

Abstract
Temperature (T) reduction increases lifespan, but the mechanisms are not understood. Because reactive oxygen species (ROS) contribute to aging, we hypothesized that lowering T might decrease mitochondrial ROS production. We measured respiratory response and ROS production in isolated mitochondria at 32, 35, and 37 °C. Lowering T decreased the rates of resting (state 4) and phosphorylating (state 3) respiration phases. Surprisingly, this respiratory slowdown was associated with an increase of ROS production and hydrogen peroxide release and with elevation of the mitochondrial membrane potential, ΔΨm. We also found that at lower T mitochondria produced more carbon-centered lipid radicals, a species known to activate uncoupling proteins. These data indicate that reduced mitochondrial ROS production is not one of the mechanisms mediating lifespan extension at lower T. They suggest instead that increased ROS leakage may mediate mitochondrial responses to hypothermia.
Keywords: Aging, ATP, Brain, Diet, Mitochondria, Temperature
Introduction
Reactive oxygen species (ROS)3 are byproducts of normal oxygen metabolism and can serve as signaling molecules (1) or mediate molecular and cellular damage (2, 3). Calorie restriction, a balanced dietary regimen demonstrated to prolong lifespan and retard aging (6, 7), reduced both oxidative damage and core body temperature (8). In addition, modest but prolonged reduction of core body temperature increased lifespan independently of calorie restriction in poikilotherms (9). Moreover, we have shown previously that a modest reduction in core temperature is associated with an increase in longevity in mice (10). Although many pathways have been shown to contribute to aging, changes in mitochondrial function and altered oxidative processes and cellular redox status are proposed as important contributors (4, 5). To investigate the effect of a graded decrease in temperature on several aspects of mitochondrial function, we subjected isolated brain mitochondria to two levels of mild hypothermia. We measured mitochondria respiratory functions, superoxide and hydrogen peroxide leakage by electron paramagnetic resonance (EPR), spin trapping spectroscopy, and polarographic oximetry. Moreover, to obtain mechanistic clues on causes and dynamics of ROS leakage we studied the effect of temperature on mitochondrial membrane potential. These assays were performed at three different temperatures selected as representative of the physiological core temperature of mice during the dark:active phase of the day (37 °C), the light:resting phase (35 °C), and the temperature reached under calorie restriction (32 °C).
MATERIALS AND METHODS
Chemicals and Reagents
All chemicals were the highest available grades and purchased from Sigma-Aldrich unless otherwise mentioned. 5-Diethoxyphosphoryl-5-methyl-1-pyrroline N-oxide (DEPMPO) and DIPPMPO (5-(diisopropyl)-5-methyl-1-pyrroline-N-oxide) were purchased from Alexis Biochemicals (AXXORA, LLC., San Diego, CA). Amplex Red and JC-1 Mito Probe assay kits were obtained from Molecular Probes.
Animals
8–12-week-old C57BL/6 male mice were purchased from The Scripps Research Institute vivarium and kept under a 12:12 h light:dark cycle with lights on at 6 a.m.; water and food (11% fat chow diet) were ad libitum. All experiments were carried out on tissues collected between 11 a.m. and 2 p.m. from 3–4-month-old animals. This study was conducted in conformity with the Guiding Principles for Research Involving Animals and Human Beings and was approved by The Scripps Institutional Animal Care and Use Committee. Animal experiments were carried out in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 80-23), revised 1996.
Analysis of Mitochondrial Respiratory Function at Different Temperatures
Cortical mitochondria were isolated from the whole brain using differential centrifugation in Percoll gradient (11). Oxygen consumption was measured using a Clark-type oxygen electrode (OxygraphTM; Hansatech, Norfolk, UK). Two Oxygraph systems were used in parallel, and the temperature of one electrode chamber was tightly controlled at the desired temperature T ± 0.1 °C in reference to the second chamber adjusted at 37 ± 0.1 °C, using PolyScience Water Bath Programmable Temperature Controllers (PolyScience, Division of Preston Industries Inc., Niles, IL), and the two oxygen consumption traces were recorded simultaneously. Respiratory steady states have been defined by Chance and Williams (12) according to a protocol for oxygraphic measurements of oxygen consumptions by isolated mitochondria as follows. State 1 is the d[O2]/dt in the presence of endogenous substrates with inorganic phosphate in the mitochondrial respiration medium. Addition of ADP induces a phase of initial oxygen consumption associated with effectively exhausting endogenous substrates, after which state 2 is a substrate-limited state of residual oxygen consumption (should also correspond to state 4). State 3 is then achieved by the addition of ADP in the presence of NAD+-linked substrates, which are excess malate and pyruvate in our case. Upon the depletion of ADP, respiration drops in the transition to state 4, which is an ADP-limited resting state (leak state). Mitochondria (∼1–2 mg of protein) were added to the two oximetry chambers in a 300 μl of solution containing 100 mm KCl, 75 mm mannitol, 25 mm sucrose, 5 mm H3PO4, 0.05 mm EDTA, and 10 mm Tris-HCl, pH 7.4. After a 2-min equilibration, 5 mm pyruvate and 5 mm malate were added, and oxygen consumption was followed for 2 min (respiratory state 2 followed by state 4 after the endogenous ADP pool expires). ADP (250 μm) was added to measure state 3 (phosphorylating) respiration. Oligomycin (2.5 μg/ml) was added 2 min later to inhibit the F0F1-ATPase and determine state 4 (resting) respiration. The maximal uncoupled respiratory rate was obtained by adding 0.2 μm carbonyl cyanide m-chlorophenylhydrazone to the mixture. Oxygen utilization traces and rate determinations were obtained using OxygraphTM software. Protein concentrations were quantified using the BCA kit (Pierce), and signals were normalized by mitochondrial protein contents obtained for each run.
