

Abstract
Mitochondria dynamically fuse and divide within cells, and the proper balance of fusion and fission is necessary for normal mitochondrial function, morphology, and distribution. Drp1 is a dynamin-related GTPase required for mitochondrial fission in mammalian cells. It harbors four distinct domains: GTP-binding, middle, insert B, and GTPase effector. A lethal mutation (A395D) within the Drp1 middle domain was reported in a neonate with microcephaly, abnormal brain development, optic atrophy, and lactic acidemia (Waterham, H. R., Koster, J., van Roermund, C. W., Mooyer, P. A., Wanders, R. J., and Leonard, J. V. (2007) N. Engl. J. Med. 356, 1736–1741). Mitochondria within patient-derived fibroblasts were markedly elongated, but the molecular mechanisms underlying these findings were not demonstrated. Because the middle domain is particularly important for the self-assembly of some dynamin superfamily proteins, we tested the hypothesis that this A395D mutation, and two other middle domain mutations (G350D, G363D) were important for Drp1 tetramerization, higher order assembly, and function. Although tetramerization appeared largely intact, each of these mutations compromised higher order assembly and assembly-dependent stimulation of Drp1 GTPase activity. Moreover, mutant Drp1 proteins exhibited impaired localization to mitochondria, indicating that this higher order assembly is important for mitochondrial recruitment, retention, or both. Overexpression of these middle domain mutants markedly inhibited mitochondrial division in cells. Thus, the Drp1 A395D lethal defect likely resulted in impaired higher order assembly of Drp1 at mitochondria, leading to decreased fission, elongated mitochondria, and altered cellular distribution of mitochondria.
Keywords: Human Genetics, Mitochondria, Neurodegeneration, Neurological Diseases, Protein Assembly, GTPase, Fission
Introduction
Mitochondria are critical organelles that generate ATP for cellular energy consumption. In addition, they are involved in redox and metabolic regulation, maintenance of calcium homeostasis, signaling, and fatty acid oxidation. Mitochondria undergo frequent fusion and fission events depending on cell type that are responsible for proper mitochondrial function as well as maintaining mitochondrial size, shape, and cellular distribution. Alterations in the balance of mitochondrial fusion and fission have been implicated in physiologic mechanisms such as cell division, chemotaxis, and neuronal dendrite development as well as in pathogenic processes such as apoptosis, autophagy, aging, and neurodegeneration (1–12). Several GTPases in the dynamin superfamily, including the mitofusins Mfn1/Mfn2, OPA1, and dynamin-related protein 1 (Drp1),6 are responsible for fusion and fission of mitochondria (13–15). Pathologic mutations in a number of these proteins cause autosomal dominant neurological disorders such as Charcot-Marie-Tooth neuropathy type 2A (Mfn2) and OPA1 (4), underscoring the importance of maintenance of mitochondrial morphology in mechanisms of neurodegeneration.
Drp1 is an evolutionally conserved, multimeric GTPase required for mitochondrial fission. Interestingly, it also has been implicated in peroxisomal division (20). Drp1 likely mediates mitochondrial and peroxisomal fission through the formation of large multimeric spirals at mitochondrial fission sites, similar to those formed by dynamin at sites of endocytosis (16–19). Similar to dynamin, Drp1 is a multidomain GTPase that consists of a GTPase domain, a middle assembly domain, a B domain of unknown function, and a GTPase-effector domain (GED). However, Drp1 lacks the pleckstrin homology domain and C-terminal proline-rich domain found in dynamin (21). Intermolecular interactions among Drp1 monomers and intramolecular interactions between the N-terminal GTP binding domain and the C-terminal GED are critical for Drp1 assembly and functional regulation (22–25).
The dynamin middle domain is critical for dynamin tetramer formation as well as higher order assembly on membranes (26). Similarly, mutations within the middle domain of Dnm1, the Drp1 ortholog in the budding yeast Saccharomyces cerevisiae, disrupt the formation of mitochondrial fission complexes (27, 28). In mammalian cells, peroxisomal and mitochondrial division are impaired by a Drp1 middle domain mutation at conserved residue 363 (G363D) on one allele (29). Together, these results suggest that middle domain-dependent assembly of Drp1 may have an important role in regulating both Drp1 function and mitochondrial morphology.
Several years ago, a de novo mutation in one allele at conserved residue 395 (A395D) within the Drp1 middle domain was reported in a neonate with microcephaly, abnormal brain development, optic atrophy, and lactic acidemia; she died at 37 days (30). Mitochondria within cultured skin fibroblasts derived from this patient were markedly elongated, but the molecular mechanisms underlying these findings were not investigated. Given the importance of the middle domain in dynamin self-assembly and activity, we hypothesized that the Drp1 A395D patient mutation may be preventing Drp1 higher order assembly, leading to defects in mitochondrial fission. We, therefore, examined the effects of this A395D mutation and two other mutations at highly conserved middle domain residues (G350D, G363D) on Drp1 self-assembly and function. Our results indicate that the Drp1 A395D lethal defect and these two other middle domain mutations impair higher order assembly of Drp1 at mitochondria, leading to markedly elongated mitochondria, presumably from decreased fission.
EXPERIMENTAL PROCEDURES
DNA Constructs
Eukaryotic expression vectors for Myc- and HA epitope-tagged pGW1-Drp1 have been described previously (24). For yeast two-hybrid assays, wild-type and mutated Drp1 fragments were cloned into pGAD10 (prey) and pBHA (bait) vectors as described previously (24, 25). Drp1 middle domain fragments (residues 332–502) were generated by PCR using Pfu Turbo DNA polymerase (Agilent Stratagene, La Jolla, CA) as EcoRI/XhoI fragments and were subcloned into pGAD10/GADT7 and pBHA. The K38A, A395D, G350D, and G363D mutations were introduced into all constructs using the QuikChange method (Agilent Stratagene).
Yeast Two-hybrid Assays
The yeast L40a strain with LexA operators upstream of HIS3 and lacZ reporter genes was used to perform yeast two-hybrid assays. Growth of transformants (serial 10-fold dilutions) on His/Leu/Trp drop out plates supplemented with 10 mm 3-amino-1,2,4-triazole (Sigma-Aldrich, St. Louis, MO) was used to measure interaction strength (34, 35). At least three independent trials were performed for each experiment, with similar results.
