

Abstract
The Root effect is a widespread property among fish hemoglobins whose structural basis remains largely obscure. Here we report a crystallographic and spectroscopic characterization of the non-Root-effect hemoglobin isolated from the Antarctic fish Trematomus newnesi in the deoxygenated form. The crystal structure unveils that the T state of this hemoglobin is stabilized by a strong H-bond between the side chains of Asp95α and Asp101β at the α1β2 and α2β1 interfaces. This unexpected finding undermines the accepted paradigm that correlates the presence of this unusual H-bond with the occurrence of the Root effect. Surprisingly, the T state is characterized by an atypical flexibility of two α chains within the tetramer. Indeed, regions such as the CDα corner and the EFα pocket, which are normally well ordered in the T state of tetrameric hemoglobins, display high B-factors and non-continuous electron densities. This flexibility also leads to unusual distances between the heme iron and the proximal and distal His residues. These observations are in line with Raman micro-spectroscopy studies carried out both in solution and in the crystal state. The findings here presented suggest that in fish hemoglobins the Root effect may be switched off through a significant destabilization of the T state regardless of the presence of the inter-aspartic H-bond. Similar mechanisms may also operate for other non-Root effect hemoglobins. The implications of the flexibility of the CDα corner for the mechanism of the T-R transition in tetrameric hemoglobins are also discussed.
Keywords: Allosteric Regulation, Evolution, Hemoglobin, Protein Structure, Spectroscopy, X-ray Crystallography
Introduction
Protein crystallography is a fundamental tool to interpret protein function at atomic level. Although this approach is usually effective in understanding the general trends of biological processes, it is often difficult to single out the structural details important for their fine regulation. The case of hemoglobin (Hb)2 is emblematic in this context. Since the pioneering studies by Perutz, the basic features of Hb function (identification of distinct quaternary states, transitions between these states, etc.) have been elucidated at atomic level. However, the identification of the structural features at the basis of the different properties of Hbs, isolated from organisms living under different conditions, has proven to be highly elusive. In this framework, the so-called Root effect represents one of the most puzzling issues.
The Root effect, first described in 1931, is a peculiar property of some fish Hbs that is associated with an extremely low affinity for oxygen at low pH values (1, 2). Notably, at oxygen partial pressures adequate to saturate most of vertebrate Hbs, Root-effect Hbs generally remain in a deoxygenated state at acidic pH values. The Root effect has been functionally related to the filling of the fish swim bladder with gas and to the supply of oxygen to the typically un-capillarized fish retina (3). Although the physiology and the structural basis of the Root effect have been addressed in a large number of studies (2–8), it remains a mystery in many aspects. Over the years, several hypotheses on the structural determinants of the Root effect have been suggested. A variety of possible mechanisms involving the replacement of Cys93β (F9) in HbA by Ser (9) in Root effect Hbs, the formation of a strong hydrogen bond between the side chains of aspartic residues in the deoxy T state (10), and the presence of a cluster of charged residues located at the β1β2 interface of the liganded R state (11), have been proposed. The characterization of Antarctic fish Hbs has played an important role in the field. Indeed, Root- and non-Root-effect Hbs isolated from Antarctic fish often display remarkable sequence identity. A paradigmatic example is the Hb isolated from Trematomus bernacchii (HbTb) (12) and the major Hb of Trematomus newnesi (Hb1Tn) (13). The sequence of the non-Root-effect Hb1Tn shows only fourteen amino acid substitutions when compared with the Root-effect HbTb (14). Moreover, Hb1Tn also shows striking similarities when compared with the Root-effect cathodic Hb from T. newnesi (HbCTn) (13). Although β chains of Hb1Tn and HbCTn exhibit significant differences (the sequence identity is 68%), these two proteins share an identical α chain. Despite the extensive characterizations of these proteins, the results have not yet provided any clues that may explain the large differences in the behavior of these Hbs toward the proton activity (13).
Here we report the crystal structure of Hb1Tn in the deoxygenated state (DeoxyHb1Tn). The structure along with canonical properties also displays some unexpected features. On the basis of these findings, an explanation of the anomalous behavior of this protein is offered. Moreover, present data reveal an intriguing mechanism for switching off the Root effect that may operate in other fish Hbs.
EXPERIMENTAL PROCEDURES
Protein Preparation and Crystallization
Hb1Tn was purified by ion-exchange chromatography on a DE52 column, equilibrated with 10 mm Tris-HCl, pH 7.6, and eluted stepwise with the same buffer (13). DeoxyHb1Tn stock solutions were prepared according to the two following protocols: (a) via photolysis, by exposing a solution of Hb1Tn in the carbomonoxy form to a strong white light under argon (sample A), or (b) through an oxidation/reduction cycle in which potassium hexacyanoferrate and sodium dithionite were alternatively added (sample B).
The formation of the deoxygenated species was tested by optical spectroscopy. The absorption spectrum showed the Soret band at 430 nm and an additional single band at 555 nm. These are markers of deoxygenated Hbs. Crystallization trials were performed at room temperature, in an inert nitrogen atmosphere provided by a glove box. The free interface diffusion technique was used: the protein, in a 100 mm sodium acetate buffer pH 6.0, 2 mm dithionite, at a final concentration of 10 mg/ml, was poured into a capillary containing 20% (w/v) MPEG 5000 (2 mm dithionite). Single crystals of deoxy-Hb1Tn, suitable for x-ray diffraction, were grown in about 12 days. Crystals grown using samples A and B displayed similar morphologies.