Measurements of Mitochondrial Superoxide Production at Different Temperatures by EPR
A Magnettech MiniScope MS-200 Benchtop EPR spectrometer was used to detect superoxide and hydroxyl radical generation by mitochondria as described (13, 14). Mitochondria suspensions (∼ 200–400 μg of protein) were incubated for 30 min with 30 mm DEPMPO or DIPPMPO and appropriate combinations of respiratory substrates in the incubators adjusted and stabilized at desired temperatures T ± 0.1 °C. The mixture was loaded into a 50-μl glass capillary and introduced into the EPR spectrometer which allowed the temperature to be maintained at T ± 0.1 °C using a biotemperature control unit during acquisitions. We confirmed that the detected EPR signals are substrate-specific and not due to redox cycling in the studied mixtures (data not shown). No EPR signals were registered when DEPMPO or DIPPMPO was mixed with combinations of substrates in the absence of mitochondria. EPR conditions were as follows: microwave power, 5 mW; modulation amplitude, 2 G; modulation frequency, 100 kHz; sweep width, 150 G centered at 3349 G; magnetic field scan rate, 7.5 G s−1; each spectrum was the average of five scans. Assignment of the observed signals from mitochondria was confirmed through computer-assisted spectral simulation using the WinSim software. Mixtures of signals due to DIPPMPO-OOH and DIPPMPO-OH adducts were detected, but complete removal of these signals was achieved upon the inclusion of superoxide dismutase (200 units/ml), which confirmed that superoxide radical was the exclusive source of the observed EPR signals. After incubation with DEPMPO for 30 min, the observed signal was due to carbon-centered radical with minor contribution from superoxide. To quantify free radicals, EPR intensities were normalized for protein contents.
Amplex Assay of H2O2 Production by Isolated Mitochondria
Mitochondrial H2O2 production was measured by the Amplex Red-horseradish peroxidase method (Molecular Probes). Horseradish peroxidase (2 units/ml) catalyzes the H2O2-dependent oxidation of nonfluorescent Amplex Red (80 μm) to fluorescent resorufin red (15). Fluorescence was kinetically followed during 30 min, at an excitation wavelength of 545 nm and an emission wavelength of 590 nm using a Tecan Infinite F500 apparatus (Tecan Systems, Inc., San Jose, CA), adjusted internally to different temperatures. Results were expressed as relative production efficiency given by slopes calculated from readings obtained along 30-min repeated measurements.
JC-1 Assay for Mitochondria Transmembrane Potential
To assess relative changes in mitochondrial membrane potential during development, 20 μg of pure mitochondria was loaded into each well of white 96-well plates together with 1.5 μm JC-1 Mito Probe (Molecular Probes) at indicated conditions (16, 17). JC-1 aggregate (red) and JC-1 monomer (green) fluorescence wavelengths were measured simultaneously during 30 min using a Tecan Infinite F500 apparatus (Tecan Systems), adjusted internally to the desired temperatures. JC-1 aggregates were detected at 535-nm excitation and 590-nm emission, and monomers were detected at 485-nm excitation and 535-nm emission.
Statistics
Data are reported as means ± S.E. Statistical analyses were carried out using OriginPro version 7.5 SR4 (OriginLab Corporation). Data were typically subjected to a one-way ANOVA followed by Tukey's post hoc test. Differences in the means were considered statistically significant when p < 0.05.