Cross-linking, Immunoprecipitation, and Immunoblotting
Chemical cross-linking studies of Myc-Drp1 overexpressed in HeLa cells were performed using bis(sulfosuccinimidyl) suberate (Pierce Biotechnology, Rockford, IL) (24). For co-immunoprecipitation studies, extracts from HeLa cells co-transfected with HA- and Myc-Drp1(wild-type or mutant, as indicated) or else transfected with Myc-Drp1 alone were immunoprecipitated using anti-HA antibodies as described previously (24). Protein samples were resolved by SDS-PAGE, transferred to nitrocellulose membranes, and immunoblotted with the following primary antibodies as described by Zhu et al. (24): mouse monoclonal anti-Myc (9E10; Santa Cruz Biotechnology, Santa Cruz, CA), rabbit polyclonal anti-HA (Y-11; Santa Cruz Biotechnology), mouse monoclonal anti-actin (AC-40; Sigma-Aldrich), and mouse monoclonal anti-HSP60 (LK1; Sigma-Aldrich).
Fusion Protein Purification
pCal-n-EK-Drp1 was transformed into Escherichia coli BL21 (DE3) cells, and pCal-n-EK-Drp1 middle domain mutant constructs were transformed into E. coli Rosetta cells (Novagen, Gibbstown, NJ). Cells were grown at 37 °C in Luria broth (LB) with carbenicillin (50 μg/ml) to an A600 of ∼0.6 while shaking at 250 rpm. Protein expression was then induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside at 14 °C for 18 h. Cells were harvested by centrifugation using a Sorvall GS3 rotor at 4000 rpm for 30 min and were resuspended in calmodulin-binding peptide (CBP) column buffer A (20 mm Tris, 250 mm NaCl, 2 mm CaCl2, 1 mm imidazole, pH 8.0) containing protease inhibitors (Complete, EDTA-free Protease Inhibitor Mixture; Roche Applied Science, Indianapolis, IN). Cells were then lysed by four passes through an EmulsiFlex C3 (Avestin, Ottawa, Canada), DNase was added to 1 μg/ml with 10 mm MgCl2, and lysates were cleared by centrifugation using a Sorvall SS34 rotor at 16,000 rpm for 30 min at 4 °C. CBP fusion proteins were isolated from the resulting supernatant by affinity chromatography using calmodulin-affinity resin (Agilent Stratagene). Bound fusion proteins were eluted with CBP-column buffer B (20 mm Tris, 250 mm NaCl, 50 mm EDTA, pH 8.0). Fractions containing CBP-Drp1 were pooled, concentrated, and dialyzed overnight at 4 °C into CBP column buffer A. Protein concentration was quantified using a Bradford assay. Purified protein was stored in CBP column buffer A at 4 °C until use.
GTPase Activity Assay
A continuous GTPase assay was used whereby the rate of GTP hydrolysis was determined through coupling to a GTP regeneration system (16). GTPase activity was assayed in 200 μl of GTPase reaction buffer (25 mm Hepes, 25 mm Pipes, 7.5 mm KCl, 5 mm MgCl2, 1 mm phosphoenolpyruvate, 20 units/ml pyruvate kinase/lactate dehydrogenase, 600 μm NADH, 1 mm GTP, pH 7.0), of which 150 μl was placed into the wells of a 96-well plate. Depletion of NADH over time was measured for 40 min at 23.5 °C by using a Molecular Devices SpectraMAX 250 96-well plate reader. GTPase assays were started by the addition of 2.5 μm Drp1 or Drp1 mutants in CBP-column buffer A, where the NaCl concentration had been adjusted to 500 mm to favor Drp1 unassembled conditions in the stock solution. For determination of salt dependence, the final NaCl concentration was varied between 50 and 500 mm as indicated. For examination of the kinetic lag associated with assembly-dependent increases in GTP hydrolysis, the activities of wild-type CBP-Drp1 or the CBP-Drp1 mutants were assayed as above at 50 mm NaCl.
Immunostaining and Confocal Microscopy
HeLa cells were grown on glass coverslips and transfected with wild-type or mutant Myc-Drp1 using Lipofectamine (Invitrogen, Carlsbad, CA). After 36–48 h, cells were washed with phosphate-buffered saline (PBS, pH 7.4) and fixed with 4% paraformaldehyde for 20 min at room temperature. Cells were incubated in blocking buffer (5% donkey serum, 0.1% bovine serum albumin, and 0.1% Triton X-100 in PBS) for 1 h. Goat anti-Myc epitope (1:500; Bethyl Laboratories, Montgomery, TX) and mouse monoclonal anti-cytochrome c (1:500; BD Biosciences, San Jose, CA) antibodies in blocking buffer were added overnight at 4 °C. Alexa Fluor 488 and Alexa Fluor 555 secondary antibodies (1:500; Invitrogen) were added for 1 h, and after three washes with PBS, coverslips were mounted using Gel/Mount (Biomeda, Foster City, CA). Images were acquired using a Zeiss LSM510 confocal microscope with a 63 × 1.4 NA Plan-APOCHROMAT lens (Carl Zeiss Microimaging, Thornwood, NY). For determination of mitochondrial morphology, three trials (n ≥ 100 transfected HeLa cells per trial) were performed, with mitochondria classified as normal, elongated, fragmented, or other.
Isolation of Mitochondria
HeLa cells transfected with wild-type or mutant Myc-Drp1 were either left untreated or else treated with 1 μm staurosporine (Sigma-Aldrich) for 9 h in the presence of 100 μm benzyloxycarbonyl-VAD-fluoromethyl ketone (Calbiochem). Mitochondria were isolated intact from HeLa cells by sucrose density gradient centrifugation and subjected to SDS-PAGE and immunoblotting as described previously (2, 36).
