content was measured for each extract using the following equation (28): μg of chlorophyll/ml of extract = 6.10(A665) + 20.04(A649). Equivalent amounts of each cell extract (as determined by chlorophyll content) were dried using a speed vacuum (Thermo Electron Corporation) at 40 °C. Samples were resuspended in 25 mm sodium acetate buffer (pH 6.0, 35% methanol, 0.02% NaN3) and clarified by centrifugation in preparation for high performance liquid chromatography (HPLC).
HPLC analyses were done essentially as described previously (16). HPLC was performed using an isocratic pump (PM-80 Solvent Delivery System, Bioanalytical Systems Inc.) using a C18 reversed phase column (AXXI Chrom octyldecyl silane, 5 μm, 4.6 mm × 25 cm) maintained at 27 °C with a LC-22C temperature controller (Bioanalytical Systems Inc.). Mobile phase (25 mm sodium acetate, 35% methanol, 0.02% NaN3, pH 6.0) was pumped at a flow rate of 0.5 ml/min. Samples (25 μl) were injected manually (Rheodyne 8125). The eluent was monitored by fluorescence detection (excitation = 420 nm; emission = 480 nm) using a Waters 2475 Multi λ Fluorescence detector. Data acquisition and analyses were performed with Clarity Lite (version 2.8) software (Data Apex). Retention times of samples were compared against a chemically synthesized FO standard (a generous gift of Dr. Robert White, Virginia Polytechnic Institute and State University, Blacksburg VA).
Determining Exon/Intron Boundaries of PHR1
Total RNA Isolation
Total RNA was isolated from Chlamydomonas cells using the RNeasy plant mini kit (Qiagen) as described by the manufacturer with the following modifications. For each isolation, 2 × 107 cells were resuspended in RLT buffer and disrupted with two cycles of flash-freezing in liquid nitrogen, thawing, and vortexing.
Poly(A) RNA Isolation
Poly(A) RNA was isolated from total RNA using the Oligotex mRNA mini kit (Qiagen) as described by the manufacturer.
5′-RACE
The FirstChoice RNA ligase-mediated RACE kit (Ambion) was used to generate the 5′-RACE product essentially as suggested by the manufacturer. Poly(A) RNA from Chlamydomonas strain cc-125 was dephosphorylated with calf intestinal alkaline phosphatase, treated with tobacco acid pyrophosphatase, and ligated to a 5′-RACE adapter oligonucleotide with T4 RNA ligase. Reverse transcription was performed using ImProm-II reverse transcriptase (Promega) and a gene-specific primer from exon 6, FB37 (5′-GGTGCTCTCCAGCATCAGGCC-3′). PCR amplification of the cDNA was performed using a gene-specific antisense primer from exon 5, FB27 (5′-CATGACGCCCGCGTTGATGTG-3′) and 5′-RACE Outer Primer (Ambion). The product of this amplification was used as template for a nested PCR with a gene-specific antisense primer from exon 4, FB35 (5′-TCCACGTACTCCAGAGTGCTG-3′) and 5′-RACE Inner Primer (Ambion). The final product was 1,043 bp long.
3′-RACE
Poly(A) RNA from Chlamydomonas strain cc-125 was reverse-transcribed using cDNA cloning primer (Integrated DNA Technologies) and ImProm-II reverse transcriptase (Promega). The resulting cDNA was PCR-amplified using a gene-specific sense primer from exon 16, FB38 (5′-CTGGCGCTGCACCCACACATC-3′) and 3′-RACE PCR primer (Integrated DNA Technologies). The product of this amplification was used as template for a nested PCR with a gene-specific sense primer from exon 17, FB30 (5′-AACGACATGGGCGGCAGCATC-3′) and 3′-RACE PCR primer. The resulting product was 840 bp in length.
RT-PCR
Poly(A) RNA from Chlamydomonas strain cc-125 was reverse-transcribed using gene-specific oligonucleotides and ImProm-II reverse transcriptase (Promega). The resulting cDNAs were subsequently used as templates for RT-PCR. RT-PCR1 spans from exon 3 to 10 and was amplified using sense primer FB24 (5′-CCGCTGCGGCTACTGCACCTTC-3′) and antisense primer FB29 (5′-CCAGGGCCGGTCCGGGTTCAC-3′). RT-PCR2 spans exons 8 to 11 and was amplified with sense primer FB26 (5′-CTTATCATCCAGAACTTCGTGGCC-3′) and antisense primer FB13 (5′-CAACATAGCTCACGTCGTCGC-3′). Sense primer FB36 (5′-GTGTGTGCTGCCGCCGACCTG-3′) and antisense primer FB17 (5′-CCAGCTGCCGCCAGCTTCTCC-3′) were used for RT-PCR3, which spans exons 11–15. RT-PCR4 was amplified using sense primer FB28 (5′-CACGTGCACGCCTTCAGCCCG-3′) and antisense primer FB31 (5′-TACAGGGTGGTGCGCTGGCGC-3′), covering exons 14–18. All amplified products were cloned into pCR2.1 (Invitrogen) and sequenced at the DNA Facility of the Iowa State University Office of Biotechnology.
PHR1p Protein Analysis
The amino acid sequence of the PHR1p protein was aligned with the amino acid sequences of FbiC from Mycobacterium bovis BCG (AAL91922) (19), Mycobacterium smegmatis (J. Craig Venter Institute-Comprehensive Microbial Resource, JCVI Locus: MSMEG_5126), and CofG (Q57888) and CofH (Q58826) from M. jannaschii (16) using MultAlin (29).
The PHR1p protein was analyzed for the presence of subcellular localization signals using TargetP 1.1 (30), iPSORT (31), and PCLR prediction (32).