RESULTS
Temperature Effects on Mitochondrial Respiratory Rates
Because changes in mitochondrial respiratory rates and coupling can directly modify ROS production, we evaluated respiration in isolated brain mitochondria of 2–4-month-old male C57BL/6 mice at different temperatures. Experiments typically involved parallel measurements of oxygen consumption by identical amounts of a given mitochondria sample using two independent Clark-type electrodes where one of them is tightly controlled at a specific temperature, T ± 0.1 °C, in reference to the second electrode which is stabilized at 37 °C. Representative oxygen consumption traces at 35 versus 37 °C were acquired as described under “Materials and Methods” and are given in Fig. 1A. At 35 °C, significant reductions in both state 4 (resting, 78.8 ± 4.8% of control at 37 °C, F(1,8) = 19.38, p < 0.01; Fig. 1B) and state 3 (phosphorylating, 71.2 ± 5.9% of control at 37 °C, F(1,8) = 6.11, p < 0.05; Fig. 1C) were observed. Maximally uncoupled respiratory rate induced by the addition of carbonyl cyanide m-chlorophenylhydrazone did not significantly or systematically depend on temperature (data not shown). This indicates that there is no significant dependence of proton pumping by the electron transport chain complexes on environmental temperature. As also shown in Fig. 1, lowering the incubational temperature to 32 °C led to a further decrease in the rates of oxygen consumption during both respiratory states; state 4 at 32 °C was 56.4 ± 7.7% of that at 37 °C, F(1,14) = 9.89, p < 0.01 (Fig. 1B), and state 3 was 57.2 ± 8.0% of that at 37 °C, F(1,14) = 5.07, p < 0.05 (Fig. 1C). The observed trend of decreased respiratory rates resembles that reported for isolated skeletal muscle mitochondria (18) and from liver mitochondria (19).
FIGURE 1.

Depression of mitochondrial metabolism at lower temperatures determined by polarographic measurements of oxygen consumption. Cortical mitochondria from 2–4-month-old C57BL/6 mice were isolated from PBS-perfused whole brain using Percoll gradient. A, representative oxygen consumption traces by mitochondria incubated at 37 °C (broken line) or 35 °C (solid line). Mitochondria (∼1–2 mg of protein) were added to the two oximetry chambers in a final volume 300-μl solution containing 100 mm KCl, 75 mm mannitol, 25 mm sucrose, 5 mm H3PO4, 0.05 mm EDTA, and 10 mm Tris-HCl, pH 7.4. BSA at 0.1% final concentration was included to eliminate free fatty acids and enhance mitochondria coupling. B and C, rates of oxygen consumptions during state 4 (B) or state 3 (C) by mitochondria freshly isolated from young mice brains. At 35 °C, significant reductions in both state 4 and state 3 are seen. Lowering the incubational temperature to 32 °C led to a further decrease in the rates of oxygen consumption during both respiratory states. Statistical significance was evaluated by a one-way ANOVA followed by Tukey's post hoc test. Differences in the means were considered statistically significant when *, p < 0.05; n = 5–8 independent runs/group.
Temperature Effects on ROS Leakage from Respiring Mitochondria
We have previously reported the detection of basal mitochondrial ROS leakage, i.e. without the inhibition of electron transport chain complexes (13, 14). Here, we used EPR spin trapping spectroscopy to follow the effect of temperature on basal ROS production during physiologically relevant respiratory states. In Fig. 2A we show representative EPR spectra of a DIPPMPO-hydroxyl radical adduct, which is originating from superoxide radical trapping and registered during state 3 or state 4 mitochondrial respirations at either 32 °C or 37 °C. Quantification of these signals and those recorded at 35 °C are shown in Fig. 2B (state 4) and Fig. 2C (state 3). At lower temperatures, mitochondria generally leaked higher superoxide concentrations which reached statistical significance at 35 °C during resting respiration (162.4 ± 17.6% of state 4 at 37 °C, F(1,8) = 6.44, p < 0.05). At 32 °C, however, mitochondria did not generate elevated amounts of superoxide during state 4 compared with physiological temperature (129.0 ± 36% of state 4 at 37 °C, F(1,18) = 0.5, p = 0.48). This trend was somewhat different for mitochondria during oxidative phosphorylation in state 3 where superoxide signals were significantly higher at both 35 °C (198.2 ± 21.4% of state 3 at 37 °C, F(1,6) = 13.58, p < 0.05) and 32 °C (225.0 ± 53% of state 3 at 37 °C, F(1,14) = 4.49, p < 0.05). These results indicate that resting and phosphorylating respiratory rates systematically decrease at lower temperatures while qualitatively leaking higher ROS levels.
FIGURE 2.

Exposure of isolated brain mitochondria to slightly lower than physiologic temperature leads to increased superoxide leakage. Whole brain of 2–4-month-old C57BL/6 mouse was harvested before mitochondria were isolated as described under “Materials and Methods.” A, EPR spectra were acquired after mixing mitochondria with 70 mm DIPPMPO, the NAD+-linked substrates malate + pyruvate in the absence (state 4 respiration) or the presence of ADP (state 3 respiration). The mixture was incubated at the target temperature for 30 min and introduced into the EPR cavity that was maintained at the targeted temperature. B and C, quantification of EPR signals at targeted temperatures during state 4 (B) or state 3 (C) respiration was carried out for five to eight independent runs/condition, and the results are shown as mean ± S.E. (error bars). *, p < 0.05.