Size-exclusion Chromatography with Detection by Multiangle Laser Light Scattering and Refractive Index
CBP-Drp1 fusion proteins were purified as above. After elution from the CBP column, fractions containing wild-type CBP-Drp1 or the indicated mutants were pooled, concentrated, and dialyzed overnight into column buffer (20 mm Tris, pH 8.0, 250 mm NaCl) at 4 °C. CBP-Drp1 proteins (500 μl of ∼0.1 mg/ml) were filtered through a 0.45-μm syringe filter and chromatographed at 0.5 ml/min at 25 °C on a Superose 6 10/30 HR (GE Healthcare, Piscataway, NJ) size-exclusion column equilibrated with column buffer. The eluate was detected using a DAWN-EOS multiangle laser light scattering instrument and the Optilab refractive index detector (Wyatt Technologies, Santa Barbara, CA). Data analysis was accomplished using the ASTRA software package (Wyatt Technologies), and traces were normalized to account for minor differences in total protein applied to the column.
Protein Assays
Protein content was determined using the bicinchoninic acid assay kit (Pierce Biotechnology) with bovine serum albumin as the standard.
Statistical Analysis
Statistical significance was determined using two-tailed, unpaired Student's t-tests, assuming unequal variance.
RESULTS
We investigated the effects of the human Drp1 A395D middle domain mutation, which was reported to cause markedly elongated mitochondrial morphology and neonatal death (30). This residue is highly conserved in Drp1 orthologs across different species as well as in the human dynamin-1 protein (Fig. 1A). Because mutations at other highly conserved middle domain residues have been shown to be critical for higher order assembly in both yeast Dnm1 and the dynamins (16, 26–28), we also investigated two of these residues in comparison studies, G350 and G363 (Fig. 1A).
FIGURE 1.

Drp1 middle domain mutations result in elongated mitochondrial morphology. A, top, shown is the schematic of human Drp1 structure (splice variant 1) with boundary amino acids shown for domains. Bottom, shown is phylogenetic sequence alignment of the depicted middle domain region of Drp1 orthologs in the indicated species. Homo sapiens (H. s.) dynamin-1 is included at the bottom. Yellow shading identifies residues identical in all proteins shown. Red arrows identify conserved residues investigated in this study (numbering is shown for human Drp1). B, HeLa cells were transfected with WT or the indicated Drp1 mutants and visualized by confocal immunofluorescence microscopy. Transfected cells are identified by arrows in the lower panels. C, mitochondrial morphology was quantified as described under “Experimental Procedures.” Bar, 10 μm.
We first examined the effects of these middle domain mutations on mitochondrial morphology. Overexpression of wild-type Drp1 in HeLa cells did not alter mitochondrial morphology (Fig. 1B), consistent with earlier findings (22, 24). In contrast, the effects of overexpression of each of the Drp1 middle domain mutants on mitochondrial morphology were very pronounced, with significantly elongated mitochondria compared with overexpression of wild-type Drp1 (Fig. 1). These results are reminiscent of the effects observed in clinical isolates form a patient with the A395D middle domain mutation (30) and of a dominant-negative K38A GTPase mutation in Drp1 that impaired mitochondrial fission (31–33), suggesting a dominant-negative effect of the middle domain mutations as well.
We suspected that elongated mitochondrial phenotype might be due to an alteration of Drp1 localization to mitochondria, retention on the mitochondrial outer membrane, or both. Thus, we examined the effect of the Drp1 middle domain mutations on mitochondrial localization/retention of Drp1 in isolated mitochondrial fractions. Although wild-type and mutant forms of Drp1 were expressed at very similar levels in HeLa cells (Fig. 2A), the levels of the mutant Drp1 proteins on isolated mitochondria were reduced as compared with wild-type Drp1. This result was observed under basal conditions as well as during programmed cell death induced by staurosporine, when wild-type Drp1 mitochondrial recruitment/retention is increased (42) (Fig. 2B). Taken together, these results suggest that each of these middle domain mutations may also have a dominant-negative effect on Drp1 localization to mitochondrial or retention on mitochondria.
FIGURE 2.
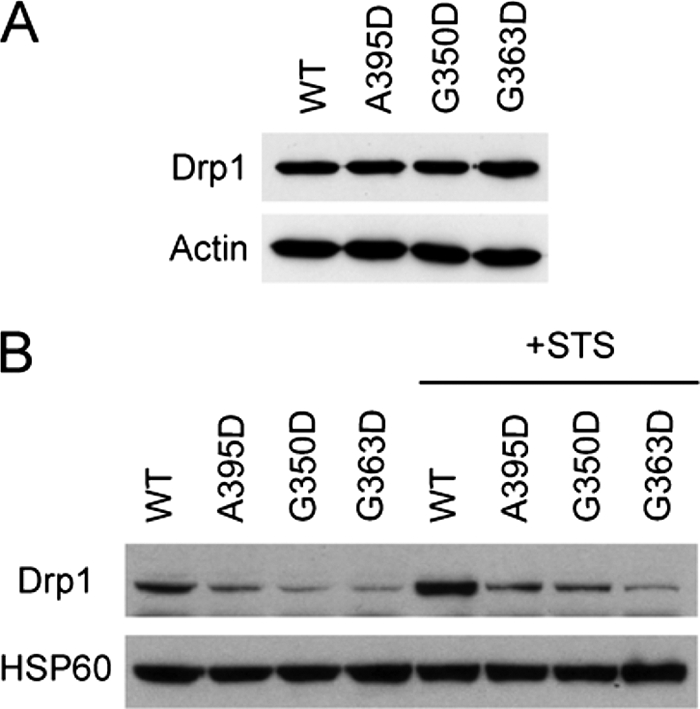
Middle domain mutations decrease Drp1 recruitment/retention on mitochondria. A, expression of the Myc-tagged wild-type (WT) and mutant forms of Drp1 was equal. Actin levels were monitored as a loading control. B, HeLa cells transfected with WT Myc-Drp1, or else the indicated Drp1 mutants were either left untreated or treated with staurosporine (STS) in the presence of the broad caspase inhibitor benzyloxycarbonyl-VAD-fluoromethyl ketone. Levels of Drp1 in the mitochondrial fraction were assessed by immunoblotting, with levels of the mitochondrial protein HSP60 monitored as a loading control.