X-ray Data Collection and Processing
X-ray diffraction data were collected using a Saturn 944 CCD detector mounted on a MicroMax 007HF rotating anode (Rigaku). Crystals were frozen at liquid-nitrogen temperature (100 K) using glycerol as a cryoprotectant (22%). Crystals from samples A and B were isomorphous and diffracted at 2.01 and 2.20 Å, respectively. The space group is tetragonal P41 with one tetramer in the asymmetric unit. Diffraction data were processed using the program DENZO and Scalepack (15). A summary of the processing statistics is reported in Table 1. The Rmerge value was 7.9 and 10.7% for crystals grown from samples A and B, respectively.
TABLE 1.
Crystal data, data collection, and refinement statistics for the two crystals of deoxyHb1Tn
| A | B | |
|---|---|---|
| Data processing | ||
| Space group | P41 | P41 |
| Cell dimensions (Å) | a = b = 62.02, c = 187.93 | a = b = 61.88, c = 187.01 |
| Unit cell volume (Å3) | 722900 | 716148 |
| Z | 4 | 4 |
| Resolution range (Å) | 39.7–2.01 (2.07–2.01)a | 26.6–2.20 (2.28–2.20) |
| Observed reflections | 148652 | 125230 |
| Unique reflections | 42455 | 32263 |
| Overall redundancy | 3.5 | 3.9 |
| Completeness (%) | 90.6 (73.1) | 90.9 (74.5) |
| Average I/σ(I) | 17.7 (4.8) | 16.7 (5.9) |
| Rmerge (%)b | 7.9 (20.8) | 10.7 (18.4) |
| Refinement | ||
| Twin fraction (see text) | 0.38 | 0.26 |
| No. of working/free reflections | 40217/2238 | 30656/1607 |
| Rwork/Rfree (%)c | 18.5/25.0 | 19.5/24.7 |
| Rfinal (all reflections) | 18.7 | 19.6 |
| No. of protein atoms | 4670 | 4670 |
| No. of water oxygen atoms | 138 | 105 |
| No. of heme atoms | 172 | 172 |
| Average ω angle/σ(ω) (°) | 180.1/3.6 | 180.1/3.0 |
a Values in parentheses refer to the highest resolution shell.
b Rmerge = Σhkl Σi |Ii(hkl) − <I(hkl)>|/ ΣhklΣ Ii(hkl), where Ii(hkl) is the intensity of the ith observation and <I(hkl)> is the average of all the observations for the reflection hkl.
c Rwork = Σhkl Io(hkl) 1/2 − Ic(hkl) 1/2/Σhkl Io(hkl) 1/2, where Io(hkl) is the observed twinned intensity of the reflection hkl and Ic(hkl) is the calculated twinned intensity obtained from the weighted sum of the calculated intensities of the reflections hkl and khl. Rfree is Rwork calculated using 5% of randomly selected reflections, omitted from refinement. Rfinal is Rwork calculated from a final refinement run in which all reflections were included.
Structure Determination and Refinement
The structure of DeoxyHb1Tn was solved by molecular replacement using the program AmoRE (16), and the structure of deoxy HbTb (Protein Data Bank code 2H8F) as a starting model. The refinement was performed using the program SHELX (17).
In the course of the refinement (see below), it became obvious that crystals were affected by merohedral twinning and the diffraction pattern was interpreted as resulting from two lattices correlated by rotation of 180° around an axis parallel to a + b diagonal. Therefore, the intensity associated to each reflection hkl is the weighted sum of two contributions in Equation 1,
where c is the twin fraction, which refined to a value of 0.38 and 0.26 for the data collected on crystals A and B, respectively.
Refinement runs were followed by manual intervention, using the molecular graphic program O (18) to correct minor errors of the side chains. Water molecules were identified by evaluating the shape of the electron density and the distance of potential hydrogen bond donors and/or acceptors. The refinement ended with an R-factor of 0.187 (Rfree 0.250) for data collected on crystal A, and to an R-factor of 0.195 (Rfree 0.247) for crystal B. A summary of the refinement statistics is reported in Table 1. Coordinates of the model derived from the crystal A have been deposited in the Protein Data Bank (3NFE).
Resonance Raman Spectroscopy and Microscopy
The deoxygenated forms of Hb1Tn, HbTb, and HbA were studied in solution by Resonance Raman (RR) spectroscopy. In addition, deoxygenated Hb1Tn and HbTb were also analyzed in the crystal state by RR microscopy. In the RR experiments carried out in solution, Hb1Tn was kept in a 100 mm NaAc pH 6.0, HbTb in 100 mm phosphate buffer pH 6.2, and HbA in 10 mm ammonium phosphate buffer pH 6.5. The heme concentration was 2 mm. Initial Hb1Tn and HbA-deoxygenated samples were prepared by deoxygenation in situ with 2 mm sodium dithionite. Because HbTb is endowed with a strong Root effect, the acidification process rapidly leads to transition from the R to the T state with release of the CO ligand; HbTb was thus prepared in situ from its CO derivative, by decreasing pH down to 6.2.
A confocal Raman microscope (Jasco, NRS-3100) was used to record Raman spectra. The 458-nm line of an air-cooled Ar+ laser (Melles Griot, 35 LAP 431–220), 125 milliwatt, was injected into an integrated Olympus microscope and focused to a spot size of ∼2 μm by a 100× or 20× objective. The laser power at the sample was 2 milliwatt. A holographic notch filter was used to reject the excitation laser line. Raman scattering was dispersed through a monochromator (2400 grooves/mm grating) and collected by a Peltier-cooled 1024 × 128 pixel CCD photon detector (Andor DU401BVI). Typically, several 10 min solution spectra were recorded and averaged (4 cm−1 resolution) by a standard software routine. Frequency shifts were calibrated by using indene and CCl4.