To confirm the dependence of mitochondrial ROS leakage on surrounding temperature, we detected hydrogen peroxide (H2O2) production by utilizing the Amplex Red assay (Fig. 3). H2O2 is a membrane-permeable ROS and is mainly produced from catalytic dismutation of superoxide by superoxide dismutase. In Fig. 3A we show that the rate of H2O2 production during state 4 is greater than state 3 and that electron transport chain inhibition by rotenone at complex I or antimycin A at complex III dramatically increase H2O2 generation. A clear trend of higher H2O2 production at lower temperatures was observed (Fig. 3B). The observation that EPR registered stronger ROS-dependence on temperature than the Amplex Red assay is probably due to the inherent limitation in the latter due to inability to detect intramitochondrial hydrogen peroxide. In contrast, the DIPPMPO spin probe is effectively distributed inside, outside, and in the membrane compartments of mitochondria (20); that is, much of the superoxide produced on the matrix side of the mitochondrial inner membrane is converted by manganese superoxide dismutase, by other matrix enzymes, or by nonenzymic processes to hydrogen peroxide, which can diffuse out of mitochondria and be registered by the Amplex Red assay. However, without exogenously added superoxide dismutase, little of the superoxide produced on the cytosolic side of the membrane is converted to hydrogen peroxide and registered by the assay because conversion of superoxide to hydrogen peroxide does not compete very effectively with other reactions not leading to hydrogen peroxide (such as the reduction of cytochrome c) (21). It is also conceivable that the topology of ROS production by both complex I and III causes the observed discrepancy between the two assays. It has been reported that although complex I-dependent superoxide is released exclusively to the matrix side, complex III releases superoxide to both sides of the inner membrane (22). This may be taken to imply that lower temperature produces more complex I-dependent superoxide than complex III.
FIGURE 3.

Hydrogen peroxide generation in phosphorylating mitochondria is greater at lower temperatures. A, hydrogen peroxide release by mitochondria incubated with various substrates, inhibitors, and/or enzymes at various temperatures was detected using Amplex Red and horseradish peroxidase. Fluorescence readings were taken at 3-min intervals for up to 30 min, and the rates of fluorescence increase were calculated for each kinetic curve. a.u., arbitrary units. B, means ± S.E. (error bars) of three or four independent runs are given. Rotenone (Rot, complex I inhibitor, 2.4 μm), and antimycin A (AA, complex III inhibitor, 10 μm) dramatically increased H2O2 release during state 3, but this increase was not temperature-dependent (data not shown). Catalase (cat, 100 units/ml) eliminated the H2O2 signal.
Lowering Temperature Increases Mitochondrial Membrane Polarization
To measure the ΔΨm of isolated mitochondria we adapted previous protocols (16, 17) using the fluorescent probe JC-1. JC-1 exists as a monomer at low values of ΔΨm (green fluorescence), whereas it forms aggregates at a high ΔΨm (red fluorescence). Thus, mitochondria with a normal ΔΨm concentrate JC-1 into aggregates (red fluorescence), but with a depolarized ΔΨm, JC-1 forms monomers (green fluorescence). We therefore treated actively respiring mitochondria with NAD+-linked substrates with JC-1 while incubated at the targeted temperature for 10 min, and the red fluorescence was recorded after washing out excess JC-1. The intensity of the red fluorescence was taken as a qualitative measure for transmembrane polarization and the higher ΔΨm values reflect more effective JC-1 uptake and aggregate formation inside mitochondria. Fig. 4 indicates that mitochondrial membrane is significantly more polarized at 32 °C than at 35 °C and 37 °C during resting (185.7 ± 5.7% of state 3 at 37 °C, F(1,22) = 43.57, p < 0.01; 181.1 ± 5.8% of state 3 at 35 °C, F(1,22) = 37.2, p < 0.01) or phosphorylating (175.4 ± 5.1% of state 4 at 37 °C, F(1,22) = 36.11, p < 0.01; 147.1 ± 5.9% of state 4 at 35 °C, F(1,22) = 37.2, p < 0.01) respiratory states. This observation is in agreement with results introduced above, including slower respiration and higher ROS production at lower temperatures; see “Discussion” below. Incubation of mitochondria with JC-1 without substrates did not significantly affect the recorded red fluorescence at any of the studied temperature, indicating that the observed effects in Fig. 4 are reflecting changes in mitochondrial metabolic activity rather than being due to temperature effects on the probe dye.
FIGURE 4.