Creating Full-length PHR1 cDNA
The full-length PHR1 cDNA was generated by combining products from RT-PCR, 5′-RACE, and 3′-RACE. The 5′- and 3′-halves of the cDNA were constructed in separate cassettes that were ultimately joined together to create the full-length PHR1 cDNA. For the 5′-end, the 5′-RACE product (A in Fig. 3) was joined with the RT-PCR1 product (B in Fig. 3) using a NotI site encoded in exon 4. This resulted in a cassette containing the 5′-UTR through a unique SmaI site encoded in exon 10. The 3′-cassette contained the unique SmaI site in exon 10 through the 3′-UTR. To generate the 3′-cassette, two additional RT-PCR products were created. RT-PCR5 (G in Fig. 3) spanned exons 8–15 and was amplified with the previously described FB26 and FB17 primers. RT-PCR6 was designed to span exons 11–18 using primers FB36 and FB31, which also have already been described. Because of PCR induced mutation in the 3′-end of RT-PCR6, the 5′-end of RT-PCR6 was joined with RT-PCR4 (E in Fig. 2A), resulting in RT-PCR6/4 (H in Fig. 3), which still covered exons 11–18. A 786-bp SphI/EagI RT-PCR6/4 fragment (spanning exons 13–18) was used to join the RT-PCR5 product and the 3′-RACE product (F in Fig. 3), completing the 3′-half of the PHR1 cDNA. The 5′ and 3′-cassettes of the PHR1 cDNA were then joined using the unique SmaI site in exon 10.
FIGURE 3.

Engineering a full-length PHR1 cDNA. Indicated by thin black lines are products generated by 5′-RACE, 3′-RACE, and RT-PCR that were used to form the full-length PHR1 cDNA. An N-terminal cassette was formed by joining products A and B using a NotI site. A C-terminal cassette was formed by joining products G, H, and F using SphI and EagI. The N- and C-terminal cassettes were joined to form the full-length PHR1 cDNA using a unique (*) SmaI site.
FIGURE 2.

Characterization of the PHR1 gene and transcript. A, exon/intron structure of the Chlamydomonas PHR1 gene. Exons are indicated by black and gray boxes, and introns are indicated with a thin black line. Below the gene is a diagram showing the 5′-RACE, 3′-RACE, and RT-PCR products used to identify the full-length sequence of the PHR1 transcript. B, PHR1 transcript has two open reading frames upstream of the FO synthase homolog ORF. The PHR1 transcript is shown with the FO synthase homolog ORF in black. Upstream ORFs are indicated by open boxes. Transcript nucleotide numbers are given.
Expression of PHR1p NoCTP and PHR1p NoCTP in Escherichia coli
For PHR1p expression in E. coli, the 5′- and 3′-ends of the PHR1 cDNA were separately modified by PCR, introduced into the 5′ and 3′-cassettes described above to generate the full-length PHR1 cDNA, and then joined together using the unique SmaI site encoded in exon 10. The 5′-end of the PHR1 cDNA was engineered by PCR using sense primer FOS 5′Bam NdeI (5′-CCGGATCCCATATGGCTGTACACAGCCGCTGCCCC-3′) and FB35 (5′-TCCACGTACTCCAGAGTGCTG-3′), an antisense primer in exon 4. The resulting 610-bp product included a 5′ NdeI site (boldface in primer) designed to facilitate cloning into the multiple cloning site of the expression plasmid and to exclude the coding region for the predicted chloroplast transit peptide (NoCTP) of PHR1p. The modified 5′-end was introduced into the 5′-cassette of the PHR1 cDNA via the NotI site from exon 4. The BamHI site included in the 5′ primer (underlined in primer) was solely to facilitate the construction of the 5′-cassette.
The 3′-end of the PHR1 cDNA was engineered by PCR with primers FB38 (5′-CTGGCGCTGCACCCACACATC-3′), a sense primer in exon 16, and FOS 3′ NoStop Xho (5′-CCCTCGAGCCGCAGTACACCGCTGCCTGG-3′). The resulting 332-bp product modified the 3′-end of PHR1p by eliminating the termination codon and introducing an XhoI site (boldface in primer). The XhoI site facilitated cloning into the expression plasmid, and removing the termination codon allowed the expression of PHR1p as a C-terminal S-tag fusion protein. The modified 3′-end was introduced into the 3′-cassette using the StuI site from the junction of exon 16 and exon 17.
The 5′- and 3′-expression cassettes were joined using the SmaI site in exon 10. The resulting construct was digested with NdeI/XhoI, and the 3,120-bp fragment containing the coding sequence for PHR1p NoCTP was cloned into pCOLADuet-1 (Novagen) that had also been cut with NdeI/XhoI.
To create the phr1p NoCTP expression construct, RT-PCR was performed with phr1 poly(A) RNA using the aforementioned primers FB26 and FB17. The resulting 1,502-bp product was cut with SmaI and SphI, and the 1,043-bp fragment spanning exons 10–13 and containing the phr1 mutation in exon 12 was cloned into the 3′-expression cassette described immediately above. As with the PHR1p NoCTP construct, the 3′-expression cassette containing the phr1 mutation was joined with the 5′-expression cassette, and the resulting 3,120-bp NdeI/XhoI fragment was introduced into pCOLADuet-1.
The PHR1p NoCTP and phr1p NoCTP expression constructs were transformed into chemically competent T7 Express E. coli cells (New England Biolabs). Transformants were cultured in Luria-Bertani (LB) media/kanamycin (33) to an A600 ≅0.6, induced to express the fusion protein with isopropyl β-d-thiogalactopyranoside (IPTG) (1.0 mm), and grown for another 3 h.
Analysis of Heterologous Protein Expression
Cells from aliquots of E. coli expression cultures removed immediately before and after 3 h of IPTG induction were resuspended in SDS-PAGE sample loading buffer with 1× Complete Protease inhibitor (Roche Applied Science) and boiled for 5 min. Protein concentration of each sample was determined using Pierce 660 nm of protein assay reagent (Thermo Scientific) in preparation for SDS-PAGE and Coomassie staining or Western blot. SDS-PAGE was performed on equivalent amounts of each sample following standard procedures (33). Coomassie staining using GelCode Blue Safe Protein Stain (Thermo Scientific) was performed per the manufacturer's recommendation. Western blots were performed following standard protocols (33). Mouse monoclonal anti-S-tag antibody (Novagen) and peroxidase-conjugated AffiniPure goat anti-mouse IgG (Jackson ImmunoResearch) were used for blotting at dilutions of 1:5,000 and 1:30,000, respectively. VisiGlo HRP Plus chemiluminescent substrate (AMRESCO) was used for detection.