Microscopy experiments were conducted on Hb1Tn deoxy and HbTbdeoxy crystals by adopting previously reported procedures. DeoxyHb1Tn crystals were obtained as indicated above, while DeoxyHbTb were grown as described (19). In microscopy experiments, crystals were transferred to a single hanging drop reactor. Experiments performed on several randomly oriented crystals did not show any significant dependence of number and position of the Raman bands on crystal orientation. Some effects on the relative band intensity in the low-frequency region were observed. Complete data sets were registered in 60 s.
RESULTS
Overall Description of the Structure
Crystallization of Hb1Tn in the deoxy state has proven to be a rather difficult task. Crystals of this protein had been previously grown, but their heavy non-merohedral twinning has prevented further crystallographic investigations (20). In the present work, highly pure samples of deoxygenated Hb1Tn were freshly prepared using two different procedures: (a) photolysis of the carbomonoxy derivative, (b) alternate oxidation and reduction steps by alternated addition of potassium hexacyanoferrate and sodium dithionite. Crystals of DeoxyHb1Tn suitable for x-ray diffraction analysis were obtained from both samples and proved to be isomorphous (see Table 1 and under “Experimental Procedures”). The refined structures are also essentially equal within the expected experimental errors; the largest differences, observed in the regions of high thermal displacement parameters, may be only marginally significant. Unless explicitly stated, only the features of the crystals obtained through the procedure (a) will be discussed.
The crystals are closely related to those of DeoxyHbTb (19). Despite the different space group symmetry (P41 for DeoxyHb1Tn and P21 for DeoxyHbTb), the two Hbs share a similar packing organization. Indeed, in both cases crystals are assembled through the stacking of layers parallel to the ac plane in DeoxyHbTb and to the ab plane in DeoxyHb1Tn; the layers are strictly isomorphous and have an almost exact C2 planar symmetry, with the 2-fold axis lying in the plane of the layer and practically coincident with the molecular dyad axis (supplemental Fig. S1). Packing interactions within the layers are fully conservative in the two crystals (supplemental Table S1 and S2). The orientation of the molecules is such that the CD and FG corners of the α and β chains as well as helix D of the β chains protrude out of the layer and mediate the inter-layer contacts. The differences between the two crystals arise from the manner the layers are repeated along the third direction: in DeoxyHb1Tn each layer is rotated 90° with respect to the previous one in the stacking, while in DeoxyHbTb this rotation is 180°. As a consequence, the packing contacts between layers are different in the crystals of the two species.
Similar to DeoxyHbTb (19), DeoxyHb1Tn crystals are merohedrally twinned. The structure was successfully refined by assuming that the diffraction pattern was generated by the scattering of two lattices correlated by a rotation of 180° around an axis parallel to a + b diagonal. As for HbTb (19), the presence of the twinning did not prevent tracing of a detailed structural model of the protein. The model was validated by the analysis of the crystallographic and stereochemical indicators (Table 1) and by the overall quality of the electron density maps (Fig. 1). In Table 1, the crystallographic data and refinement parameters of both crystals of DeoxyHb1Tn obtained with the (a) and (b) procedures are reported. All crystals hitherto reported of deoxygenated Hbs isolated from Antarctic fishes are either merohedrally or non-merohedrally twinned.
FIGURE 1.
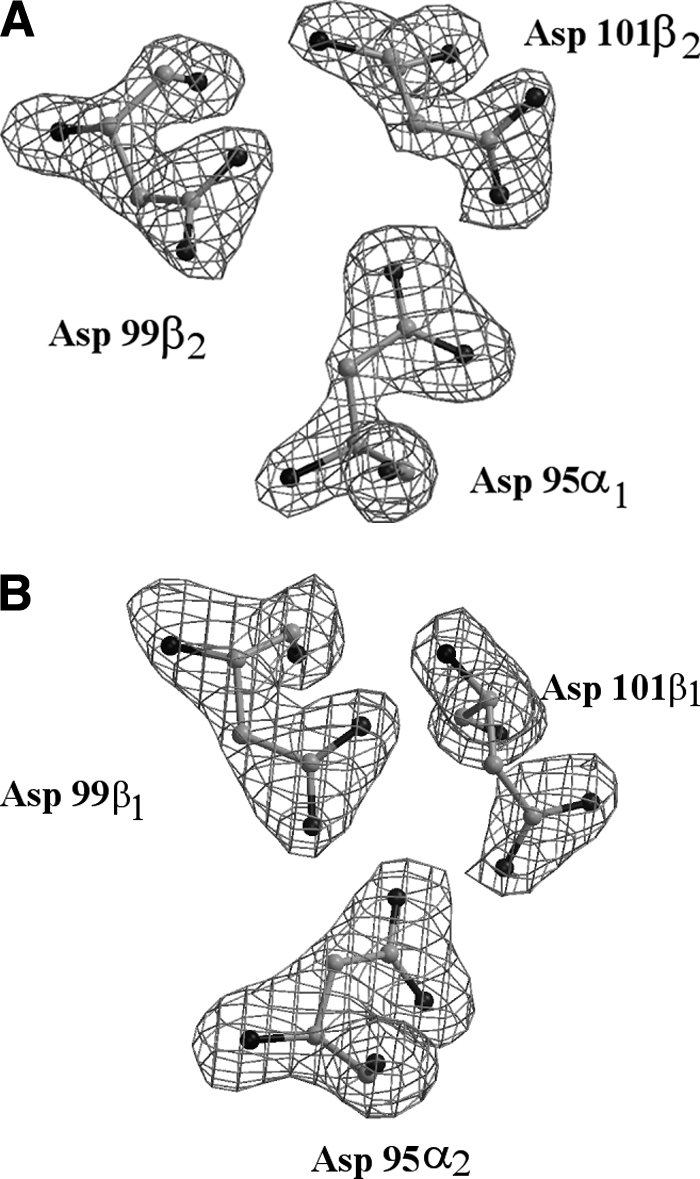
Omit Fo-Fc electron density map of the inter-aspartic triad at the α1β2 (A) and α2β1 (B) interface contoured at 3.8 σ and at 3.0 σ, respectively.
