

Abstract
Francisella tularensis contains four putative acid phosphatases that are conserved in Francisella novicida. An F. novicida quadruple mutant (AcpA, AcpB, AcpC, and Hap [ΔABCH]) is unable to escape the phagosome or survive in macrophages and is attenuated in the mouse model. We explored whether reduced survival of the ΔABCH mutant within phagocytes is related to the oxidative response by human neutrophils and macrophages. F. novicida and F. tularensis subspecies failed to stimulate reactive oxygen species production in the phagocytes, whereas the F. novicida ΔABCH strain stimulated a significant level of reactive oxygen species. The ΔABCH mutant, but not the wild-type strain, strongly colocalized with p47phox and replicated in phagocytes only in the presence of an NADPH oxidase inhibitor or within macrophages isolated from p47phox knockout mice. Finally, purified AcpA strongly dephosphorylated p47phox and p40phox, but not p67phox, in vitro. Thus, Francisella acid phosphatases play a major role in intramacrophage survival and virulence by regulating the generation of the oxidative burst in human phagocytes.
Francisella tularensis has been classified by the Centers for Disease Control and Prevention as a category A pathogen. Inhalation of <10 CFU of F. tularensis subspecies tularensis (hereafter F. tularensis) can have fatal consequences (1). F. tularensis can enter and multiply in a wide range of host cell types (2–9); however, in vivo, its primary target is the macrophage (10). F. tularensis enters host macrophages by asymmetric pseudopod loops, and this uptake is dependent on serum complement and host cell receptors, including C3, mannose, and scavenger (3, 11–13). After entering the host cell, the bacterium arrests maturation of the phagosome, and the phagosome is transiently acidified. This acidification is reported to be essential for the subsequent escape of F. tularensis into the cytosol of the macrophage (14, 15), but more recent work has contradicted this (15, 16). Eventually, the phagosomal membrane is compromised by unknown mechanisms, and the bacteria escape into the cytoplasm and replicate (15, 17, 18). Subsequently, it was reported that the bacteria re-enter the endocytic pathway by an autophagy-like process, residing in large multiple-membrane bound vesicles (17, 19). Bacterial release from host cells is thought to occur following Francisella-induced apoptosis (20–22) and pyroptosis (21). However, the final stages of the intracellular cycle are not well understood.
The intracellular life cycle of F tularensis is complex, and the genes involved with all stages are not well understood. However, the key genes involved in Francisella survival in host cells are found on the Francisella pathogenicity island (FPI). This island contains 19 genes, and bioinformatic analysis revealed that the FPI encodes a putative type VI secretion system, similar to the systems involved in the virulence of Pseudomonas aeruginosa (23) and Vibrio cholerae (24). Genes within the FPI are regulated by MglA, SspA, PmrA, MigR, Hfq, and FevR (25–31). Transcriptional analysis indicated that many genes outside of the FPI are also affected by these regulators (27, 30).
In addition to the FPI genes, Francisella acid phosphatase AcpA was shown to play a key role in intracellular survival in macrophages (19, 32, 33). Bioinformatic analysis revealed that Francisella carries four or five acid phosphatases in its genome, depending on the species. Deletion of acpA resulted in a strain that was more susceptible than wild-type (WT) Francisella tularensis subspecies novicida (F. novicida) to killing by human and murine macrophages and had decreased phosphatase activity (32). Additionally, the mutant showed a decreased ability to escape from the phagosome (32). Deletion of three additional acid phosphatases (AcpA, AcpB, AcpC, and Hap [ΔABCH]) in F. novicida resulted in an attenuated strain that was 100% protective against homologous challenge in the mouse model. This mutant did not escape from the macrophage phagosome (19). Transcriptional analysis demonstrated that AcpA and Hap expression are increased during the early stages of macrophage infection (19).