As a dynamin family member, Drp1 self-assembly is likely governed by a dynamic equilibrium of different assembly states. To test whether these middle domain mutations affect Drp1 assembly, we performed co-immunoprecipitation studies using Drp1 differentially tagged with either HA or Myc epitope. Consistent with previous investigations (24), we observed that wild-type HA-Drp1 co-immunoprecipitated with wild-type Myc-Drp1 (Fig. 3A). This result was not altered by the presence of any of the Drp1 middle domain mutations, suggesting that these Drp1 mutant proteins are able to assemble to the extent measured by co-immunoprecipitation experiments.
FIGURE 3.

Drp1 middle domain mutants do not affect Drp1 self-association. A, co-immunoprecipitation of Myc- and HA-tagged Drp1 proteins is shown. HeLa cells expressing the indicated WT or mutant Drp1 proteins were immunoprecipitated with anti-HA antibodies (αHA IP) and immunoblotted with anti-Myc antibodies. Input represents 20% of the starting lysate. The IgG heavy chain from the precipitating antibodies is noted with an asterisk (*). B, cytosolic extracts from cells overexpressing wild-type or mutant Myc-Drp1 were cross-linked with the indicated concentrations of bis(sulfosuccinimidyl) suberate (BS3), resolved by SDS-PAGE and immunoblotted with anti-Myc antibodies. Migrations of molecular mass standards are indicated at the left. A prominent cross-linked product at ∼280 kDa is indicated with an arrowhead in B.
To further address whether middle domain mutants affect assembly, we pursued chemical cross-linking studies in cell lysate using bis(sulfosuccinimidyl) suberate and immunoblotted for Drp1. Consistent with previous studies (22, 24, 29), SDS-PAGE analysis of cross-linked products of wild-type Myc-Drp1 revealed a prominent species at a molecular mass of ∼280 kDa, which we interpret to be a tetramer. Similar cross-linked products were seen for the Myc-tagged Drp1 G350D and G363D mutants (Fig. 3B), indicating that these mutations do not dominantly interfere with tetramer formation. However, although Myc-Drp1 A395D also exhibited a major cross-linked product consistent with the size of a tetramer, a larger proportion of monomeric A395D was clearly evident at the highest concentration of cross-linker relative to the other mutations, suggesting that the A395D mutation, but not the G350D or G363D mutations, may interfere with Drp1 tetramerization.
To examine the effects of these mutations on both intra-and intermolecular Drp1 self-interactions, we conducted a comprehensive series of yeast two-hybrid tests. We first examined yeast two-hybrid interactions between the N-terminal GTPase/middle domains of Drp1 (residues 1–489, Fig. 1) with the C-terminal B insert/GED domains (residues 502–736) as a measure of intramolecular interactions. We observed a strong yeast-two hybrid interaction between the N-terminal and C-terminal portions of Drp1, consistent with previous reports (22, 24) and highly reminiscent of similar studies investigating dynamin and the dynamin-like GBP and Mx proteins (17, 37–41). The Drp1 middle domain mutations modestly decreased these intramolecular associations regardless of whether the N- or C-terminal portions were used as bait or prey (Fig. 4A). In one reciprocal bait/prey pair with the A395D mutation, this mutation had a more pronounced disruption than others (Fig. 4A, arrowhead). These results suggest that each of the middle domain mutations may cause a decrease in Drp1 intramolecular interactions and that the A395D mutation has the most deleterious effect.
FIGURE 4.

Middle domain mutations disrupt intra- and intermolecular interactions of full-length Drp1. Yeast two-hybrid assays using the HIS3 reporter (sequential 10-fold yeast dilutions are shown) with the indicated bait and prey constructs are shown for intramolecular (A) and intermolecular (B) Drp1 interactions. Drp1 mutations across the top are present in constructs identified as mut. Arrows, arrowheads, and asterisks (*) are referenced under “Results.”
A markedly different situation was observed for the Drp1 intermolecular interactions using yeast two-hybrid assays. The middle domain mutations in all cases inhibited the interactions of the full-length Drp1 proteins and in some cases abolished them, particularly when both bait and prey Drp1 constructs harbored mutations (Fig. 4B, arrowhead and asterisk). This might seem incongruous with the co-immunoprecipitation and chemical cross-linking results in Fig. 3 that demonstrated self-association. However, Nunnari and co-workers (28) reported a similar result for yeast Dnm1 and suggested that yeast two-hybrid tests with full-length protein are predominantly assaying interactions important for higher order assembly; our data are consistent with this interpretation.
We next wanted to explore the affect of adding a mutation that is known to stabilize Drp1 higher order complexes. A dominant-negative Drp1 K38A mutation has previously been shown to favor a higher order of Drp1 assembly, most likely from impaired GTP hydrolysis that is thought necessary for disassembly of Drp1 complexes (24). Consistent with this idea, the addition of the K38A mutation had little effect on the intramolecular interactions (Fig. 4A, asterisk and arrow). By contrast, the simultaneous addition of the K38A mutation to both bait and prey of the full-length constructs with middle domain mutations (A395D, G350D, G363D) partially restored the interaction between full-length Drp1 proteins (Fig. 4B, arrow). This is consistent with the notion that our yeast two-hybrid tests are reporting on Drp1 higher order assembly. Moreover, these results suggest that all of the Drp1 middle domain mutants are deficient in higher order assembly and that these associations are stabilized by the K38A mutation.
We next conducted yeast two-hybrid tests between truncated middle domain proteins (residues 323–502) to explore whether the A395D, G350D, and G363D mutations alter Drp1 self-interactions on a global level or specifically alter middle domain-dependent Drp1 assembly. As expected, we observed a strong yeast two-hybrid interaction between wild-type middle domains (Fig. 5A, arrowhead), similar to the interaction between the full-length wild-type proteins (Fig. 5A, arrow). The addition of any of the middle domain mutations markedly decreased this interaction when one bait/prey pair contained a mutation and completely abolished the interaction when both bait and prey contained a mutation (Fig. 5B). These results are consistent with Drp1 assembly being mediated in part by the middle domains, which is impaired by these mutations.