Quaternary Structure of DeoxyHb1Tn
The overall structure of DeoxyHb1Tn displays all the features typically associated to Hbs in the T state. The root mean square deviation (RMSD) computed on the Cα atoms with respect to the structures of DeoxyHbTb (19) and DeoxyHbCTn (21) is 0.48 Å and 0.68 Å, respectively. These values are similar to the RMSD computed between the DeoxyHbTb and DeoxyHbCTn tetramers (0.58 Å). As generally found in tetrameric Hbs, DeoxyHb1Tn significantly differs from the fully or partially liganded states of Hb1Tn (14, 22). For comparison, the carbomonoxy derivative of Hb1Tn was used, since for this Hb, as well as for other Antarctic fish Hbs, the oxygenated crystals cannot be obtained due to their high autoxidation rate (22). The superposition, based on the BGH core (23, 24), of α2β2 (α1β1) dimers between the carbomonoxy (R state) (14) and deoxy (T state) models of Hb1Tn, after superposition of the α1β1 (α2β2) dimers, requires a rotation χ = 9.3°. This value and the position of the rotation axis compare very well with the superposition parameters obtained by a similar analysis carried out on the liganded (12) and unliganded (10, 19) forms of HbTb, underlining the strict similarity in the quaternary-structure organization of these Hbs. In comparison to human HbA (24), the rotation angle is considerably smaller; but this smaller value is somewhat compensated by the position of the rotation axis, which is displaced toward the periphery of the molecule and passes through the N termini of the α chains. Thus, in going from the R to the T state the relative shift at the switch region of the α1β2 (α2β1) interface remains essentially the same in HbTb and Hb1Tn with respect to HbA. The superposition parameters in Antarctic fish Hbs are also similar to those displayed by trout and tuna Hbs (6, 25). The small differences with respect to HbA may be correlated to the acetylation of the N-terminal residues in the α chains (11), a structural feature common to all teleost Hbs. Specific interactions, typical of the HbA T state, are maintained in the present structure and include the salt bridge between Lys40α (C5) and the carboxyl group of C-terminal His146β (HC3), the hydrogen bond between the charged chain of Arg142α (HC3) and the backbone oxygen atom of Val34β (B16) and the hydrogen bonds between the side chain of Asp99β (G1) and those of Tyr42α (C6)and Asn98α (G4). Unexpectedly, the tetramer is also stabilized by the hydrogen-bonding interactions between aspartic side chains, that are usually associated to the T state of Root-effect Hbs (6, 19, 21). Indeed, the omit map clearly reveals the spatial proximity of the side chains of Asp95α (G1), Asp99β (G1), and Asp101β(G3) at both the α1β2 and α2β1 interfaces (Fig. 1). The distance between the Asp95α1 (α2) and Asp101β2 (β1) carboxylic oxygens (2.46 Å and 2.30 Å in the two cases, respectively) is indicative of a strong hydrogen bond, which requires one of the two Asp (presumably Asp95α) to be protonated. Thus, the quaternary structure of the deoxy state of the non-Root-effect Hb1Tn is stabilized by interactions typically associated to Root effect Hbs.
Tertiary Structure of DeoxyHb1Tn and Heme Pocket
The analysis of the tertiary structure of the α and β chains within the tetramer highlights analogies and differences with respect to the previously characterized Hbs in their T state. The β chains present an overall structure and coordination of the heme iron atom that are in line with those generally observed in tetrameric Hbs in their deoxygenated state. Despite the relatively high values of the displacement thermal parameters, evenly distributed along the chain (35.4 ± 7.6 Å2 averaged over all the Cα,C,O main chain atoms), the electron density is sufficiently well defined to provide a clear picture of the β-heme pocket (Fig. 2A). In this region a canonical arrangement of the relevant structural elements lining the prosthetic group is observed. Moreover, in agreement with the T structure of the Root-effect HbTb (19), the side chain of C-terminal His146β (HC3) also experiences an elevated thermal motion and is not hydrogen bonded to the side chain of Glu94β (FG1). It is important to mention that the formation of the ion pair between His146β and Asp94β (FG1), has been invoked to explain part of the pH dependence of the oxygen affinity for mammalian Hbs endowed with Bohr effect (26, 27).
FIGURE 2.

Omit Fo-Fc electron density maps of the β1 (A), α1 (B), and α2 (C) heme regions contoured at 2 σ. The electron density at the β2 heme is identical to that observed at β1.