Human neutrophils play a key role in host defense against invading pathogens and are major effectors of the acute inflammatory response. In response to a variety of agents, neutrophils produce a large amount of superoxide anion (O2−), which is essential for bacterial killing and amplifies the inflammatory response (34, 35). The NADPH oxidase complex is responsible for O2− production, and its activation is induced by receptor ligation and uptake of microbial pathogens or chemical or particulate stimuli, such as PMA, fMLP, or zymosan. NADPH oxidase induction in neutrophils or macrophages requires the translocation of phosphorylated p47phox, p40phox, p67phox, and Rac1/2 from the cytoplasm to the phagosomal membrane (34). However, F. tularensis live vaccine strain (LVS) enters neutrophils without triggering the respiratory burst and inhibits NADPH oxidase assembly by an unknown mechanism (36). Earlier studies showed that purified AcpA isolated from Francisella inhibited the respiratory burst of fMLP-induced porcine neutrophils in vitro (33). This suggests that Francisella may alter the function of the NADPH oxidase complex in a manner involving acid phosphatase dephosphorylation of key cellular components. We show in this study that the Francisella acid phosphatases are required for inhibiting NADPH oxidase assembly and function and may do so by dephosphorylation of NADPH oxidase components. This process aids the intracellular survival and proliferation of the bacterium.
Materials and Methods
Isolation of human monocyte-derived macrophages and neutrophils for infection with Francisella spp
Using an Ohio State University-approved Institutional Review Board protocol, heparinized blood samples were collected from normal human donors. The heparinized blood samples from normal human donors were diluted 1:1 with normal saline, and the fraction containing the PBMCs was obtained following centrifugation at 800 × g for 40 min at 20°C over a Ficoll-Hypaque density cushion (Amersham Biosciences, Piscataway, NJ). This fraction was diluted 1:1 with RPMI 1640, and the PBMCs were collected again by centrifugation. The cells were washed twice with RPMI 1640, resuspended in the same media, and counted in a hemocytometer. The cell numbers were adjusted to 2 ± 106 PBMCs/ml in RPMI 1640 containing 20% autologous human serum. The monocytes were allowed to differentiate into monocyte-derived macrophages (MDMs) by incubation for 5 d at 37°C in 5% CO2 in sterile screw cap Teflon wells. PBMCs (MDMs plus lymphocytes) were recovered and resuspended in RPMI 1640 with 10% autologous serum, and MDMs were plated in 24-well tissue culture plates or on coverslips at a density of 2 × 106 cells/well in 0.5 ml culture media, resulting in 2 × 105 MDMs/monolayer. For the respiratory burst experiments, the MDMs were placed in monolayer culture at 105 cells/well in 0.2 ml culture media in 96-well plates. After 2 h of incubation at 37°C in 5% CO2, nonadherent cells were removed by washing the monolayers three or four times with prewarmed RPMI 1640 and replenished with fresh culture medium with 10% autologous serum. For intramacrophage survival assays, MDMs were incubated for an additional 7 d in fresh culture medium with 20% autologous serum to stabilize the MDM monolayer prior to infection (37). Macrophages were incubated with Francisella at a multiplicity of infection (MOI) ∼50:1, as described earlier (12). At various time points, macrophages were lysed with 0.05% SDS and plated on Chocolate II plates to enumerate the CFU. This lysis protocol does not result in death of the bacteria (38).
After PBMC separation on Ficoll-Hypaque, the RBC-containing pellet was further fractionated by dextran sedimentation to separate the neutrophils. Briefly, RBCs were diluted 1:1 with normal saline, and an equal amount of 3% dextran was added to sediment the RBCs. After 30 min incubation on ice, the top layer of cells was transferred to a new tube and centrifuged at 800 × g for 15 min to enrich for the neutrophils. The erythrocytes that remained in the cell pellet were lysed with sterile distilled water for 15 s, and an equal amount of HBSS (without calcium and magnesium ions) containing 0.9% normal saline was added to prevent the lysis of neutrophils. Neutrophils were separated by centrifugation at 800 × g for 5 min. The cell pellet was washed, resuspended in HBSS media, and kept on ice for experimentation.