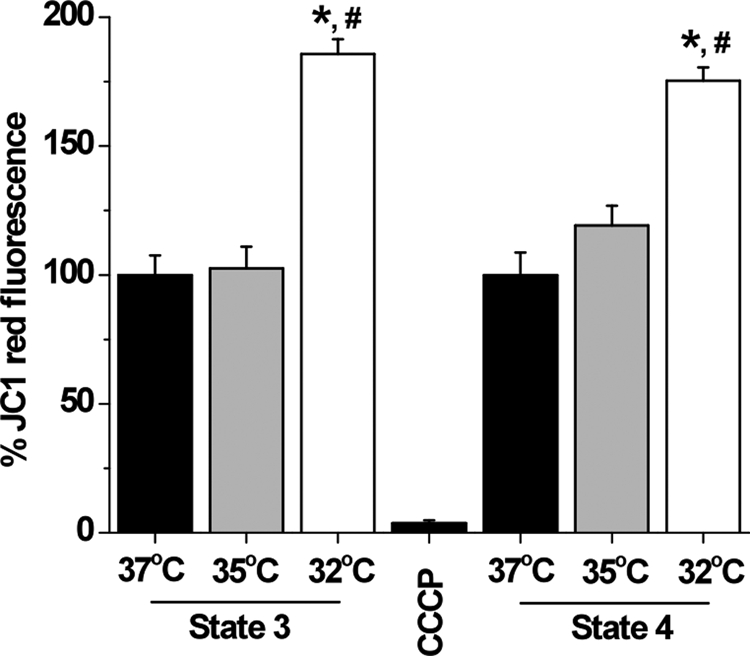
Cold-induced hyperpolarization of mitochondrial membrane potential. Fluorescence measurements of changes in mitochondrial membrane potential are probed by following JC-1 red fluorescence due to JC-1 aggregation in mitochondria. Addition of the uncoupler control carbonyl cyanide m-chlorophenylhydrazone (CCCP; 1 μm) rapidly depolarized the mitochondrial potential. Phosphorylating and resting mitochondria incubated at lower temperatures exhibited hyperpolarized membrane potential that reached statistical significance at 32 °C; *, p < 0.05 compared with state 3 at 37 °C; #, p < 0.05 compared with state 4 at 37 °C, n = 3 per group.
Lowering Temperature Increased Carbon-centered Radicals (CCRs)
Because superoxide was demonstrated to activate uncoupling proteins (UCPs) by generating CCRs (23), we investigated the effect of temperature on the DEPMPO/CCR EPR signal. It was shown previously that in a lipid-rich environment, a DEPMPO/superoxide adduct rapidly decays and a lipid-derived CCR prevails (20, 24). DIPPMPO/CCR did not contribute significantly to the observed signals in Fig. 2 due to the long half-life time of DIPPMPO adducts with superoxide, 23 min (25), which is almost twice as long as that of DEPMPO, 14 min (26). After a 30-min incubation at the targeted temperatures, we therefore detected major contributions from DEPMPO/CCR adduct with minor signal from DEPMPO/superoxide adduct as revealed by computer simulations using previously reported EPR line parameters (Fig. 5A) (20). Mitochondria incubated at 32 °C produced significantly greater CCR than those incubated at 37 °C both during state 3 (164.8 ± 7.44% of state 3 at 37 °C, F(1,6) = 20.27, p < 0.01, n = 4) and state 4 (199.0 ± 14.4% of state 4 at 37 °C, F(1,6) = 11.52, p < 0.05, n = 4). This result indicates that the ROS increase at lower temperature may trigger the activation of UCPs via their interaction with CCRs.
FIGURE 5.

Cold increases the production of lipid-derived carbon-centered radicals in brain mitochondria. A, EPR spectra acquired after a 30-min incubation of mitochondria (0.2–0.4 mg of protein) at 32 °C or 37 °C, with 70 mm DEPMPO during state 4 or state 3 respirations. The top trace is a computer-generated spectrum of 95% carbon-centered radical + 5% superoxide radical using the following hyperfine splitting constants: DEPMPO-OOH, ap = 50 G, aN = 13.11 G, aH = 11.05 G; aH = 0.9 G; aH = 0.60 G; aH(2) = 0.44 G; aH(3) = 0.38 G; DEPMPO/CCR, ap = 48.1 G, aN = 15.0 G, aH = 22.3 G. B, quantification of the EPR signals of the adduct between DEPMPO and a lipid-derived CCR (mean ± S.E. (error bars)). One-way ANOVA indicated that mitochondria incubated at 32 °C produced significantly greater CCRs than those incubated at 37 °C both during state 3 and state 4, n = 4 per condition.