Fluorescence of E. coli Expression Culture Media
LB media from E. coli expression cultures induced with IPTG for 3 h were clarified by centrifugation. Fluorescence (excitation 420 nm; emission 480 nm) of clarified culture media was measured using a BioTek Synergy 4 hybrid multimode microplate reader and Gen5 version 1.05 software. The culture media from each sample was measured in triplicate for each experiment. Values were calculated using noncultured medium for background subtraction. Results are reported as the mean of the means from three separate experiments.
Isolation and Characterization of FO
Isolation and characterization of FO was performed essentially as described by Graham et al. (16).
Cell Extraction
Cell pellets from 20-ml expression cultures induced for 3 h with IPTG were resuspended in 50% methanol (3 ml) and heated at 70 °C for 5 min. Following centrifugation, the supernatant was reserved, and an identical extraction was performed on the pellet. Extraction supernatants were combined and dried by speed vacuum. Samples were resuspended in 50% methanol for TLC.
TLC
Samples were spotted on pre-equilibrated silica gel 60 F254 TLC plates (EMD Chemical, Inc.) and separated using acetonitrile/water/formic acid (88%), 40:10:5 by volume, as the developing solvent. Plates were visualized by illumination with long wavelength UV light. Images were taken with a Canon digital SLR camera with shutter priority set to 1/60 s. FO migrated with an Rf = 0.43 and was identifiable by its blue fluorescence. Spots containing FO were scraped from the TLC plate, and FO was eluted with 50% methanol and dried by speed vacuum in preparation for HPLC analysis.
HPLC
HPLC analyses were done as described above.
Gene Bank Accession Number
The DNA sequence for the PHR1 gene has been assigned GenBankTM accession number HM156042.
RESULTS
Cloning and Characterizing the PHR1 Gene
The phr1 strain of Chlamydomonas has a defect in photoreactivation, a blue light-dependent repair mechanism for DNA damaged by UV light (20). To better understand the role of PHR1p in photoreactivation, a forward genetics approach known as molecular mapping (24) was used to clone the PHR1 gene. The principle of map-based cloning is to identify the genetic basis of a particular phenotype based on cosegregation of the mutant phenotype and markers located throughout the genome of the organism. The phr1 mutation was mapped to within ∼6.9 centimorgans using the FLA10 marker on the left arm of LG XIX. A search of the Chlamydomonas genome sequence in this region of LG XIX for candidate genes involved in photoreactivation revealed a gene model (e_gwH.60.32.1) predicted to encode an FO synthase homolog (Fig. 1A). Given that FO is a known chromophore for DNA photolyases, e_gwH.60.32.1 was deemed a promising candidate for the PHR1 gene.
FIGURE 1.

DNA constructs containing the e_wgH.60.32.1 gene model from Chlamydomonas LG XIX complement the photoreactivation deficiency of phr1 as well as a deficiency in an FO-like molecule. A, Chlamydomonas gene model e_wgH.60.32.1 DNA constructs. The thick line indicates the genomic region in which gene model e_wgH.60.32.1 (gray arrow) resides. The thin lines indicate four DNA constructs, each containing e_wgH.60.32.1. B, Chlamydomonas photoreactivation complementation assay. 10-μl aliquots of 1–1.5 serial dilutions of cell cultures grown to the approximate same cell density were spotted on media plates. One plate received no UV dose and was placed immediately under normal growth conditions. Two additional plates received 450 J/m2 UV light. Of these, one was immediately placed under normal growth conditions, which includes photoreactivating light. The other was placed in the dark for 18 h to prevent photoreactivation and then placed under normal growth conditions. C, HPLC elution profiles of an FO standard and Chlamydomonas cell extracts made from wild type, phr1, and phr1 strains transformed with e_gwH.60.32.1 constructs.
As part of the Chlamydomonas genome sequencing project, BAC end sequences were mapped to the Chlamydomonas genome (34, 35). Based on these efforts, BACs 27E15 and 36E21 were each predicted to contain a wild type copy of e_gwH.60.32.1 (Fig. 1A). To determine whether the phr1 photoreactivation deficiency was caused by a mutation in the putative FO synthase gene, complementation studies of the phr1 strain were performed using 27E15 and 36E21. phr1 cells that were transformed with either of the two BAC clones were tested for photoreactivation proficiency via a UV spot test. The basis for the UV spot test is that the ability of cells to survive after UV challenge is indicative of the ability to repair UV-induced DNA damage. As shown in Fig. 1B, phr1 cells transformed with either BAC 27E15 or 36E21 showed an increased ability to survive a given dose of UV light when subsequently supplied with photoreactivating light. In the absence of photoreactivating light, the complemented strains did not survive, indicating that the mechanism for increased survival is photoreactivation and not an alternative mechanism. A 13.2-kb ApaI/ClaI fragment and an 11.3-kb HindIII/ClaI fragment (Fig. 1A) were subcloned from BAC 27E15 and confirmed to contain the putative e_gwH.60.32.1 gene through DNA sequencing. As shown in Fig. 1B, phr1 was restored to photoreactivation proficiency when transformed with the 13.2-kb fragment, as well as with the 11.3-kb HindIII/ClaI subclone.