Growth and opsonization of Francisella spp
F. novicida U112, ΔacpA, and ΔABCH mutants were routinely cultured, as previously described (19). For phagocyte infection studies, Francisella strains were grown on Chocolate II plates overnight at 37°C and collected in HBSS buffer or RPMI 1640 (without phosphate). Cell density was determined spectrophotometrically at 600 nm; bacteria were opsonized with 50% autologous serum for 30 min at 37°C and subsequently washed three times with HBSS buffer to remove excess serum. F. tularensis subspecies tularensis (Schu S4, Type A) and F. tularensis subspecies holarctica (OR96-0246 from Biodefense and Emerging Infections Research Resources Repository, Type B, Manassas, VA) were cultivated and opsonized similarly to F. novicida strains. Opsonized Francisella were resuspended in appropriate buffer and kept on ice for experiments. Formalin-killed Francisella were prepared as described earlier (11). Zymosan beads were opsonized with 50% autologous serum, washed twice in HBSS without divalent cations, and kept on ice for the experiments.
Neutrophils (2 × 105 cells/well) were seeded onto human serum-coated 24-well plates in RPMI 1640, and Francisella were added at an MOI ∼50:1. At time points ≤2 h postinfection, cells were washed and treated with 50 μg/ml gentamicin for 30 min, followed by washing with HBSS to remove the extracellular bacteria. Neutrophils were lysed by the addition of 0.05% SDS, and lysates were diluted in HBSS and plated on Chocolate II plates to enumerate the CFU.
Microscopy of Francisella association with and uptake by polymorphonuclear leukocytes
The association with and uptake of F. novicida and acid phosphatase mutants by polymorphonuclear leukocytes (PMNs) were performed as described earlier (12). In brief, neutrophils (2 × 105 cells/well) were seeded onto human serum-coated 24-well plates in RPMI 1640, and opsonized Francisella was added at an MOI ∼50:1 and incubated for 10 min at 37°C in 5% CO2. After incubation, the cells were washed extensively with RPMI 1640 to remove nonadherent bacteria and fixed in 3.5% paraformaldehyde without permeabilization or permeabilized after paraformaldehyde fixation with chilled 100% methanol for 15–30 s. The coverslips were washed and allowed to dry. Phagocyte-associated bacteria were visualized by indirect immunofluorescence microscopy. In this assay, PMNs on coverslips were incubated with a monoclonal mouse anti-F. novicida LPS primary Ab (Immuno-Precise Antibodies Limited, Victoria, British Columbia, Canada) and diluted 1:100 in blocking buffer composed of 20% human AB serum (Cambrex, East Rutherford, NJ) and 5% BSA (Sigma-Aldrich, St. Louis, MO) for 4 h at room temperature with gentle rotation. After being washed extensively, PMNs were incubated with Alexa Fluor 488-conjugated rabbit anti-mouse IgG (Molecular Probes, Eugene, OR) (diluted 1:1000 in blocking buffer) for 90 min at room temperature. Coverslips were mounted on glass slides. In all assays, the average number of bacteria per PMN on each coverslip was determined by counting a minimum of 200 cells per coverslip using an ×100 oil-immersion objective with a wide-bandwidth 570-nm dichroic mirror on a BX51 Olympus fluorescence microscope. Pictures were taken with a Color 3 digital camera (Olympus, Melville, NY). Triplicate coverslips were used for each test group. Attached bacteria were assessed by scoring nonpermeabilized PMNs, and total associated bacteria were assessed by scoring permeabilized PMNs. The number of bacteria taken up (internalized) was calculated by subtracting the number of attached bacteria from the number of associated bacteria.