DISCUSSION
The present study was carried out hypothesizing that decreasing environmental temperature might cause mitochondria to spew fewer free radicals by virtue of slowing down the biochemistry of superoxide production and hence explaining the increased lifespan due to reduced oxidative damage. We followed changes in respiratory activity and ROS production as well as membrane potential in freshly isolated cortical mitochondria that were incubated at 37 °C, 35 °C, and 32 °C. These temperatures were chosen as representative of the physiological core temperature of mouse during the dark/active phase of the day, the light/resting phase, and of torpor (often induced by calorie restriction), respectively. Mitochondrial respiratory response to lower temperatures consistently involved decreased rates of resting (state 4) as well as phosphorylating (state 3) respirations. Brooks and colleagues reported similar slow-down in respiratory activities of isolated skeletal muscle mitochondria when cooled from 45 °C to 25 °C (18). A similar temperature dependence was seen also in liver mitochondria (19). This trend in which mitochondria isolated from different organs respond to lower environmental temperature by slowing their rate of oxygen consumption may be explained in terms of decreased ATPase activity at lower temperature (27). This may result from the ability of mitochondria to modulate the rate of metabolism to cope with cellular and organismal demands and challenges. For instance, metabolic depression is a phenotypic response to lower temperature in hibernating mammals (28).
Contrary to our expectations, we found that at lower T mitochondria increased superoxide leakage and hydrogen peroxide production. Because these measurements were carried out on isolated mitochondria, the findings do not conclusively demonstrate that reducing temperature is not associated with reduction of oxidative damage. In fact, mitochondria are not the only cellular source of ROS, and the role of endogenous cellular antioxidants was not assessed. Also, the possibility that mitochondrial ROS production responded differently in a cellular context cannot be excluded. Yet, these data do not support our initial hypothesis that mitochondrial ROS production was decreased at lower temperature, indicating that this is not a likely mechanism of temperature-dependent increased lifespan. Instead, these observations suggest that increased ROS production at lowered T may have a biological significance other than the mediation of oxidative damage.
Decreased respiration and increased ROS leakage at lower T were associated with a significant increase in mitochondrial membrane potential, ΔΨm, suggesting that hyperpolarized membrane might be instrumental in the regulation of mitochondrial response to hypothermicity. These observations are consistent with the following scenario (12, 29, 30). The generation of superoxide radical ( ) as a byproduct of oxidative phosphorylation is known to involve ubisemiquinone (CoQ
) as a byproduct of oxidative phosphorylation is known to involve ubisemiquinone (CoQ ) due to the highly reducing midpoint potential (−160 mV) of the redox couple (CoQ/CoQ). CoQ occurs at three different sites along the electron transport chain: one site at complex I in addition to two sites at complex III. As a result, complexes I and III are the main sources of superoxide leakage in mitochondria. Factors increasing the lifetime of CoQ, i.e. populating the reduced state, are expected to increase the chance of electron transfer from CoQ to oxygen (31). Decreased activity of ATPase at lower temperature (27) is expected to polarize the proton gradient across the inner membrane and, in effect, increase transmembrane potential as observed. This results in reduced oxygen consumption by electron transport complexes, which in turn leads to increased local [O2]. Because of increased proton gradient and impeded electron transfers, the reduced forms of CoQ, flavins, cytochromes, and non-heme iron proteins accumulate which, in addition to increased [O2], fulfills the redox couples required to generate . Hyperpolarized ΔΨ may also retard electron transfer from cytochrome b to CoQ, which results in an apparent increase in the lifetime of CoQ and increased superoxide production at lower temperatures. It is well established that a strong positive correlation exists between membrane potential and ROS production in isolated mitochondria. It was reported that, at high membrane potentials, even a small increase in membrane potential gave rise to a large stimulation of H2O2 production (32–34). Mitochondrial ROS production is particularly sensitive to changes in membrane potential ΔΨm at the highest range of values (i.e. 170–185 mV). To maximize mitochondrial coupling we included bovine serum albumin in the incubation medium (0.1% final concentration), which helps mitochondria maintain high membrane potentials. Bovine serum albumin binds free fatty acids that can be generated from membrane lipids and that can uncouple mitochondria (35, 36).
) due to the highly reducing midpoint potential (−160 mV) of the redox couple (CoQ/CoQ). CoQ occurs at three different sites along the electron transport chain: one site at complex I in addition to two sites at complex III. As a result, complexes I and III are the main sources of superoxide leakage in mitochondria. Factors increasing the lifetime of CoQ, i.e. populating the reduced state, are expected to increase the chance of electron transfer from CoQ to oxygen (31). Decreased activity of ATPase at lower temperature (27) is expected to polarize the proton gradient across the inner membrane and, in effect, increase transmembrane potential as observed. This results in reduced oxygen consumption by electron transport complexes, which in turn leads to increased local [O2]. Because of increased proton gradient and impeded electron transfers, the reduced forms of CoQ, flavins, cytochromes, and non-heme iron proteins accumulate which, in addition to increased [O2], fulfills the redox couples required to generate . Hyperpolarized ΔΨ may also retard electron transfer from cytochrome b to CoQ, which results in an apparent increase in the lifetime of CoQ and increased superoxide production at lower temperatures. It is well established that a strong positive correlation exists between membrane potential and ROS production in isolated mitochondria. It was reported that, at high membrane potentials, even a small increase in membrane potential gave rise to a large stimulation of H2O2 production (32–34). Mitochondrial ROS production is particularly sensitive to changes in membrane potential ΔΨm at the highest range of values (i.e. 170–185 mV). To maximize mitochondrial coupling we included bovine serum albumin in the incubation medium (0.1% final concentration), which helps mitochondria maintain high membrane potentials. Bovine serum albumin binds free fatty acids that can be generated from membrane lipids and that can uncouple mitochondria (35, 36).