FIGURE 5.

Middle domain mutations disrupt intermolecular interactions mediated by Drp1 middle domains. A and B, yeast two-hybrid assays using the HIS3 reporter (sequential 10-fold yeast dilutions are shown) with the indicated bait and prey constructs are shown. Arrows and arrowheads are referenced under “Results.”
Our cross-linking and yeast two-hybrid results suggest that the A395D, G350D, and G363D middle domain mutations do not significantly inhibit Drp1 tetramer formation but likely affect higher order assembly. We wanted to determine whether this assembly defect was coupled to changes in the catalytic activity of Drp1, which is stimulated upon self-assembly. Specifically, we pursued a functional analysis of Drp1 GTPase activity in vitro using affinity-purified CBP-Drp1 fusion proteins to determine whether these middle domain mutations affect the assembly profile of GTP hydrolysis. We utilized a coupled GTPase assay in which GTP was continuously regenerated from GDP (16). The presence of the CBP tag did not alter the catalytic activity of wild-type Drp1 or any of the middle domain mutants (data not shown). All recombinant CBP-Drp1 middle domain mutants had a decreased rate of GTP hydrolysis as compared with the wild-type Drp1 (Table 1). Although the G350D and G363D mutants showed a 2-fold decrease in the kcat of GTP hydrolysis, the A395D mutant had the most severe reduction in GTPase activity (Table 1), consistent with our cross-linking and yeast two-hybrid results that suggest that the A395D mutation has the most deleterious effect on Drp1 assembly.
TABLE 1.
Kinetic characterization of wild-type Drp1 and middle domain mutants
kcat values were calculated for 2.5 μm Drp1 at the indicated salt concentrations by linear least-squares analysis from the 10–20 min period for at least three independent enzyme preparations.
| Drp1 | 50 mm NaCl |
500 mm NaCl |
||
|---|---|---|---|---|
| kcat | Kinetic lag | kcat | Kinetic lag | |
| min−1 | min | min−1 | min | |
| WT | 0.85 ± 0.14 | 4.8 ± 0.3 | 0.20 ± 0.02 | 0.1 ± 0.3 |
| A395D | 0.29 ± 0.02 | 0.3 ± 0.3 | 0.16 ± 0.01 | 0.0 ± 0.0 |
| G350D | 0.36 ± 0.02 | 0.5 ± 0.4 | 0.18 ± 0.01 | 0.0 ± 0.0 |
| G363D | 0.41 ± 0.03 | 3.3 ± 0.3 | 0.22 ± 0.02 | 0.1 ± 0.3 |
Higher order assembly of dynamins is required for maximal GTPase activity, which can be induced by lowering the ionic strength. Therefore, we investigated Drp1-dependent GTP hydrolysis by diluting wild-type or mutant CBP-Drp1 fusion proteins from non-assembly conditions (500 mm NaCl) into conditions that favor higher order self-assembly (50 mm NaCl). For wild-type Drp1 we observed a kinetic lag that is indicative of Drp1 self-assembly before steady-state GTP hydrolysis rates were achieved (Fig. 6), consistent with previous studies investigating human dynamin-1 (26) and yeast Dnm1p (16). Interestingly this rate-limiting nucleation event was reduced in all three CBP-Drp1 middle domain mutants (Table 1), with the kinetic lag being completely abolished in the A395D and G350D mutants. This suggests that the decreased catalytic activity of these mutants is due to a defect in higher order assembly that is essential for maximal GTPase activity.
FIGURE 6.

Drp1 middle domain mutations decrease assembly-dependent GTP hydrolysis. GTP hydrolysis by 2.5 μm CBP fusions of WT Drp1, and the indicated Drp1 mutants were measured in a coupled, substrate-regenerative GTPase assay as described under “Experimental Procedures.” Each trace shown is representative of experiments comprising at least three independent preparations of Drp1 performed in triplicate.
To investigate the assembly properties of these Drp1 proteins in more detail, we exploited the fact that high ionic strength buffers decrease higher order assemblies of yeast Dnm1p as well as dynamin-1 (16, 26). Consistent with those studies, we observed a clear inverse relationship of increasing NaCl concentration on the GTPase activity of human Drp1 (Fig. 7). Conversely, there was a diminished effect of ionic strength on GTPase activity for A395D, G350D, and G363D mutant Drp1 proteins. Although the G363D mutant retained some salt dependence to its activity, the extent of this salt dependence was clearly reduced compared with wild-type Drp1. The A395D and G350D mutant proteins showed markedly decreased salt dependence to their activity, which mirrors the loss of a kinetic lag for these mutants in Fig. 6. Moreover, the activity of wild-type Drp1 at the highest ionic strength (500 mm NaCl) was comparable with the activity of the middle domain mutants at the lowest ionic strength (50 mm NaCl), consistent with the idea that the unassembled form of wild-type Drp1 at high ionic strength is comparable with the assembly state of the mutants at low ionic strength (Table 1). Taken together, these results support the notion that these mutations impair higher order assembly necessary for efficient GTP hydrolysis.
FIGURE 7.

Drp1 middle domain mutations alter salt-dependent GTPase activities. The total amount of GTP hydrolyzed by 2.5 μm Drp1 or Drp1 mutants was assayed as a function of [NaCl] for 40 min. Data shown are averages of three independent preparations of CBP fusions of WT Drp1 and Drp1 A395D (A), Drp1 G350D (B), and Drp1 G363D (C) measured in triplicate ± S.E. **, p < 0.005; *, p < 0.05.
To establish directly whether the middle domain mutations affected Drp1 higher order assembly, wild-type and mutant Drp1 proteins were individually resolved by size-exclusion chromatography (SEC; Fig. 8), and the eluate was analyzed by multiangle laser light scattering (MALS), which when combined with refractive index detection allows a direct determination of molecular weight (44–46). Unlike conventional estimations of molecular weight by SEC alone, which relies on relative standards and assumptions on molecular shape, a combination of SEC and MALS (SEC-MALS) determines the average molecular weight of the eluate directly and is independent of elution volume. This technique is particularly well suited for analysis of complex assembly profiles, where molecules may exchange between different assembly states during elution.