Respiratory burst assays
Neutrophils (106/well) were added to human serum-coated microtiter wells in HBSS containing 10 mM glucose, 1% human serum albumin, and 50 μm luminol (Invitrogen, Carlsbad, CA) and left on ice for 15 min. Subsequently, serum-opsonized Francisella spp. were added at an MOI ∼50:1. The microtiter plate was centrifuged at 400 × g for 2 min at 12°C to synchronize the infection. The relative amount of reactive oxygen species (ROS) generated by neutrophils over time was detected by measuring the luminescence by addition of luminol as a substrate in an ELISA reader (Spectramax M5, Molecular Devices, Sunnyvale, CA) after warming the cells to 37°C. In similar fashion, MDMs (105/well) were added in 0.2 ml culture media in 96-well plates. After 2 h of incubation at 37°C in 5% CO2, nonadherent cells were removed by washing the monolayers three or four times with prewarmed RPMI 1640 and replenished with fresh culture medium containing 10% autologous serum, and the ROS production in MDMs in response to serum-opsonized Francisella (MOI ∼50:1) was detected using the Diogenes enhanced luminescence system for superoxide detection (National Diagnostics, Atlanta, GA) with lucigenin as the substrate. Human serum-opsonized zymosan particles (MOI of 20:1) and PMA (200 nM) were used as positive controls for ROS production in neutrophils and MDMs. The inhibition of ROS production by Francisella spp. was tested by incubating phagocytes with Francisella for 10 min at 37°C prior to adding opsonized zymosan. ROS production was detected, as described above, in an ELISA reader.
ELISA to detect complement component deposition on Francisella strains
C5-depleted fresh human serum (CompTech, Tyler, TX) was used to evaluate complement component C3 deposition on Francisella strains, as described (39). Briefly, after preblocking microcentrifuge reaction tubes for 30 min in PBS with 0.1% HSA (ZLB Plasma, Boca Raton, FL), 3 × 108 bacteria/reaction were incubated in 10% or 50% serum for 30 min at 37°C. Reactions were stopped, and samples were washed twice in blocking buffer and once in PBS. A total of 3 × 107 bacteria in suspension were added to medium-binding polystyrene wells in triplicate (Costar, Cambridge, MA) and left to dry overnight. Wells were blocked overnight at 4°C with 3% OVA. After extensive washing with PBS, primary Ab (goat antisera to human C3 diluted 1:10,000 in 0.3% OVA [Quidel, San Diego, CA]) was added for 1 h at room temperature. HRP-conjugated rabbit anti-goat IgG (H+L) Ab (Bio-Rad, Hercules, CA) diluted to 1:2000 was used as the secondary Ab and was added for 1 h at room temperature. Substrate was added for 10 min at room temperature (Bio-Rad), and the reaction was stopped with 2% oxalic acid. Absorbance at 415 nm was measured on a 96-well plate reader (Molecular Devices). Values obtained from reactions with heat-inactivated control serum were subtracted in each case.
Confocal microscopy
Neutrophils (106/well) were plated onto human serum-coated glass coverslips in HBSS in a 24-well plate. Serum-opsonized Francisella spp. were added at an MOI ∼50:1, and the infection was synchronized at 12°C by centrifugation at 400 × g for 2 min. At different time intervals, coverslips were washed with HBSS to remove extracellular bacteria, and the cells were fixed with 3.5% paraformaldehyde for 30 min (40). After fixation, cells were washed three times with HBSS and permeabilized with chilled methanol for 15 s (41). Cells were washed again in HBSS and blocked in HBSS containing 20% normal human serum (Cambrex) and 5% BSA (blocking solution) for 2 h. Infected cells were treated with primary Ab consisting of mouse monoclonal anti-F. novicida (1:2000 dilution; Immunoprecise, Victoria, British Columbia, Canada) or mouse monoclonal anti-F. tularensis LPS (1:2000 dilution; Abcam, Cambridge, MA) and/or rabbit anti-p47phox Ab (1:1000 dilution; Molecular Probes) for 2 h in blocking solution. Coverslips were washed three times in blocking solution and secondary Ab [goat anti-mouse Alexa Fluor 488 or donkey anti-rabbit Alexa Fluor 546, 1:1000 dilution (Invitrogen)] was added for 1 h. Coverslips were washed and mounted with Prolong anti-fade reagent (Invitrogen) and viewed with a Zeiss 510 META confocal microscope. Resting neutrophils or cells activated with 200 nM PMA for 5 min were used as negative and positive controls, respectively. Using the same protocol, ∼105 human MDMs in monolayer culture on coverslips were incubated with serum-opsonized F. novicida at an MOI ∼50:1 to determine the colocalization of the bacterium with p47phox by confocal microscopy. Slides were examined using an ×63 oil-immersion objective on a Zeiss 510 META laser-scanning confocal microscope (Carl Zeiss Microimaging, Thornwood, NY). Quantification of colocalization of the F. novicida acid phosphatase mutants with p47phox at different time intervals was achieved by counting 1000 infected cells from triplicate coverslips in three independent experiments. Several attempts to perform colocalization experiments with Abs to gp91 and p40phox were unsuccessful because the rabbit anti-p40phox and rabbit anti-gp91 Abs did not recognize these NADPH oxidase components well.