To determine whether the photoreactivation deficiency of phr1 was caused by a defect in FO biosynthesis, a method was developed to measure FO levels from Chlamydomonas cells using HPLC. Cell extracts were prepared from the complemented strains described above as well as from wild type and phr1 cells. Based on chlorophyll content, an equivalent amount of extract from each strain was run on HPLC. The wild type and complemented strains all produced a well defined peak with a retention time of 9.5 min, directly matching the retention time for a synthesized FO standard (Fig. 1C). In contrast, the phr1 strain produced only a slight increase in the base line at the same retention time, likely indicating a deficit in FO levels. These results, in combination with the photoreactivation complementation assays, indicate that e_gwH.60.32.1 is the PHR1 gene.
An additional component of the Chlamydomonas genome sequencing project aligned over 30,000 expressed sequence tags with their respective genes in the Chlamydomonas genome (36, 37). However, at the initiation of this project, no EST clones of PHR1 had been identified. Therefore, the exon/intron boundaries of the PHR1 gene were determined by comparing genomic sequence with the sequences of products from 5′-RACE, 3′-RACE, and RT-PCR (Fig. 2A). To be certain that the 5′-RACE, 3′-RACE, and RT-PCR products were generated from cDNA and not from contaminating genomic DNA, all products spanned at least one exon/intron boundary. These studies revealed that the PHR1 gene is composed of 18 exons and covers ∼9.5 kb of genome sequence (Fig. 2A).
The deduced PHR1 transcript is 4,169 nt in length and contains two open reading frames (ORFs) that are upstream of the FO synthase homolog ORF (Fig. 2B). The larger upstream ORF initiates at nucleotide 9. It encodes a putative protein of 71 amino acids (7.6 kDa) and, of the two upstream ORFs, terminates closest (41 nt upstream) to the FO synthase homolog ORF. The smaller upstream ORF initiates at nt 80 and encodes a putative protein of 28 amino acids (3.1 kDa). A BLAST search revealed that the larger putative protein did not have significant similarity with known proteins, whereas the smaller had only low scoring similarity with known proteins. The ORF encoding the FO synthase homolog initiates at nt 266 and results in a predicted protein of 1,106 amino acids (114.1 kDa).
To determine the nature of the phr1 mutation, the phr1 allele was amplified by PCR and sequenced. An amber (UAG) nonsense mutation was identified (and confirmed by a separate PCR amplification) in exon 12 of the FO synthase homolog gene. This mutation truncates the phr1p protein after amino acid 783 (supplemental Fig. S1).
PHR1 Protein
FO synthases from Euryarchaea and cyanobacteria consist of two paralogous subunits, CofG and CofH. In actinomycetes, the genes for the two subunits have fused and encode the FO synthase in a single bifunctional polypeptide (16, 19). The PHR1 protein from Chlamydomonas is most similar to the bifunctional FO synthases. For instance, PHR1p has 34% amino acid sequence identity with the FO synthases from M. bovis BCG and M. smegmatis (supplemental Fig. S1). Moreover, the N-terminal half of the bifunctional FO synthases has the most similarity with CofG, whereas the C-terminal half is most similar to CofH. This is also the case for PHR1p. Finally, members of the radical AdoMet superfamily of proteins are characterized by a cysteine-rich motif (CXXXCXXC) and a glycine-rich motif that is ∼30 amino acids downstream of the cysteine-rich motif (17). CofG and CofH are members of this superfamily, and as a result, the bifunctional FO synthases have two copies of both the cysteine- and glycine-rich motifs, one in the N-terminal half and one in the C-terminal half. PHR1p also has these characteristic motifs in its N- and C-terminal halves (supplemental Fig. S1). Despite the similarities that PHR1p has with the bifunctional FO synthases, it also has some traits that are unique. For instance, in PHR1p the intervening sequence between the regions of CofG and CofH homology is ∼170 amino acids in length, although it is only ∼75 amino acids in the FO synthases from Mycobacteria. Also, in both the N- and C-terminal halves of PHR1p, there is an insert of ∼20 amino acids that is not found in the other characterized FO synthases. Finally, based on the alignment shown in supplemental Fig. S1, PHR1p has an N-terminal extension of ∼60 residues compared with the other FO synthases. Computer modeling predicts that this N-terminal extension contains a chloroplast transit peptide (30–32).
Heterologous Expression of PHR1p in E. coli
E. coli are unable to synthesize FO (38). However, E. coli strains that heterologously expressed the FO synthase from M. smegmatis and coexpressed the two subunits for the FO synthase from M. jannaschii were capable of generating measurable amounts of FO (16). Therefore, to provide further support that the PHR1 gene encodes a functional FO synthase, a full-length PHR1 cDNA was generated (Fig. 3). PCR was then used to engineer the PHR1 cDNA for heterologous expression in E. coli. The 5′-end of the PHR1 cDNA was engineered so the PHR1 protein would begin at amino acid 52, excluding the predicted chloroplast targeting sequence (NoCTP). The 3′-end was engineered to remove the termination codon for the protein allowing PHR1p NoCTP to be expressed as a C-terminal S-tag fusion protein. Cells induced to express the PHR1p NoCTP construct produced a protein of approximately the correct size (∼111.5 kDa) (Fig. 4A, lane 4) that was recognized by anti-S-tag antibody (Fig. 4B, lane 4) and was not found in the vector-only control (Fig. 4, A and B, lane 2).
FIGURE 4.