FIGURE 8.

Drp1 middle domain mutants are higher order assembly-deficient dimers and tetramers. CBP-Drp1 WT and mutant fusion proteins were analyzed in 250 mm NaCl by SEC-MALS using a Superose 6 10/30 HR column as described under “Experimental Procedures.” Refractive index is plotted as a function of elution volume (ml). A molecular weight scale based on MALS analysis for eluted protein is shown for WT Drp1 (top panel; red) and is similar for each of the mutant proteins. WT Drp1 eluted primarily as 3 peaks, one that corresponds to a molecular mass range of 31–16 MDa that eluted in the void volume and two peaks that corresponded to molecular mass ranges of 420–325 and 243–180 kDa. This is consistent with an equilibrium between dimer-tetramer and higher order species given that the expected molecular mass for Drp1 dimer is ∼180 kDa and tetramer is ∼360 kDa. Mutant proteins predominantly comprise dimer and tetramer species, with little to no assembled protein in the column void volume, consistent with a defect in higher order assembly.
Wild-type Drp1 predominantly eluted in 3 broad peaks, the first of which corresponds to a molecular mass range of 31–16 MDa. This peak eluted in the void volume of the Superose 6 column, which we interpret to represent an ensemble of self-assembled Drp1 molecules because the relative size of this peak was sensitive to the loading concentration of Drp1 (data not shown) in a manner consistent with mass-action assembly. The two other predominant peaks corresponded to molecular mass ranges of 450–325 and 243–180 kDa. Because the expected molecular mass of a Drp1 dimer is 180 kDa and that of a tetramer is 360 kDa, these results are consistent with the wild-type protein (at 250 mm NaCl) populating dimeric, tetrameric, and higher order species. This suggests that, like dynamin (19, 26), the minimal assembly unit of Drp1 is a dimer and that tetrameric Drp1 is composed of a dimer of dimers. We interpret the range of molecular weights observed for each of the dimeric, tetrameric, and higher order species to be due to exchange occurring between these species during elution.
Each Drp1 middle domain mutant decreased the amount of higher ordered species (eluting at 33 ml) compared with wild-type Drp1, indicating a defect in higher order assembly. By contrast, the relative populations of dimeric and tetrameric species were different for each mutant as were their peak shapes. This data suggests that each mutant differentially affects the exchange rate between dimeric, tetrameric, and possibly higher order species. For example, the A395D lethal mutant was mainly dimeric, with a clearer separation between dimeric and tetrameric species than observed for either the G350D or G363D mutant (Fig. 8). This suggests that the A395D mutant is able to form a tetramer but that the exchange rate between dimer and tetramer is slower than observed for the other middle domain mutants. In contrast, the G350D mutant was mainly tetrameric, with a broad elution profile that obscures the presence of a distinct dimeric species, suggesting that the exchange rate between dimer and tetramer may be faster for this mutant compared with the A395D mutant (Fig. 8). The presence of a very small amount of higher ordered species suggests that the G350D mutant, like the A395D lethal mutant, is impaired for assembly past the tetrameric state. By contrast to the other mutants, the G363D mutant appeared more impaired for higher ordered assembly, as suggested by the disappearance of the peak eluting at 33 ml. Moreover, the broad peak between dimer and tetramer suggests faster exchange between these two species than for the other mutants (Fig. 8). Taken together, these results suggest that each of the Drp1 middle domain mutants is impaired in higher order assembly compared with wild-type Drp1, most likely by altering the exchange rates between dimeric, tetrameric, and higher ordered Drp1.
DISCUSSION
In this study we have explored the molecular mechanisms of middle domain-dependent assembly of the dynamin-related GTPase Drp1 and its importance for mitochondrial fission. These investigations were spurred by a very prominent, elongated mitochondrial phenotype present in cells from a patient with a neonatal lethal syndrome resulting from a de novo missense mutation in one DRP1 allele, resulting in an A395D substitution in the Drp1 protein (30). We also evaluated two other highly conserved middle domain residues (Gly-350, Gly-363; Fig. 1) important for assembly of the mammalian Drp1 ortholog in yeast, Dnm1 (27–29).
Our principal finding is that all of these mutations cause prominent lengthening of mitochondria, primarily by decreasing assembly of higher order Drp1 complexes at the mitochondrial surface in a dominant-negative manner. Initial Drp1 tetramerization as analyzed by co-immunoprecipitation and chemical cross-linking of differentially tagged Drp1 proteins appeared normal for the G350D and G363D mutants, consistent with previous reports for the G363D mutant in CHO cells (29). The A395D mutant showed a cross-linked product at the size of a tetramer but also exhibited increased levels of monomeric species on SDS-PAGE gels, indicating that this mutation may also weaken interactions important for tetramer formation. On the other hand, our yeast two-hybrid results suggest that higher order assembly is markedly impaired in these mutant forms. These results are confirmed by our SEC-MALS analysis, which demonstrates that the A395D mutant has a lower proportion of tetrameric and higher ordered species than wild-type Drp1. Furthermore, our SEC-MALS analysis indicates that both the G350D and G363D mutants are able to form tetramers but are deficient in higher ordered assembly, which is consistent with results seen in our co-immunoprecipitation and cross-linking assays.
This defect in higher ordered assembly is directly related to the GTPase activity of Drp1, as suggested by the loss of both a kinetic lag and salt-dependent changes in GTPase activity of each Drp1 middle domain mutant. We note that the G363D mutant appears less impaired for kinetic lag than the other mutants (Fig. 6) yet appears to have a faster exchange between dimer and tetramer species (Fig. 8). These differences suggest that these mutants affect Drp1 assembly in different manners than previously anticipated. For wild-type Drp1, the catalytic activity is consistent with the basal activity levels seen for other human dynamins (26). Although the kcat of our recombinant Drp1 protein is ∼60-fold lower than that reported for the yeast mitochondrial dynamin-related protein, Dnm1 (16), we see a similar kinetic lag and salt dependence of Drp1 activity compared with Dnm1, suggesting that the human Drp1 protein behaves in a similar manner. Our SEC-MALS data allow a more quantitative description of assembly than previously possible and indicate that the wild-type Drp1 protein self-assembles into 16–31 MDa species, which corresponds to higher order species consisting of 44–88 tetramers or 88–176 dimers. In fact, Dnm1 has been shown to assemble into higher order rings and spirals, with cooperative increases in GTPase activity that are inhibited with middle domain mutations orthologous to those that we investigated in Drp1 (16, 28). Thus, impairments in higher order assembly are the likely cause for the observed decreases in GTPase activity of the Drp1 middle domain mutants.