Isolation and infection of bone marrow-derived macrophages from p47phox knockout mice
All of the mouse experiments were performed according to animal protocols approved by The Ohio State University. C57BL/6 WT and p47phox knockout (C57BL/6 background) mice were used in this study (generously provided by Dr. Chandan Sen, Ohio State University) (42). Three mice from each group were sacrificed, and the bone marrow-derived macrophages (BMDMs) were obtained from cultures of bone marrow stem cells, as previously described (43). In brief, the marrow was flushed from femurs with supplemented DMEM containing 10% FCS. The marrow plugs were disrupted into single-cell suspensions and cultured at a cell density of 1 × 106 nucleated cells/ml in 100-mm polystyrene tissue culture dishes containing 20 ml DMEM supplemented with 10% l cell-conditioned medium, 10% FCS. After 2 d of incubation at 37°C in 5% CO2, the nonadherent cells were decanted. The remaining adherent monolayers were supplemented with fresh media, and cells were harvested after 2 d using cold HBSS buffer. P47phox+/+ and p47phox−/− BMDMs were incubated with F. novicida strains at an MOI ∼50:1. At 30 min or 2 h postinfection, cells were washed twice and incubated with 50 μg/ml gentamicin for 30 min at 37°C and 5% CO2. The cells were subsequently washed twice and replenished with fresh media containing 10 μg/ml gentamicin. Cells were lysed in 0.05% SDS at different time intervals and plated on Chocolate II agar plates to enumerate the CFU. Assays were performed in triplicate for each test group.
Detection of phosphorylated p40phox and p47phox in cell lysates of neutrophils and MDMs
Neutrophils (106/well) or MDMs (106/well) were added to a 12-well plate in RPMI 1640. Serum-opsonized F. novicida strains were added at an MOI ∼50:1, and the infection was synchronized by centrifugation at 400 × g for 2 min at 12°C. At different time intervals, uninfected and infected cells were lysed in TN1 buffer (50 mM Tris [pH 8.0], 10 mM EDTA, 10 mM Na4P2O7, 10 mM NaF, 1% Triton-X 100, 125 mM NaCl, 10 mM Na3VO4, 10 μg/ml each aprotinin and leupeptin). The cell lysates were boiled in Laemmli sample buffer, and equal amounts of proteins in the different test groups were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and incubated with a primary Ab against phospho-p40phox (Cell Signaling Technology, Beverly, MA; 1:500 dilution in Tris-buffer saline with 5% milk) or phospho-p47phox, produced as described previously (1:1000 dilution) (44), overnight at 4°C. This was followed by a goat anti-rabbit HRP-conjugated secondary Ab (Bio-Rad; 1:1000 dilution for 2 h at room temperature) and development by ECL (Amersham/GE Healthcare Bio-Sciences, Piscataway, NJ). The ECL signal was quantified using a scanner and densitometry (Scion Image, Frederick, MD), as previously described (45).