We propose that increased ROS production by mitochondria is a physiological rather than pathological response to hypothermia. One interesting possibility is that mitochondria are capable, at least to a certain degree, of autonomous thermoregulatory responses and that ROS may represent important signals for its modulation. For instance, increased ROS occurring at lowered temperature might stimulate thermogenesis by activating UCPs. This hypothesis is supported by our findings that reduced temperature is associated with increased levels of CCRs, a species previously demonstrated to mediate the superoxide-dependent activation of UCPs (23, 41). Interestingly, UCP activation can reduce ROS formation (37) providing negative feedback regulatory signals. This response might serve to enhance ATP synthesis through glycolysis to compensate for the decreased activity of ATPase and to generate heat in response to cold exposure. In the brain this might also affect neuronal energy metabolism and synaptic transmission both proposed to be regulated by UCPs (38–40).
Acknowledgment
We thank Jacinta Lucero for technical assistance in optimizing protocol for mitochondria isolation.
This work was supported, in whole or in part, by NIA/National Institutes of Health Grants K25AG026379 (to S. S. A.) and AG028040. This work was also supported by The Ellison Medical Foundation.
- ROS
- reactive oxygen species
- CCR
- carbon centered radical
- ΔΨm
- mitochondrial membrane potential
- DEPMPO
- 5-diethoxyphosphoryl-5-methyl-1-pyrroline N-oxide
- DIPPMPO
- 5-(diisopropyl)-5-methyl-1-pyrroline-N-oxide
- JC-1
- 5,59,6,69-tetrachloro-1,19,3,39-tetraethylbenzimidazole carbocyanide iodide
- UCP
- uncoupling protein.
REFERENCES
- 1.D'Autréaux B., Toledano M. B. (2007) Nat. Rev. Mol. Cell Biol. 8, 813–824 [DOI] [PubMed] [Google Scholar]
- 2.Wallace D. C., Fan W. (2009) Genes Dev. 23, 1714–1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wallace D. C. (2005) Annu. Rev. Genet. 39, 359–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harman D. (1992) Mutat. Res. 275, 257–266 [DOI] [PubMed] [Google Scholar]
- 5.Sohal R. S., Mockett R. J., Orr W. C. (2002) Free Radic. Biol. Med. 33, 575–586 [DOI] [PubMed] [Google Scholar]
- 6.Gredilla R., Barja G. (2005) Endocrinology 146, 3713–3717 [DOI] [PubMed] [Google Scholar]
- 7.Merry B. J. (2004) Aging Cell 3, 7–12 [DOI] [PubMed] [Google Scholar]
- 8.Lane M. A., Baer D. J., Rumpler W. V., Weindruch R., Ingram D. K., Tilmont E. M., Cutler R. G., Roth G. S. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 4159–4164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu R. K., Walford R. L. (1972) Gerontologia 18, 363–388 [DOI] [PubMed] [Google Scholar]
- 10.Conti B., Sanchez-Alavez M., Winsky-Sommerer R., Morale M. C., Lucero J., Brownell S., Fabre V., Huitron-Resendiz S., Henriksen S., Zorrilla E. P., de Lecea L., Bartfai T. (2006) Science 314, 825–828 [DOI] [PubMed] [Google Scholar]
- 11.Sims N. R. (1990) J. Neurochem. 55, 698–707 [DOI] [PubMed] [Google Scholar]
- 12.Chance B., Williams G. R. (1955) J. Biol. Chem. 217, 429–438 [PubMed] [Google Scholar]
- 13.Ali S. S., Xiong C., Lucero J., Behrens M. M., Dugan L. L., Quick K. L. (2006) Aging Cell 5, 565–574 [DOI] [PubMed] [Google Scholar]
- 14.Quick K. L., Ali S. S., Arch R., Xiong C., Wozniak D., Dugan L. L. (2008) Neurobiol. Aging 29, 117–128 [DOI] [PubMed] [Google Scholar]