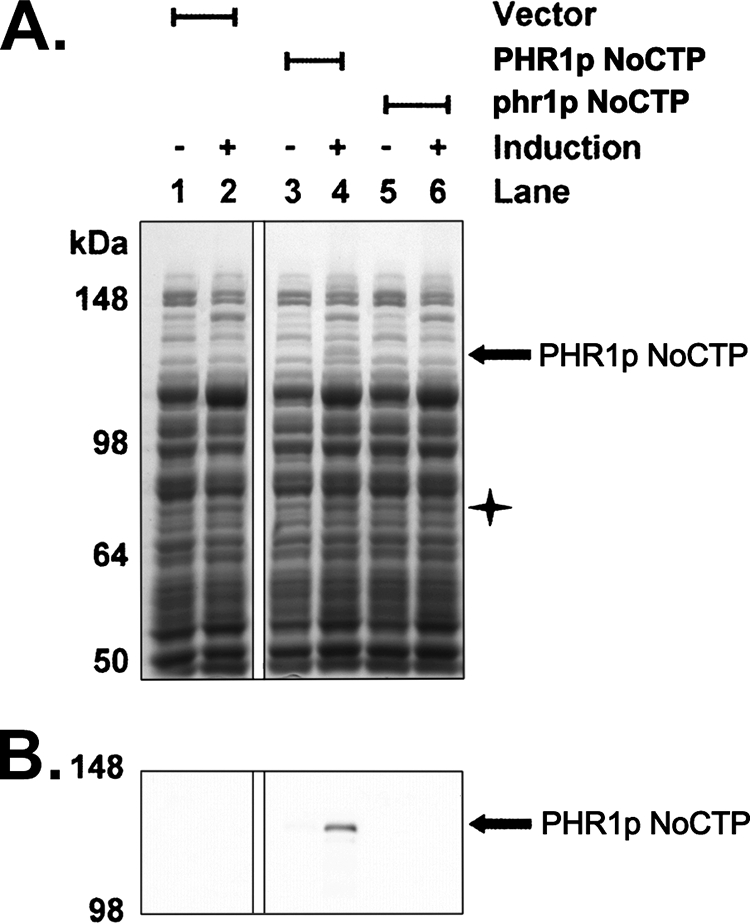
Heterologous expression of Chlamydomonas PHR1p NoCTP in E. coli. E. coli cells were transformed with either an empty expression vector (lanes 1 and 2), a PHR1p NoCTP expression plasmid (lanes 3 and 4), or a mutant phr1p NoCTP expression plasmid (lanes 5 and 6). Equivalent amounts of cell lysate from before (lanes 1, 3, and 5) or after (lanes 2, 4, and 6) 3 h of IPTG induction for each strain were run on SDS-polyacrylamide gels and either stained with GelCode Blue Safe Protein Stain (A) or transferred to nitrocellulose for Western blot with anti-S-tag antibody (B). The PHR1p NoCTP protein is indicated with black arrows. The star indicates the approximate mass of the undetectable phr1p NoCTP protein. Images shown for this figure are from the same gel and the same nitrocellulose membrane with unrelated lanes removed for clarity.
A nearly identical construct was created that differed only in that it contained the phr1 mutation. This construct was also transformed into E. coli and induced for expression. As anticipated, the 111.5-kDa protein that was detected by Coomassie staining, and anti-S-tag Western blot in the PHR1p NoCTP sample was undetectable in the phr1p NoCTP sample (Fig. 4, A and B, lane 6). Because the phr1p NoCTP protein is truncated prior to the translation of the C-terminal S-tag, it is undetectable by anti-S-tag Western blot at the predicted molecular mass of 75.4 kDa (data not shown). Unfortunately, the truncated phr1p NoCTP protein is also undetectable by Coomassie staining (Fig. 4A, lane 6). Because the phr1p NoCTP construct is identical to the PHR1p NoCTP except for the single nucleotide phr1 mutation, it is highly likely that phr1p NoCTP is expressed. Whether phr1p NoCTP is stably expressed and just undetectable by Coomassie staining or whether phr1p NoCTP is unstable and degraded is unknown.
Heterologously Expressed PHR1p Produces FO
E. coli cells that heterologously express FO synthase generate FO and secrete it into the culture medium. This secreted FO can be detected by measuring the fluorescence (excitation wavelength, 420 nm; emission wavelength, 480 nm) of the culture medium (16). Consequently, comparing the fluorescence intensity of culture medium from cells that are and are not heterologously expressing FO synthase can serve as an indicator of FO synthase activity. Therefore, a comparison was made between the fluorescence intensity of culture media from cells expressing PHR1p NoCTP and control cells expressing empty vector. Fig. 5A shows that medium from cells expressing PHR1p NoCTP had fluorescence measurements that were nearly 3.5 times higher than the empty vector controls, indicating that PHR1p is a functional FO synthase. Moreover, the culture medium from cells expressing phr1p NoCTP produced fluorescence levels similar to empty vector controls, suggesting that the phr1p protein produced in E. coli retains little or no residual ability to synthesize FO.
FIGURE 5.

E. coli cells that heterologously express Chlamydomonas PHR1p NoCTP produce a molecule with similarity to FO. A, culture medium from E. coli cells heterologously expressing the Chlamydomonas PHR1p NoCTP protein has increased fluorescence. Shown are fluorescent measurements (excitation 420 nm; emission 480 nm) of culture supernatant from E. coli cells transformed with either an empty expression vector, a PHR1p NoCTP expression plasmid, or a phr1p NoCTP expression plasmid and induced for expression with IPTG for 3 h. Values are presented as the mean of the means of three separate experiments measured in triplicate. Error bars indicate S.D. B, thin layer chromatography was performed on E. coli cell extracts from cells transformed with either an empty expression vector (lane 1), a PHR1p NoCTP expression plasmid (lane 2), or a phr1p NoCTP expression plasmid (lane 3) and induced for expression with IPTG for 3 h. Results were visualized with long wavelength UV light. TLC matrix was physically removed from lanes 1–3 at level indicated by arrow and extracted in preparation for HPLC. C, HPLC profiles of FO standard and spots extracted from TLC lanes 1–3.