In vitro phosphorylation and dephosphorylation of p47phox, p40phox, and p67phox
Recombinant p47phox, p67phox, and p40phox (5 μg each) were expressed in Escherichia coli using pGEX plasmids (gift from Bernard M. Babior, The Scripps Research Institute, La Jolla, CA) and then phosphorylated by active protein kinase C (PKC; Promega, Madison, WI) or AKT (Calbio-chem, San Diego, CA) in the presence of 1 μCi [32P]-[γ]-ATP for 30 min using published protocols (46). The reaction was terminated by adding the same volume of 2× Laemmli sample buffer, boiled, and resolved by SDS-PAGE using 10% polyacrylamide gels. The separated proteins were transferred to a nitrocellulose membrane. Proteins were stained by ponceau red (data not shown), and [32P]-labeled p47phox, p67phox, and p40phox were detected by a phosphoimager. The nitrocellulose areas containing [32P]-labeled p47phox, p67phox, and p40phox were cut and incubated for 30 min at 37°C with polyvinylpyrrolidone, washed, and then incubated with 10 μg purified AcpA, AcpC, or Hap (47–50). All three proteins were separated and visualized by SDS-PAGE and showed functional acid phosphatase activity (data not shown). Supernatants containing released [32P] were spotted onto a thin-layer cellulose plate (Merck, Whitehouse Station, NJ), and [32P] was detected by autoradiography. To further confirm that the signal was not due to the release of the protein from the nitrocellulose, the supernatant was subjected to SDS-PAGE using 15% polyacrylamide gels and analyzed by autoradiography. Experiments were performed in duplicate and repeated twice.
Results
Production of ROS in human neutrophils and macrophages by F. novicida acid phosphatase mutants
Previous work showed that F. tularensis LVS is unable to stimulate an oxidative burst in infected human neutrophils and macrophages (36, 51). In addition, acid phosphatases of organisms, including Francisella spp., have been implicated in respiratory burst inhibition (33, 52–55). To determine whether F. novicida acid phosphatase mutants are no longer able to suppress the respiratory burst in human phagocytes, we measured the generation of ROS in infected human neutrophils (30-min time course) and MDMs (60-min time course) using the luminescence probe, luminol or lucigenin, respectively. F. novicida induced minimal amounts of ROS in neutrophils and macrophages (Fig. 1A, 1B), demonstrating that F. novicida, like F. tularensis LVS (36), does not generate significant ROS production. Similar inhibitory effects were observed with Type A F. tularensis SchuS4 (Fig. 1C, 1D). Formalin-killed Francisella subspecies effectively induced a respiratory burst in MDMs and neutrophils, suggesting that ROS suppression was an active process of live bacteria (Fig. 1). Opsonized zymosan and PMA stimulated the production of ROS in neutrophils and MDMs, as expected (Fig. 1). However, the addition of opsonized zymosan after MDM or PMN infection by F. novicida or Schu S4 dramatically reduced ROS induction compared with opsonized zymosan alone (Fig. 1), demonstrating that Francisella subspecies are able to suppress ROS production from human phagocytes.
FIGURE 1.
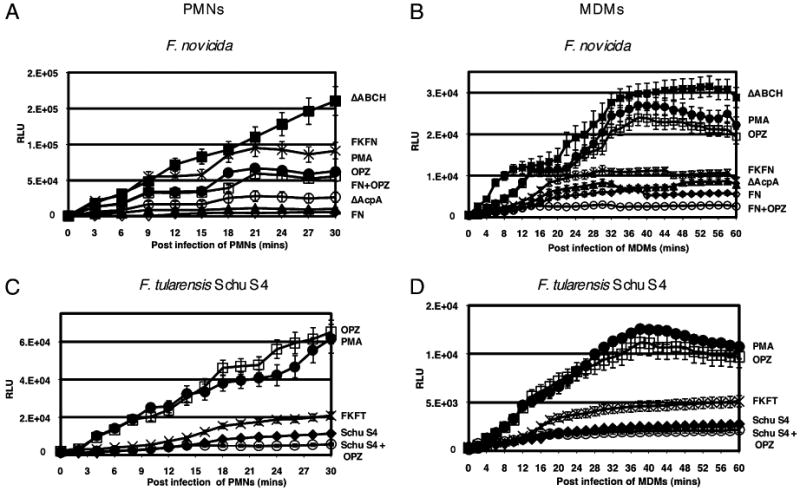
Detection of ROS production in human neutrophils and MDMs. The luminescence was measured over a 30-min (A, C, neutrophils) or 60-min (B, D, MDMs) time period in these human phagocytes, and the baseline ROS luminescence was determined by measuring the luminescence of control cells treated with medium only. A and B, F. novicida (FN; ◊), ΔacpA (▲), ΔABCH (■), formalin-killed F. novicida (FKFN; X), and F. novicida followed by serum-opsonized zymosan (FN+OPZ; ○), serum-opsonized zymosan (OPZ; □), or 200 nM PMA (●). C and D, F. tularensis subspecies tularensis SchuS4 (Schu S4; ◆), formalin-killed F. tularensis subspecies tularensis (FKFT; X), and F. tularensis subspecies tularensis followed by serum-opsonized zymosan (Schu S4 + OPZ; ○), serum-opsonized zymosan (OPZ; □), or 200 nM PMA (●). Data are the mean ± SD of triplicate samples from a representative experiment (n = 7).