On average, the α chain also displays a similarly elevated displacement thermal parameters (35.1 ± 9.9 Å2). This contrasts with the usual finding that the α chain has a lower flexibility with respect to the partner β chain (28, 29). However, as it can be deduced by the inspection of Fig. 3 and from the value of the mean square fluctuation of the B factors, the larger values are unevenly distributed along the chain and are particularly associated to the CD loop, to a substantial part of the helix E that includes distal His, to the region 80–95, which embodies the whole helix F and the FG corner at the proximal site of the heme pocket, and to the heme group itself. This observation is corroborated by the interruptions observed locally in the electron density profile that reveals an unusually high level of static and/or dynamic disorder (Fig. S2 in supplementary material).
FIGURE 3.

B factors as a function of the residue number for DeoxyHb1Tn and DeoxyHbTb are reported for the α (A) and β (B) chain. For each residue the average value calculated over the main chain atoms has been reported. Stars indicate positions of distal and proximal His residues.
In addition, the structural details at the heme pocket are also atypical. The bond length of proximal His to the α iron (2.3 Å) is slightly longer than that usually found in tetrameric Hbs (Fig. 2, B and C), (30) and, most surprising, the Nϵ of distal His is only 2.94 Å (3.0 Å and 2.9 Å for α1 and α2, respectively) away from the iron ion. The displacement of the iron from the heme plane (0.2 Å) is also lower than that normally found in Fe(II) penta-coordinated forms of Hb (30). In addition, the Cα-Cα distance between distal and proximal His (13.1 Å) is significantly shorter than the usual values (22). Indeed, for all the structurally characterized Hb heme pockets, in both the oxy and deoxy state, the distribution of this distance is highly peaked around a mean value of 14.5 Å but shrinks to a much smaller value (12.4 Å) when both residues are bound to the central iron, forming a bis-histidyl adduct (31). This anomalous set of the refined stereochemical parameters and the disordering observed in the electron density maps may find an explanation in the presence of alternate structures around the heme group (see “Discussion”). Interestingly, the unusual features of the E/F fork, the V-shaped two-helix motif that holds the heme group in place, is correlated to an enhanced disorder of the CD corner. In DeoxyHbTb, the protonated His55α (E3) is salt-bridged to Asp48α (CD6) and makes a stacking interaction with Trp46α (CD4); both interactions are important to maintain the structure organization of this segment (19). The change on the protonation state of His55α upon oxygenation has been proposed to be important in the modulation of the Root-effect strength (19). The replacement of His by Asn deprives the Hb1Tn CDα corner of an important interaction and destabilizes the entire region (Fig. 4). In one of the subunits, the destabilization of the region also results in the break of electrostatic interaction between one of the heme propionates and His45α (CD2) that is present in both the T and R state of Hb1Tb (10, 12, 19), and in the R state of Hb1Tn (14). In the second subunit this interaction is also present, but the density of the imidazole moiety as well as that of the propionate group is poorly defined.
FIGURE 4.

Superimposition of the CD corner and the distal heme pocket of the α chain of DeoxyHb1Tn (red) and DeoxyHbTb (green). Residues of helices B (21–35), G (97–113), and H (120–138) of the subunit have been used for superimposition. The salt bridges (His 45-heme propionate and His55-Asp48) that stabilize the local structure of DeoxyHbTb are also shown.
All peculiarities observed for the α chain were essentially confirmed by the refined structure B of the crystals obtained by treating the carbomonoxy form of Hb1Tn with potassium hexacyanoferrate and sodium dithionite. It must be stressed that in both A and B structures the stereochemical restraints regarding the interactions with the heme group of proximal and distal His were not included in the refinement protocols. Even though their final values may be subject to larger errors resulting from the fact that the electron density map is poorer than in the remaining part of the α chain, in principle they are not biased against any a priori assumption.
Resonance Raman Spectroscopy and Microscopy Experiments
Resonance micro-Raman spectra were measured in the high-frequency (1300–1700 cm−1) and low-frequency (200–450 cm−1) regions. The former includes the porphyrin in-plane vibrational modes sensitive to the electron density of the macrocycle, to the oxidation, coordination and spin state, whereas the latter contains bands with contributions from deformation of the various angles of the heme group, as well as the stretching of the bonds from the pyrrole N atoms to the central metal atom (32). For comparative purposes, the spectra were registered on Hb1Tn, HbTb, and HbA. As shown in Fig. 5, the spectra of the proteins, including Hb1Tn, are characteristic of a ferrous penta-coordinated high-spin state. Indeed, the absence of bands at 1361 and 1496 cm−1 (ν4 and ν3, respectively), indicates that hexa-coordinated low-spin bis-histidyl ferrous adducts are not present in a detectable amount (>5%) both in solution and in the crystal deoxy state. In the high-frequency region the spectra of Hb1Tn and HbTb are different from those of HbA and show evidence of a second ν(C=C) vinyl stretching mode at 1625 cm−1 in addition to that at 1617 cm−1. In agreement with this observation, two δ(CβCαCβ) bending modes of the vinyl groups are observed in the low frequency region at 405 and 412 cm−1 (Fig. 6), respectively, that are characteristic of Hbs of high-Antarctic (22, 33–35) and sub-Antarctic (36) Nototenioidei. The analysis of the RR ν(Fe-Im) stretching mode reveals that Hb1Tn has a distinct behavior when compared with HbA and HbTb. In solution DeoxyHb1Tn displays an RR ν(Fe-Im) stretching mode at 208–210 cm−1 that is to be compared with the value of 215 cm−1 observed in HbTb and HbA, the latter in full agreement with previous reports (37) (Fig. 6, solid lines). A deconvolution of this band suggests that at least two Fe-Im bands are present, centered at 205 and 215 cm−1, almost equally populated (45 and 55%, respectively). This overall picture was confirmed by the microRaman spectra collected on Hb1Tn and HbTb in the crystal state (Fig. 6, dashed lines). The downshift of the ν(Fe-Im) stretching mode observed in Hb1Tn in the deoxy state supports the crystallographic data that indicate a weaker coordination of proximal His. Moreover, the broadening of the band, when compared with HbTb and HbA, suggests that at least two species, distinguishable for their binding properties at the heme group, can be accessed by DeoxyHb1Tn both in solution and in the crystal state.