As an additional means to verify that PHR1p NoCTP expression in E. coli was producing FO, thin layer chromatography was performed on extracts from cells expressing PHR1p NoCTP, phr1p NoCTP, and empty vector. Fig. 5B shows the results of the TLC visualized with long wavelength UV light, which causes FO to fluoresce green-blue. Indeed, a green-blue fluorescent spot with a Retention factor (Rf) = 0.43 was identified in the cells expressing PHR1p NoCTP (Fig. 5B, lane 2), but not in the vector control (lane 1) or in the cells expressing phr1p NoCTP (lanes 3). At nearly the same Rf, there migrated a yellow-orange spot, but this spot was found in all three lanes. The green-blue spot from the PHR1p NoCTP sample was physically removed from the TLC plate and extracted from the TLC silica. HPLC analysis of the extracted TLC spot was performed and compared with a synthesized FO standard run under the same conditions. As Fig. 5C shows the blue-green fluorescent molecule extracted from the TLC plate eluted from the HPLC column with a retention time matching that of the synthesized FO standard. HPLC analysis of TLC silica extracts from the vector control and phr1p NoCTP sample revealed no fluorescent signal at the same retention time as FO (Fig. 5C).
DISCUSSION
The phr1 strain of Chlamydomonas was initially characterized with a severe deficit in photoreactivation (20). In this work, we have shown that this deficit is the result of a defect in FO biosynthesis, caused by a mutation in the PHR1 gene, which encodes an FO synthase. The PHR1 gene is composed of 18 exons and encodes a protein of 1,106 amino acids. The PHR1p protein is the first eukaryotic FO synthase to be characterized.
We hypothesize that the role of PHR1p in photoreactivation is to produce FO, which serves as the light-harvesting chromophore for PHR2p, a known CPD-specific DNA photolyase in Chlamydomonas. Studies are currently underway to identify the chromophore complement of PHR2p. If FO serves as the photoantenna chromophore for PHR2p, which is almost certain because PHR2p requires functional PHR1p for full activity (22), then the phr1 strain of Chlamydomonas is the first strain in any organism to show that a defect in the biosynthesis of a DNA photolyase chromophore can cause a photoreactivation deficiency. This discovery would present an exception to the existing paradigm that second chromophores are nonessential, except under limited light conditions (4, 5, 11, 12).
Understanding why photoreactivation in Chlamydomonas relies so heavily on FO is of interest. Previous work has demonstrated that photolyase is able to bind FAD even in the absence of a second chromophore (5, 39). It appears that this is also the case for PHR2p given the following two results. 1) The ability to bind UV-irradiated DNA of extracts from phr1 cells overexpressing an epitope-tagged version of PHR2p was enhanced following the addition of FAD (40). 2) PHR2p expressed as a fusion protein with maltose-binding protein was able to complement a photoreactivation-deficient E. coli strain that presumably does not produce FO (21). Given these data indicating PHR2p can bind FAD in the absence of FO, it will be informative to determine whether PHR2p binds FAD in the phr1 mutant, and if so, to determine whether it binds with the same affinity as in cells with normal levels of FO. An important consideration for these studies is that the phr1 mutant does retain a limited ability to photoreactivate pyrimidine dimers (20), suggesting that PHR2p binds at least some FAD in the phr1 background. The outcomes of these studies are important for understanding photoreactivation in Chlamydomonas and perhaps for photoreactivation in other organisms as well.
The fact that no other photoreactivation mutants have been identified that are the result of a defect in chromophore biosynthesis is not entirely surprising given that FAD is essential for several metabolic processes, and mutations in the biosynthesis of FAD would likely be lethal, as they are in Saccharomyces cerevisiae (41). Still, it would be of interest to determine whether mutants for photoantenna biosynthesis in organisms other than Chlamydomonas are photoreactivation-deficient. FO synthase mutants have been identified in mycobacteria (19, 42), but although mycobacteria have been shown to perform photoreactivation (43, 44), it is unknown whether FO serves as the photoantenna for mycobacteria photolyases. 5,10-Methenyltetrahydrofolate (5,10-MTHF) serves as the photoantenna in many DNA photolyases, including for S. cerevisiae and E. coli (45). Although studies with purified photolyase have indicated that 5,10-MTHF is not essential for repairing CPDs, except under limited light conditions, the in vivo requirement may be different. Mutants in the synthesis of 5,10-MTHF are available and could readily be tested. For instance, the S. cerevisiae strain MWY3 has disrupted MTD1 and ADE3 genes (46). ADE3 encodes the trifunctional C1-tetrahydrofolate (THF) synthase (47). The C1-THF synthase generates 5,10-MTHF from 10-formyl-THF via its MTHF cyclohydrolase activity or from 5,10-methylene-THF via its NADP-dependent methylene-THF dehydrogenase activity. MTD1 generates 5,10-MTHF from 5,10-methylene-THF via its NAD-dependent methylene-THF dehydrogenase activity. The level of 10-formyl-THF, which is in chemical equilibrium with 5,10-MTHF, was tested in MWY3 and was found to be undetectable, suggesting that the level of 5,10-MTHF is also low or nonexistent (46).
In some organisms FO serves as the precursor for coenzyme F420, and in other organisms FO appears to be produced as an end product (16). FbiA and FbiB are enzymes that catalyze steps in the biosynthesis of F420 downstream of FO synthase (48). Searches of the Chlamydomonas genome revealed no genes encoding putative homologs of FbiA and FbiB. This, as well as the absence of additional peaks unique to the wild type and complemented strains in the HPLC profiles of Chlamydomonas extracts (Fig. 1C), suggests that Chlamydomonas synthesizes FO as an end product. It has been shown that FO can act as a substitute for coenzyme F420 in F420-requiring enzymes (49–51). Perhaps, in the absence of F420, FO serves as the primary coenzyme for unknown enzymatic reactions in Chlamydomonas. The phr1 strain could provide a resource for identifying these putative functions.
Supplementary Material
Acknowledgments
We are grateful to Dr. Robert H. White (Virginia Polytechnic Institute and State University, Blacksburg, VA) for providing the synthesized FO standard; Dr. Dean R. Appling (University of Texas, Austin) for discussions regarding 5,10-MTHF in S. cerevisiae; Dr. Ken Renner (University of South Dakota, Vermillion) for use of the HPLC fluorescence detector, and to Dr. Gary Small for helpful comments regarding the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant P20RR015567 from the NCRR (to P. J. R.). This work was also supported by Avera Research Institute (Sioux Falls, SD), the Great Plains Medical Research Foundation Grant GPMRF-001-2009 (to J. L. P.), and with resources and the use of facilities at the Sioux Falls Veterans Affairs Medical Center (Sioux Falls, SD).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
The nucleotide sequence(s) reported in this paper has been submitted to the GenBankTM/EBI Data Bank with accession number(s) HM156042.