Phagocytes infected with the ΔacpA mutant produced a small amount of ROS, which was particularly evident in MDMs. However, neutrophils and MDMs infected with the ΔABCH strain produced significantly higher levels of ROS, with an overall 28-fold increase in neutrophils at 30 min postinfection and a 5.5-fold increase in MDMs at 60 min postinfection versus infection with F. novicida. The magnitude of ROS production in MDMs was significantly less than that observed in neutrophils during F. novicida strain infections (compare vertical axis of Fig. 1A with Fig. 1B). The ΔABCH strain was complemented with a plasmid expressing acpA (19). Additional complementation was too difficult to pursue because of the multiple antibiotic resistances of the ΔABCH strain. The results demonstrate that expression of the plasmid-borne acpA in the ΔABCH mutant complemented and dramatically reduced the amount of ROS produced by this strain, but not to the level of the WT F. novicida strain (Supplemental Fig. 1). Thus, these data suggest that loss of acid phosphatases in Francisella spp. results in a failure to suppress the production of ROS.
The above experiments were repeated with the addition of diphenylene iodonium (DPI; flavoprotein inhibitor) 15 min prior to bacterial infection. Luminescence was completely abrogated in neutrophils and MDMs postinfection with all bacterial strains as well as with PMA and opsonized zymosan (data not shown).
To confirm that differences in cell association or uptake were not responsible for the observed disparity in ROS production between acid phosphatase mutants and WT F. novicida, strains were examined by microscopy upon infection of MDMs and neutrophils in the presence of various concentrations of autologous serum. No significant differences in cell association or uptake were observed (data not shown). Additionally, there were no differences in the deposition of the complement component C3, a central complement factor whose cleavage leads to the deposition of C3bi, a major opsonin, on the surface of the F. novicida WT strain or the acid phosphatase mutants (data not shown).
Decreased survival of F. novicida acid phosphatase mutants in human macrophages and neutrophils is associated with enhanced ROS production
The ΔABCH mutant of F. novicida was reported to have an impaired ability to replicate intracellularly in J774.1 murine and THP-1 macrophages (19). In this study, we measured CFU in neutrophils (15 and 30 min) and MDMs (30 and 60 min) infected with the ΔacpA, ΔABCH, and WT strains of F. novicida (Fig. 2). At 30 and 60 min postinfection of neutrophils and MDMs, respectively, there was a 19- and 46-fold decrease in survival of the ΔABCH versus the F. novicida WT strain (Fig. 2A, 2C). In the presence of DPI, the intracellular growth of WT F. novicida, ΔacpA, and ΔABCH strains was nearly identical up to 30 min postinfection in neutrophils and 60 min postinfection in MDMs (Fig. 2B, 2D). Although growth at these time points was nearly identical, a nearly 2-log increase in survival was noted in the presence versus the absence of DPI. This is likely due to functional bacterial killing by residual ROS production upon infection by the F. novicida WT strain (incomplete suppression of ROS production). These results suggest that the reduction in intracellular growth in phagocytes seen with the mutant strains was due to the generation of ROS.
FIGURE 2.