FIGURE 5.

Resonance Raman spectra in solution in the high-wavenumber region of Hb1Tn, HbTb, and HbA in the deoxy state.
FIGURE 6.

Resonance Raman spectra in the low-wavenumber region of the deoxy form of Hb1Tn and HbTb in solution (solid lines) and in the crystal state (dashed lines) the spectrum of the deoxy form of HbA is also shown.
DISCUSSION
Recent investigations have highlighted that the major Hb of T. newnesi undergoes an unusual oxidation process characterized by the presence of states, such as hemichromes and penta-coordinated ferric states (22), which are atypical for tetrameric Hbs in their folded states (22). From the functional point of view, its oxygen affinity is only marginally dependent on pH despite the remarkable sequence similarity to Hb of T. bernacchii that possesses a strong Root effect (13). This similarity also extends to tertiary and quaternary assembly in the R state. HbTb and Hb1Tn both present, in their R state, a cluster of positive charges similar to that found in Spot Hb and suggested to be one of the major determinants of the Root effect (11). In the lack of any data on the T structure of Spot Hb, the results on the R state of HbTb and Hb1Tn indicate that the differences in the functional properties of these two Hbs should find an explanation in the comparison of their T state (14). The present data further support this earlier suggestion (14, 38).
The crystal structure of Hb1Tn in its deoxygenated state was obtained by refining two independent data sets collected on crystals prepared by different protocols (structures A and B). Except for few minor differences in the most disordered regions, the two models reveal the same unexpected features: (i) the presence of the inter-aspartic H-bond at the α1β2 (α2β1) interface, so far strictly associated to Root-effect Hbs; (ii) the high level of thermal displacement parameters that in the α chain is particularly evident for the CD corner, helices E and F, the FG corner and the heme group; (iii) the geometrical parameters of the E and F interactions with the α heme, that are somewhat intermediate between the usual penta-coordinated unliganded state and a bis-His coordination at iron atom (hemochrome species).
The latter observation suggests that the observed crystal structure at the α chain could be an average of at least two competitive states, as the observed stereochemistry cannot be fully accounted by a single model. This hypothesis is also supported by the large values of the thermal displacement parameters observed for the entire E/F fork, the FG corner and the α heme. However, a possible contribution to the crystallographic model by a structure possessing a bis-histidyl coordination at the heme iron is ruled out by the RR spectra (Fig. 5). Evidences for the presence of more than one conformation can also be spotted on the electron density maps presented in Fig. 2, B and C. This finding together with the observed downshift of the Fe-Im stretching frequency and the broadening of the band both in solution and in the crystal state (Fig. 6) suggest the possibility of a competitive penta-coordination to the Hb1Tn α heme involving distal His. However, due to the medium resolution of the diffraction pattern and to the presence of twinning, modeling the disorder was not attempted.
Are the observed differences between Hb1Tn and HbTb intrinsic to the molecular structure or could they be caused by the crystal environment? It should be recalled that in both cases the crystals are built by layers that are practically isomorphous; therefore the intermolecular interactions within layers are strictly preserved. Differences between the two crystal forms arise from the way the layers are stacked on top of each other and this must be related to differences in the interlayer interactions. As these interactions mainly involve the CD and FG corners and the helices E and F of the α and β chains, as well as the helix D of the β chains, the difference in the stacking between the two Hbs seems to be a consequence of the different properties of these segments, rather than vice versa.
The present findings undermine the current paradigm that relates the presence of the inter-aspartic H-bond in the T state with the occurrence of the Root effect. In HbTn the formation of the H-bond appears to be counterbalanced by an extensive weakening of the CD and α-heme regions that is not observed in Root-effect Hbs so far characterized. In the latter, the absence of cooperativity at low pH values is likely caused by overstabilization of the T state versus the R state, whereas in Hb1Tn this stabilization effect is opposed by the disordered regions. Thus, the accessibility to both R and T states together with the different oxygen affinities of the two forms can account for the observed cooperativity of Hb1Tn at acidic pH. Analysis of the R (14) and T states of Hb1Tn also provides an indication for the absence of proton uptake upon deoxygenation. Indeed, the protonation of the Asp at the α1β2 (α2β1) interfaces, which occurs upon oxygen release, is in part balanced by the proton release that result from the weaker interaction between His45α, located in the disordered region, and a heme propionate. Therefore, the absence of proton release upon deoxygenation appears to be the net result of opposing events and the inter-aspartic salt bridge is presumably a necessary, but not sufficient motif in determining the Root effect (22). A comparative analysis of the amino acid substitutions between Hb1Tn and HbTb provides further clues (supplemental Fig. S3). The replacement of Asn by His in position 55α (E3) plays an important role in determining the observed structural differences between the two Hbs, as Asn is unable to establish the stabilizing interactions formed by charged His55α in HbTb (22). With a more general statement, we suggest that the activation of an order-disorder transition in the hot region at the CD corner of the α chain, occurring along the R to T transition, could play a major role in switching the Root effect on and off. Along the evolutionary pathway several different structural motifs could in principle activate or deactivate this general mechanism. With regards to HbCTn, cathodic Hb of T. newnesi, endowed with a mild Root effect (the number of protons exchanged at constant pH in the R/T transition is only half of that exchanged by HbTb (12, 13)), the disordering in the CDα region is limited, despite the presence of Asn in position 55. In this case, however, the relevant number of substitutions in the β chain with respect to Hb1Tn prevents a clear identification of residues responsible for these effects (supplemental Fig. S3). In a more general framework, as the residues involved in the formation of the inter-aspartic H-bond are universally conserved in fish Hbs, it can be surmised that in many non-Root effect Hbs the perturbation of the T-state compensates the stabilization produced by the inter-aspartic H-bond, thus reducing in this way the impact of the pH on the oxygen affinity of these Hbs.