- CPD
- cyclobutane pyrimidine dimer
- FO
- 7,8-didemethyl-8-hydroxy-5-deazariboflavin
- AdoMet
- S-adenosylmethionine
- BAC
- bacterial artificial chromosome
- NoCTP
- no chloroplast targeting sequence
- 5,10-MTHF
- 5,10-methenyltetrahydrofolate
- THF
- tetrahydrofolate
- RACE
- rapid amplification of cDNA ends
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- nt
- nucleotide.
REFERENCES
- 1.Sinha R. P., Häder D. P. (2002) Photochem. Photobiol. Sci. 1, 225–236 [DOI] [PubMed] [Google Scholar]
- 2.Mitchell D. L., Nairn R. S. (1989) Photochem. Photobiol. 49, 805–819 [DOI] [PubMed] [Google Scholar]
- 3.Beukers R., Eker A. P., Lohman P. H. (2008) DNA Repair 7, 530–543 [DOI] [PubMed] [Google Scholar]
- 4.Sancar A. (2003) Chem. Rev. 103, 2203–2237 [DOI] [PubMed] [Google Scholar]
- 5.Jorns M. S., Wang B. Y., Jordan S. P., Chanderkar L. P. (1990) Biochemistry 29, 552–561 [DOI] [PubMed] [Google Scholar]
- 6.Eker A. P., Hessels J. K., van de Velde J. (1988) Biochemistry 27, 1758–1765 [Google Scholar]
- 7.Kiener A., Husain I., Sancar A., Walsh C. (1989) J. Biol. Chem. 264, 13880–13887 [PubMed] [Google Scholar]
- 8.Eker A. P., Kooiman P., Hessels J. K., Yasui A. (1990) J. Biol. Chem. 265, 8009–8015 [PubMed] [Google Scholar]
- 9.Eker A. P., Dekker R. H., Berends W. (1981) Photochem. Photobiol. 33, 65–72 [DOI] [PubMed] [Google Scholar]
- 10.Glas A. F., Maul M. J., Cryle M., Barends T. R., Schneider S., Kaya E., Schlichting I., Carell T. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 11540–11545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sancar A. (2008) J. Biol. Chem. 283, 32153–32157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamm-Alvarez S., Sancar A., Rajagopalan K. V. (1989) J. Biol. Chem. 264, 9649–9656 [PubMed] [Google Scholar]
- 13.Jorns M. S., Wang B., Jordan S. P. (1987) Biochemistry 26, 6810–6816 [DOI] [PubMed] [Google Scholar]
- 14.Heelis P. F., Payne G., Sancar A. (1987) Biochemistry 26, 4634–4640 [DOI] [PubMed] [Google Scholar]
- 15.Kleiner O., Butenandt J., Carell T., Batschauer A. (1999) Eur. J. Biochem. 264, 161–167 [DOI] [PubMed] [Google Scholar]
- 16.Graham D. E., Xu H., White R. H. (2003) Arch. Microbiol. 180, 455–464 [DOI] [PubMed] [Google Scholar]
- 17.Sofia H. J., Chen G., Hetzler B. G., Reyes-Spindola J. F., Miller N. E. (2001) Nucleic Acids Res. 29, 1097–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang S. C., Frey P. A. (2007) Trends Biochem. Sci. 32, 101–110 [DOI] [PubMed] [Google Scholar]
- 19.Choi K. P., Kendrick N., Daniels L. (2002) J. Bacteriol. 184, 2420–2428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cox J. L., Small G. D. (1985) Mutat. Res. 146, 249–255 [DOI] [PubMed] [Google Scholar]
- 21.Petersen J. L., Lang D. W., Small G. D. (1999) Plant Mol. Biol. 40, 1063–1071 [DOI] [PubMed] [Google Scholar]
- 22.Petersen J. L., Small G. D. (2001) Nucleic Acids Res. 29, 4472–4481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gross C. H., Ranum L. P., Lefebvre P. A. (1988) Curr. Genet. 13, 503–508 [DOI] [PubMed] [Google Scholar]
- 24.Rymarquis L. A., Handley J. M., Thomas M., Stern D. B. (2005) Plant Physiol. 137, 557–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kindle K. L. (1990) Proc. Natl. Acad. Sci. U.S.A. 87, 1228–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Debuchy R., Purton S., Rochaix J. D. (1989) EMBO J. 8, 2803–2809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gorman D. S., Levine R. P. (1965) Proc. Natl. Acad. Sci. U.S.A. 54, 1665–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elizabeth H, Harris (1989) The Chlamydomonas Sourcebook: A Comprehensive Guide to Biology and Laboratory Use, p. 608, Academic Press, Inc., San Diego: [DOI] [PubMed] [Google Scholar]
- 29.Corpet F. (1988) Nucleic Acids Res. 16, 10881–10890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Emanuelsson O., Nielsen H., Brunak S., von Heijne G. (2000) J. Mol. Biol. 300, 1005–1016 [DOI] [PubMed] [Google Scholar]
- 31.Bannai H., Tamada Y., Maruyama O., Nakai K., Miyano S. (2002) Bioinformatics 18, 298–305 [DOI] [PubMed] [Google Scholar]