We suggest that defects in the formation of Drp1 higher order complexes result in the decrease in Drp1 levels observed at mitochondria both under steady-state conditions and when mitochondrial recruitment of Drp1 is stimulated by the addition of staurosporine, which induces programmed cell death (42). Even so, we considered other possible explanations. For instance, the middle domain mutations could conceivably impair interactions with other proteins, analogous to the yeast adaptor protein Mdv1 that is important for stable retention of Dnm1 structures on mitochondria (27, 28). However, there are no known functional orthologs of Mdv1 in mammals. Last, in dynamin some mutations in this region interfere with lipid binding (26), and it is conceivable that altered interactions of mutant Drp1 with lipid membranes could decrease mitochondrial recruitment, retention, or both. However, the dramatic changes in yeast two-hybrid interactions and salt dependence of GTP hydrolysis in vitro prefigure a clear effect on Drp1 middle domain-dependent self-interactions independent of any effect on lipid interactions. Moreover, unlike dynamin, which contains a pleckstrin homology domain and binds readily to lipid membranes, Drp1 lacks a clear lipid binding domain and has not been shown to have lipid binding ability. Perhaps Drp1 assembly is necessary for lipid binding, which would represent a new mechanism for protein amphitropism.
Taken together, our cellular data are highly consistent with the mitochondrial phenotype reported in fibroblasts from the patient with the Drp1 A395D mutation (30). We suggest that the markedly elongated mitochondria in these cells are a consequence of decreased fission, likely because higher order Drp1 complexes do not form or are unstable at mitochondria, leading to decreased levels of Drp1 at mitochondria and impaired formation of fission complexes. In an analogous situation, the G363D mutation on one DRP1 allele in CHO cells similarly caused mitochondrial and peroxisome elongation (29). In fact, these authors further noted that levels of the mutant protein were higher than for the wild type (29), perhaps reflecting the fact that a recruitment/assembly step coupled with disassembly and subsequent degradation may have been bypassed.
Our study further emphasizes the importance of maintaining a balance between mitochondrial fission and fusion. A number of studies have suggested increased cell survival when mitochondrial fission is inhibited, and fission is increased during programmed cell death (9, 42). However, decreases in mitochondrial fission are not always protective. Indeed, ganglioside-induced differentiation-associated protein 1 (GDAP1) mutations underlying Charcot-Marie-Tooth type 4A have also been suggested to impair mitochondrial fission selectively (43). Many disorders characterized by disruption of the mitochondrial fission and fusion balance primarily affect the nervous system (4, 8), and neurons in general may be most susceptible to such insults because of their extreme polarity. Indeed, ATP generation may be needed at locales far distant from the cell body, and mitochondria comprise parts of signaling cascades where positioning may also be critically important (4, 5, 8). Given the increasing emphasis on disruption of mitochondrial fission and fusion in common neurodegenerative disorders such as Alzheimer and Parkinson diseases, rare genetic presentations such as the Drp1 A395D mutation that impact proteins involved in mitochondrial fission and fusion will provide important insights into the pathogenesis of the much more common acquired neurodegenerative disorders.
Acknowledgments
We thank J. Nagle and D. Kauffman (NINDS DNA Sequencing Facility, National Institutes of Health) for DNA sequencing.
This work was supported, in whole or in part, by the National Institutes of Health Intramural Research Program of the NINDS (to C.-R. C., J. S., and C. B.), Training Grant 5T32GM080189 (to C. M. M.), Training Grant 5T32GM008403 (to A. E. P.), and Grant RO1GM067180 (to R. B. H.).
- Drp1
- dynamin-related protein 1
- CBP
- calmodulin-binding peptide
- GED
- GTPase-effector domain
- MALS
- multiangle laser light scattering
- SEC
- size-exclusion chromatography.