Historically, Root effect has been physiologically related to the presence of the swim bladder and choroid rete (3), two organs that have been frequently acquired and lost during fish evolution. The weakening of the Root effect has been noticed in lineages where the choroid rete has been lost, whereas the loss of swim bladder does not affect the strength of the Root effect when the choroid rete is still present (4). Because high-Antarctic notothenioids still have Hbs endowed with Root effect also when the choroid rete is absent, this function may undergo neutral selection not representing a disadvantage for the species. In contrast to T. bernacchii, T. newnesi has lost choroid rete, very well developed in the most basal lineage of the suborder. In this scenario, the presence in DeoxyHb1Tn of the inter-Asp H-bond at α1β2 interface leads to the intriguing hypothesis that Hb1Tn was originally endowed with Root effect. Antarctic fish are undergoing dynamic changes in response to temperature adaptation with tendency toward reduction of Hb multiplicity, functionality, and concentration from basal families to more derived clades.
For Hb1Tn the evolution may be in action with the Root effect disappearing as a consequence of the loss of the choroid rete. This or similar mechanisms may have taken place for several other non-Root-effect Antarctic fish Hbs that have remarkable similarity with Root-effect Hbs.
As a concluding remark, it should be underlined that the observed flexibility of CDα and EFα in DeoxyHb1Tn may also have a fundamental implication for the transition of vertebrate Hbs from the T to the R state. Indeed, the order-to-disorder transition of these key regions in Hb tetramer, that in DeoxyHb1Tn destabilizes the overall structure of the T state, may also play a role in the conversion of the low-oxygen affinity T state to the high affinity R-state.
Supplementary Material
Acknowledgments
We thank Giosuè Sorrentino and Maurizio Amendola for technical assistance. CIMCF (Centro Interdipartimentale di Metodologie Chimico-Fisiche) and Consorzio Regionale di Competenza in Biotecnologie Industriali (BioTekNet) are also acknowledged.
This work was financially supported by PNRA (Italian National Programme for Antarctic Research) and is in the framework of the programme Evolution and Biodiversity in the Antarctic (EBA), sponsored by the Scientific Committee for Antarctic Research (SCAR). This study was also supported in part by the Ministero Italiano dell'Università e della Ricerca Scientifica (PRIN 2007 “Struttura, funzione ed evoluzione di emoproteine da organismi marini artici ed antartici: meccanismi di adattamento al freddo e acquisizione di nuove funzioni”).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1—S3 and Tables S1 and S2.
The atomic coordinates and structure factors (code 3NFE) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- Hb
- hemoglobin
- DeoxyHbTb
- deoxy hemoglobin of T. bernacchii
- DeoxyHb1Tn
- deoxy form of the major hemoglobin from T. newnesi
- HbTb
- Hb of T. bernacchii
- Hb1Tn
- major Hb of T. newnesi
- HbCTn
- cathodic Hb of T. newnesi
- HbA
- adult human hemoglobin
- PDB
- Protein Data Bank
- RMSD
- root mean square deviations
- RR
- resonance Raman.
REFERENCES
- 1.Brittain T. (1987) Comp. Biochem. Physiol. Part B: Biochem. Mol. Biol. 86, 473–481 [DOI] [PubMed] [Google Scholar]
- 2.Brittain T. (2005) J. Inorg. Biochem. 99, 120–129 [DOI] [PubMed] [Google Scholar]
- 3.Verde C., Vergara A., Mazzarella L., di Prisco L. (2008) Curr. Prot. Pept. Sci. 9, 578–590 [DOI] [PubMed] [Google Scholar]
- 4.Berenbrink M., Koldkjaer P., Kepp O., Cossins A. R. (2005) Science 307, 1752–1757 [DOI] [PubMed] [Google Scholar]
- 5.Bonaventura C., Crumbliss A. L., Weber R. E. (2004) Acta Physiol. Scand. 182, 245–258 [DOI] [PubMed] [Google Scholar]
- 6.Yokoyama T., Chong K. T., Miyazaki G., Morimoto H., Shih D. T., Unzai S., Tame J. R., Park S. Y. (2004) J. Biol. Chem. 279, 28632–28640 [DOI] [PubMed] [Google Scholar]
- 7.Unzai S., Imai K., Park S.-Y., Nagai K., Brittain T., Tame J. R. (eds) (2008) Mutagenic Studies on the Origins of the Root Effect, Springer, Milan [Google Scholar]