- 32.Schein A. I., Kissinger J. C., Ungar L. H. (2001) Nucleic Acids Res. 29, E82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sambrook J., Fritsch E. F., Maniatis T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 34.Grossman A. R., Harris E. E., Hauser C., Lefebvre P. A., Martinez D., Rokhsar D., Shrager J., Silflow C. D., Stern D., Vallon O., Zhang Z. (2003) Eukaryot. Cell 2, 1137–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Merchant S. S., Prochnik S. E., Vallon O., Harris E. H., Karpowicz S. J., Witman G. B., Terry A., Salamov A., Fritz-Laylin L. K., Maréchal-Drouard L., Marshall W. F., Qu L. H., Nelson D. R., Sanderfoot A. A., Spalding M. H., Kapitonov V. V., Ren Q., Ferris P., Lindquist E., Shapiro H., Lucas S. M., Grimwood J., Schmutz J., Cardol P., Cerutti H., Chanfreau G., Chen C. L., Cognat V., Croft M. T., Dent R., Dutcher S., Fernández E., Fukuzawa H., González-Ballester D., González-Halphen D., Hallmann A., Hanikenne M., Hippler M., Inwood W., Jabbari K., Kalanon M., Kuras R., Lefebvre P. A., Lemaire S. D., Lobanov A. V., Lohr M., Manuell A., Meier I., Mets L., Mittag M., Mittelmeier T., Moroney J. V., Moseley J., Napoli C., Nedelcu A. M., Niyogi K., Novoselov S. V., Paulsen I. T., Pazour G., Purton S., Ral J. P., Riaño-Pachón D. M., Riekhof W., Rymarquis L., Schroda M., Stern D., Umen J., Willows R., Wilson N., Zimmer S. L., Allmer J., Balk J., Bisova K., Chen C. J., Elias M., Gendler K., Hauser C., Lamb M. R., Ledford H., Long J. C., Minagawa J., Page M. D., Pan J., Pootakham W., Roje S., Rose A., Stahlberg E., Terauchi A. M., Yang P., Ball S., Bowler C., Dieckmann C. L., Gladyshev V. N., Green P., Jorgensen R., Mayfield S., Mueller-Roeber B., Rajamani S., Sayre R. T., Brokstein P., Dubchak I., Goodstein D., Hornick L., Huang Y. W., Jhaveri J., Luo Y., Martínez D., Ngau W. C., Otillar B., Poliakov A., Porter A., Szajkowski L., Werner G., Zhou K., Grigoriev I. V., Rokhsar D. S., Grossman A. R. (2007) Science 318, 245–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shrager J., Hauser C., Chang C. W., Harris E. H., Davies J., McDermott J., Tamse R., Zhang Z., Grossman A. R. (2003) Plant Physiol. 131, 401–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jain M., Shrager J., Harris E. H., Halbrook R., Grossman A. R., Hauser C., Vallon O. (2007) Nucleic Acids Res. 35, 2074–2083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takao M., Oikawa A., Eker A. P., Yasui A. (1989) Photochem. Photobiol. 50, 633–637 [DOI] [PubMed] [Google Scholar]
- 39.Payne G., Wills M., Walsh C., Sancar A. (1990) Biochemistry 29, 5706–5711 [DOI] [PubMed] [Google Scholar]
- 40.Petersen J. L. (2001) Chlamydomonas Genes Involved in Photoreactivation, Doctoral Dissertation, Ann Arbor, MI [Google Scholar]
- 41.Giaever G., Chu A. M., Ni L., Connelly C., Riles L., Véronneau S., Dow S., Lucau-Danila A., Anderson K., André B., Arkin A. P., Astromoff A., El-Bakkoury M., Bangham R., Benito R., Brachat S., Campanaro S., Curtiss M., Davis K., Deutschbauer A., Entian K. D., Flaherty P., Foury F., Garfinkel D. J., Gerstein M., Gotte D., Güldener U., Hegemann J. H., Hempel S., Herman Z., Jaramillo D. F., Kelly D. E., Kelly S. L., Kötter P., LaBonte D., Lamb D. C., Lan N., Liang H., Liao H., Liu L., Luo C., Lussier M., Mao R., Menard P., Ooi S. L., Revuelta J. L., Roberts C. J., Rose M., Ross-Macdonald P., Scherens B., Schimmack G., Shafer B., Shoemaker D. D., Sookhai-Mahadeo S., Storms R. K., Strathern J. N., Valle G., Voet M., Volckaert G., Wang C. Y., Ward T. R., Wilhelmy J., Winzeler E. A., Yang Y., Yen G., Youngman E., Yu K., Bussey H., Boeke J. D., Snyder M., Philippsen P., Davis R. W., Johnston M. (2002) Nature 418, 387–391 [DOI] [PubMed] [Google Scholar]
- 42.Guerra-Lopez D., Daniels L., Rawat M. (2007) Microbiology 153, 2724–2732 [DOI] [PubMed] [Google Scholar]
- 43.David H. L., Jones W. D., Jr., Newman C. M. (1971) Infect. Immun. 4, 318–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peccia J., Hernandez M. (2001) Appl. Environ. Microbiol. 67, 4225–4232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson J. L., Hamm-Alvarez S., Payne G., Sancar G. B., Rajagopalan K. V., Sancar A. (1988) Proc. Natl. Acad. Sci. U.S.A. 85, 2046–2050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.West M. G., Horne D. W., Appling D. R. (1996) Biochemistry 35, 3122–3132 [DOI] [PubMed] [Google Scholar]
- 47.Paukert J. L., Williams G. R., Rabinowitz J. C. (1977) Biochem. Biophys. Res. Commun. 77, 147–154 [DOI] [PubMed] [Google Scholar]
- 48.Choi K. P., Bair T. B., Bae Y. M., Daniels L. (2001) J. Bacteriol. 183, 7058–7066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Michel R., Massanz C., Kostka S., Richter M., Fiebig K. (1995) Eur. J. Biochem. 233, 727–735 [DOI] [PubMed] [Google Scholar]
- 50.Jacobson F. S., Daniels L., Fox J. A., Walsh C. T., Orme-Johnson W. H. (1982) J. Biol. Chem. 257, 3385–3388 [PubMed] [Google Scholar]
- 51.Fox J. A., Livingston D. J., Orme-Johnson W. H., Walsh C. T. (1987) Biochemistry 26, 4219–4227 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.