REFERENCES
- 1.Li Z., Okamoto K., Hayashi Y., Sheng M. (2004) Cell 119, 873–887 [DOI] [PubMed] [Google Scholar]
- 2.Arnoult D., Rismanchi N., Grodet A., Roberts R. G., Seeburg D. P., Estaquier J., Sheng M., Blackstone C. (2005) Curr. Biol. 15, 2112–2118 [DOI] [PubMed] [Google Scholar]
- 3.Campello S., Lacalle R. A., Bettella M., Mañes S., Scorrano L., Viola A. (2006) J. Exp. Med. 203, 2879–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan D. C. (2006) Cell 125, 1241–1252 [DOI] [PubMed] [Google Scholar]
- 5.McBride H. M., Neuspiel M., Wasiak S. (2006) Curr. Biol. 16, R551–R560 [DOI] [PubMed] [Google Scholar]
- 6.Yoon Y. S., Yoon D. S., Lim I. K., Yoon S. H., Chung H. Y., Rojo M., Malka F., Jou M. J., Martinou J. C., Yoon G. (2006) J. Cell Physiol. 209, 468–480 [DOI] [PubMed] [Google Scholar]
- 7.Lee S., Jeong S. Y., Lim W. C., Kim S., Park Y. Y., Sun X., Youle R. J., Cho H. (2007) J. Biol. Chem. 282, 22977–22983 [DOI] [PubMed] [Google Scholar]
- 8.Rismanchi N., Blackstone C. (2007) in Molecular Neurology (Waxman S. G. ed.) pp. 29–41, Elsevier Science Publishers, San Diego, CA [Google Scholar]
- 9.Suen D. F., Norris K. L., Youle R. J. (2008) Genes Dev. 22, 1577–1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee Y. J., Jeong S. Y., Karbowski M., Smith C. L., Youle R. J. (2004) Mol. Biol. Cell 15, 5001–5011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tan F. J., Husain M., Manlandro C. M., Koppenol M., Fire A. Z., Hill R. B. (2008) J. Cell Sci. 121, 3373–3382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rolland S. G., Lu Y., David C. N., Conradt B. (2009) J. Cell Biol. 186, 525–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okamoto K., Shaw J. M. (2005) Annu. Rev. Genet. 39, 503–536 [DOI] [PubMed] [Google Scholar]
- 14.Cerveny K. L., Tamura Y., Zhang Z., Jensen R. E., Sesaki H. (2007) Trends Cell Biol. 17, 563–569 [DOI] [PubMed] [Google Scholar]
- 15.Westermann B. (2008) J. Biol. Chem. 283, 13501–13505 [DOI] [PubMed] [Google Scholar]
- 16.Ingerman E., Perkins E. M., Marino M., Mears J. A., McCaffery J. M., Hinshaw J. E., Nunnari J. (2005) J. Cell Biol. 170, 1021–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hinshaw J. E. (1999) Curr. Opin. Struct. Biol. 9, 260–267 [DOI] [PubMed] [Google Scholar]
- 18.Danino D., Hinshaw J. E. (2001) Curr. Opin. Cell Biol. 13, 454–460 [DOI] [PubMed] [Google Scholar]
- 19.Zhang P., Hinshaw J. E. (2001) Nat. Cell Biol. 3, 922–926 [DOI] [PubMed] [Google Scholar]
- 20.Schrader M. (2006) Biochim. Biophys. Acta 1763, 531–541 [DOI] [PubMed] [Google Scholar]
- 21.van der Bliek A. M. (1999) Trends Cell Biol. 9, 96–102 [DOI] [PubMed] [Google Scholar]
- 22.Shin H. W., Takatsu H., Mukai H., Munekata E., Murakami K., Nakayama K. (1999) J. Biol. Chem. 274, 2780–2785 [DOI] [PubMed] [Google Scholar]
- 23.Pitts K. R., McNiven M. A., Yoon Y. (2004) J. Biol. Chem. 279, 50286–50294 [DOI] [PubMed] [Google Scholar]
- 24.Zhu P. P., Patterson A., Stadler J., Seeburg D. P., Sheng M., Blackstone C. (2004) J. Biol. Chem. 279, 35967–35974 [DOI] [PubMed] [Google Scholar]
- 25.Chang C. R., Blackstone C. (2007) J. Biol. Chem. 282, 21583–21587 [DOI] [PubMed] [Google Scholar]
- 26.Ramachandran R., Surka M., Chappie J. S., Fowler D. M., Foss T. R., Song B. D., Schmid S. L. (2007) EMBO J. 26, 559–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhar D., Karren M. A., Babst M., Shaw J. M. (2006) J. Biol. Chem. 281, 17312–17320 [DOI] [PubMed] [Google Scholar]
- 28.Naylor K., Ingerman E., Okreglak V., Marino M., Hinshaw J. E., Nunnari J. (2006) J. Biol. Chem. 281, 2177–2183 [DOI] [PubMed] [Google Scholar]
- 29.Tanaka A., Kobayashi S., Fujiki Y. (2006) Exp. Cell Res. 312, 1671–1684 [DOI] [PubMed] [Google Scholar]
- 30.Waterham H. R., Koster J., van Roermund C. W., Mooyer P. A., Wanders R. J., Leonard J. V. (2007) N. Engl. J. Med. 356, 1736–1741 [DOI] [PubMed] [Google Scholar]
- 31.Pitts K. R., Yoon Y., Krueger E. W., McNiven M. A. (1999) Mol. Biol. Cell 10, 4403–4417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smirnova E., Griparic L., Shurland D. L., van der Bliek A. M. (2001) Mol. Biol. Cell 12, 2245–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoon Y., Pitts K. R., McNiven M. A. (2001) Mol. Biol. Cell 12, 2894–2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rismanchi N., Soderblom C., Stadler J., Zhu P. P., Blackstone C. (2008) Hum. Mol. Genet. 17, 1591–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang D., Rismanchi N., Renvoisé B., Lippincott-Schwartz J., Blackstone C., Hurley J. H. (2008) Nat. Struct. Mol. Biol. 15, 1278–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arnoult D., Grodet A., Lee Y. J., Estaquier J., Blackstone C. (2005) J. Biol. Chem. 280, 35742–35750 [DOI] [PubMed] [Google Scholar]
- 37.Muhlberg A. B., Warnock D. E., Schmid S. L. (1997) EMBO J. 16, 6676–6683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schumacher B., Staeheli P. (1998) J. Biol. Chem. 273, 28365–28370 [DOI] [PubMed] [Google Scholar]
- 39.Di Paolo C., Hefti H. P., Meli M., Landis H., Pavlovic J. (1999) J. Biol. Chem. 274, 32071–32078 [DOI] [PubMed] [Google Scholar]
- 40.Smirnova E., Shurland D. L., Newman-Smith E. D., Pishvaee B., van der Bliek A. M. (1999) J. Biol. Chem. 274, 14942–14947 [DOI] [PubMed] [Google Scholar]
- 41.Prakash B., Praefcke G. J., Renault L., Wittinghofer A., Herrmann C. (2000) Nature 403, 567–571 [DOI] [PubMed] [Google Scholar]
- 42.Youle R. J., Karbowski M. (2005) Nat. Rev. Mol. Cell Biol. 6, 657–663 [DOI] [PubMed] [Google Scholar]
- 43.Niemann A., Ruegg M., La Padula V., Schenone A., Suter U. (2005) J. Cell Biol. 170, 1067–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wen J., Arakawa T., Philo J. S. (1996) Anal. Biochem. 240, 155–166 [DOI] [PubMed] [Google Scholar]
- 45.Andersson M., Wittgren B., Wahlund K. G. (2003) Anal. Chem. 75, 4279–4291 [DOI] [PubMed] [Google Scholar]
- 46.Ye H. (2006) Anal. Biochem. 356, 76–85 [DOI] [PubMed] [Google Scholar]