- 8.Vergara A., Franzese M., Merlino A., Bonomi G., Verde C., Giordano D., di Prisco G., Lee H. C., Peisach J., Mazzarella L. (2009) Biophys. J. 97, 866–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perutz M. F. (1996) Nat. Struct. Biol. 3, 211–212 [DOI] [PubMed] [Google Scholar]
- 10.Ito N., Komiyama N. H., Fermi G. (1995) J. Mol. Biol. 250, 648–658 [DOI] [PubMed] [Google Scholar]
- 11.Mylvaganam S. E., Bonaventura C., Bonaventura J., Getzoff E. D. (1996) Nat. Struct. Biol. 3, 275–283 [DOI] [PubMed] [Google Scholar]
- 12.Camardella L., Caruso C., D'Avino R., di Prisco G., Rutigliano B., Tamburrini M., Fermi G., Perutz M. F. (1992) J. Mol. Biol. 224, 449–460 [DOI] [PubMed] [Google Scholar]
- 13.D'Avino R., Caruso C., Tamburrini M., Romano M., Rutigliano B., Polverino de Laureto P., Camardella L., Carratore V., di Prisco G. (1994) J. Biol. Chem. 269, 9675–9681 [PubMed] [Google Scholar]
- 14.Mazzarella L., D'Avino R., di Prisco G., Savino C., Vitagliano L., Moody P. C., Zagari A. (1999) J. Mol. Biol. 287, 897–906 [DOI] [PubMed] [Google Scholar]
- 15.Otwinowski Z., Minor W. (1997) Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 16.Navaza J. (1994) Acta Crystallogr. A50, 157–163 [Google Scholar]
- 17.Sheldrick G., Schneider T. (1997) Methods Enzymol. 277, 319–343 [PubMed] [Google Scholar]
- 18.Jones T. A., Zou J. Y., Cowan S. W., Kjedgaard M. (1991) Acta Crystallogr. Sect. D: Biol. Cryst. 56, 714–721 [Google Scholar]
- 19.Mazzarella L., Vergara A., Vitagliano L., Merlino A., Bonomi G., Scala S., Verde C., di Prisco G. (2006) Proteins: Struct. Funct. Bioinformatics 65, 490–498 [DOI] [PubMed] [Google Scholar]
- 20.Riccio A., Vitagliano L., di Prisco G., Zagari A., Mazzarella L. (2001) Acta Cryst. D: Biol. Cryst. 57, 1144–1146 [DOI] [PubMed] [Google Scholar]
- 21.Mazzarella L., Bonomi G., Lubrano M. C., Merlino A., Riccio A., Vergara A., Vitagliano L., Verde C., di Prisco G. (2006) Proteins: Struct. Funct. Bioinformatics 62, 316–321 [DOI] [PubMed] [Google Scholar]
- 22.Vitagliano L., Vergara A., Bonomi G., Merlino A., Verde C., di Prisco G., Howes B. D., Smulevich G., Mazzarella L. (2008) J. Am. Chem. Soc 130, 10527–10535 [DOI] [PubMed] [Google Scholar]
- 23.Baldwin J., Chothia C. (1979) J. Mol. Biol. 129, 175–220 [DOI] [PubMed] [Google Scholar]
- 24.Park S. Y., Yokoyama T., Shibayama N., Shiro Y., Tame J. R. (2006) J. Mol. Biol. 360, 690–701 [DOI] [PubMed] [Google Scholar]
- 25.Tame J. R., Wilson J. C., Weber R. E. (1996) J. Mol. Biol. 259, 749–760 [DOI] [PubMed] [Google Scholar]
- 26.Perutz M. (1970) Nature 228, 734–73916058681 [Google Scholar]
- 27.Perutz M. F. (1980) Proc. Royal Society London, Series B: Biol. Sci. 208, 135–162 [DOI] [PubMed] [Google Scholar]
- 28.Giordano D., Boechi L., Vergara A., Martí M. A., Samuni U., Dantsker D., Grassi L., Estrin D. A., Friedman J. M., Mazzarella L., di Prisco G., Verde C. (2009) FEBS J. 276, 2266–2277 [DOI] [PubMed] [Google Scholar]
- 29.Merlino A., Vergara A., Sica F., Aschi M., Amedei A., Di Nola A., Mazzarella L. (2010) J. Phys. Chem. B 114, 7002–7008 [DOI] [PubMed] [Google Scholar]
- 30.Merlino A., Vergara A., Sica F., Mazzarella L. (2009) Marine Genomics 2, 51–56 [DOI] [PubMed] [Google Scholar]
- 31.Riccio A., Vitagliano L., di Prisco G., Zagari A., Mazzarella L. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 9801–9806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi S., Spiro T. G., Langry K. C., Smith K. M., Budd D. L., La Mar G. N. (1982) J. Am. Chem. Soc. 104, 4345–4351 [Google Scholar]
- 33.Merlino A., Vitagliano L., Howes B. D., Verde C., di Prisco G., Smulevich G., Sica F., Vergara A. (2009) Biopolymers 91, 1117–1125 [DOI] [PubMed] [Google Scholar]
- 34.Merlino A., Verde C., di Prisco G., Mazzarella L., Vergara A. (2008) Spectroscopy 22, 143–152 [Google Scholar]
- 35.Vergara A., Vitagliano L., Verde C., di Prisco G., Mazzarella L. (2008) Methods Enzymol. 436, 421–440 [DOI] [PubMed] [Google Scholar]
- 36.Verde C., Howes B. D., de Rosa M. C., Raiola L., Smulevich G., Williams R., Giardina B., Parisi E., di Prisco G. (2004) Protein Sci. 13, 2766–2781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smulevich G., Possenti M., D'Avino R., di Prisco G., Coletta M. (1998) J. Raman Spectroscopy 29, 57–65 [Google Scholar]
- 38.Giangiacomo L., D'Avino R., di Prisco G., Chiancone E. (2001) Biochemistry 40, 3062–3068 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.