- 15.Zhou M., Diwu Z., Panchuk-Voloshina N., Haugland R. P. (1997) Anal. Biochem. 253, 162–168 [DOI] [PubMed] [Google Scholar]
- 16.Scaduto R. C., Jr., Grotyohann L. W. (1999) Biophys. J. 76, 469–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zamzami N., Kroemer G. (2004) Methods Mol. Biol. 282, 103–115 [DOI] [PubMed] [Google Scholar]
- 18.Brooks G. A., Hittelman K. J., Faulkner J. A., Beyer R. E. (1971) Am. J. Physiol. 220, 1053–1059 [DOI] [PubMed] [Google Scholar]
- 19.Dufour S., Rousse N., Canioni P., Diolez P. (1996) Biochem. J. 314, 743–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stolze K., Udilova N., Nohl H. (2000) Free Radic. Biol. Med. 29, 1005–1014 [DOI] [PubMed] [Google Scholar]
- 21.Miwa S., St.-Pierre J., Partridge L., Brand M. D. (2003) Free Radic. Biol. Med. 35, 938–948 [DOI] [PubMed] [Google Scholar]
- 22.Muller F. L., Liu Y., Van Remmen H. (2004) J. Biol. Chem. 279, 49064–49073 [DOI] [PubMed] [Google Scholar]
- 23.Murphy M. P., Echtay K. S., Blaikie F. H., Asin-Cayuela J., Cocheme H. M., Green K., Buckingham J. A., Taylor E. R., Hurrell F., Hughes G., Miwa S., Cooper C. E., Svistunenko D. A., Smith R. A., Brand M. D. (2003) J. Biol. Chem. 278, 48534–48545 [DOI] [PubMed] [Google Scholar]
- 24.Anzai K., Aikawa T., Furukawa Y., Matsushima Y., Urano S., Ozawa T. (2003) Arch. Biochem. Biophys 415, 251–256 [DOI] [PubMed] [Google Scholar]
- 25.Villamena F. A., Zweier J. L. (2004) Antioxid. Redox Signal. 6, 619–629 [DOI] [PubMed] [Google Scholar]
- 26.Chalier F., Tordo P. (2002) J. Chem. Soc. Perkin Trans. 2, 2110–2117 [Google Scholar]
- 27.Swegert C. V., Dave K. R., Katyare S. S. (1999) Mech. Ageing Dev. 112, 27–42 [DOI] [PubMed] [Google Scholar]
- 28.Carey H. V., Andrews M. T., Martin S. L. (2003) Physiol. Rev. 83, 1153–1181 [DOI] [PubMed] [Google Scholar]
- 29.Cadenas E., Boveris A., Ragan C. I., Stoppani A. O. (1977) Arch. Biochem. Biophys. 180, 248–257 [DOI] [PubMed] [Google Scholar]
- 30.Boveris A., Chance B. (1973) Biochem. J. 134, 707–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Papa S., Skulachev V. P. (1997) Mol. Cell. Biochem. 174, 305–319 [PubMed] [Google Scholar]
- 32.Starkov A. A., Fiskum G. (2003) J. Neurochem. 86, 1101–1107 [DOI] [PubMed] [Google Scholar]
- 33.Korshunov S. S., Skulachev V. P., Starkov A. A. (1997) FEBS Lett. 416, 15–18 [DOI] [PubMed] [Google Scholar]
- 34.Votyakova T. V., Reynolds I. J. (2001) J. Neurochem. 79, 266–277 [DOI] [PubMed] [Google Scholar]
- 35.Skulachev V. P. (1991) FEBS Lett. 294, 158–162 [DOI] [PubMed] [Google Scholar]
- 36.Dedukhova V. I., Mokhova E. N., Skulachev V. P., Starkov A. A., Arrigoni-Martelli E., Bobyleva V. A. (1991) FEBS Lett. 295, 51–54 [DOI] [PubMed] [Google Scholar]
- 37.Krauss S., Zhang C. Y., Lowell B. B. (2005) Nat. Rev. Mol. Cell. Biol. 6, 248–261 [DOI] [PubMed] [Google Scholar]
- 38.Horvath T. L., Warden C. H., Hajos M., Lombardi A., Goglia F., Diano S. (1999) J. Neurosci. 19, 10417–10427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andrews Z. B., Diano S., Horvath T. L. (2005) Nat. Rev. Neurosci. 6, 829–840 [DOI] [PubMed] [Google Scholar]
- 40.Liu D., Chan S. L., de Souza-Pinto N. C., Slevin J. R., Wersto R. P., Zhan M., Mustafa K., de Cabo R., Mattson M. P. (2006) Neuromol. Med. 8, 389–414 [DOI] [PubMed] [Google Scholar]
- 41.Echtay K. S., Roussel D., St.-Pierre J., Jekabsons M. B., Cadenas S., Stuart J. A., Harper J. A., Roebuck S. J., Morrison A., Pickering S., Clapham J. C., Brand M. D. (2002) Nature 415, 96–99 [DOI] [PubMed] [Google Scholar]