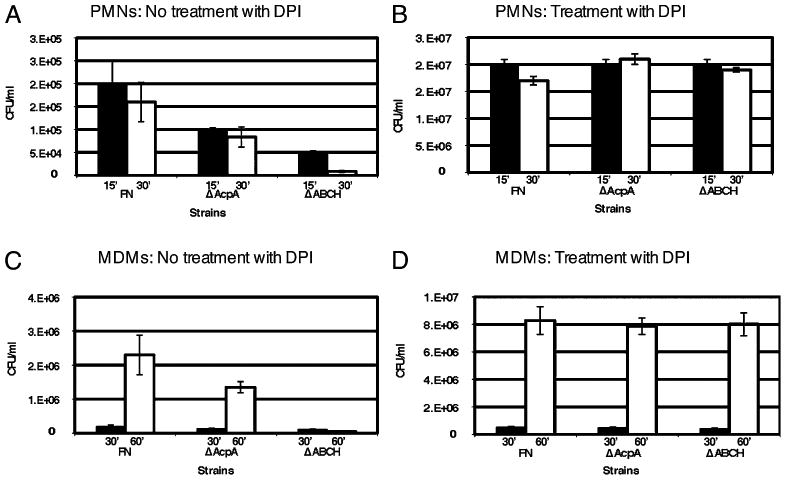
Intracellular survival assays performed in neutrophils or MDMs in the absence or presence of DPI (10 μM). Macrophages were infected with F. novicida or the mutant derivatives ΔacpA or ΔABCH in the absence (A, C) or presence (B, D) of DPI (10 μM). Black bars represent 15 min in neutrophils (A, B) and 30 min in MDMs (C, D), and white bars represent 30 min in neutrophils (A, B) and 60 min postinfection in MDMs (C, D). Data are the mean ± SD of triplicate samples from one representative experiment (n = 5).
To confirm whether ROS production was responsible for the decreased survival of acid phosphatase-deficient strains, we examined the survival of Francisella acid phosphatase mutants in BMDMs isolated from p47phox knockout mice. These mice were shown to be more susceptible than WT mice to infection by F. tularensis LVS (56), demonstrating a role of ROS in controlling bacterial dissemination and growth. WT and p47phox−/− BMDMs were incubated with F. novicida WT, ΔacpA, and ΔABCH strains, and the CFU were compared at different time points. At 12 h postinfection, the ΔABCH strain showed a slightly less than 3-log decrease in survival compared with the WT strain (Fig. 3A). This decrease was even greater at 24 h. The ΔacpA strain demonstrated intermediate survival, with a >1.5-log decrease in survival at 12 h postinfection and ∼1-log decrease in survival at 24 h. In contrast, the F. novicida WT, ΔacpA, and ΔABCH strains grew equally well in infected BMDMs isolated from p47phox−/− mice (Fig. 3B). Thus, there is a strong correlation of intracellular survival with the induction of ROS by the NADPH oxidase. These data, combined with the results in the presence of DPI, provide strong evidence that the production of ROS in phagocytes by the NADPH oxidase is responsible for the reduced intracellular survival seen with the F. novicida acid phosphatase mutants.
FIGURE 3.
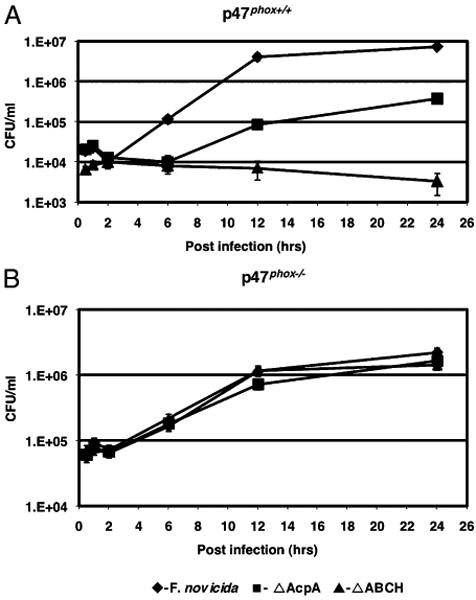
Intramacrophage survival of F. novicida acid phosphatase mutants in BMDMs from p47phox+/+ and p47phox−/− knockout mice. BMDMs were isolated from p47phox+/+ WT C57BL/J mice (A) and p47phox−/− knockout mice C57BL/J (B) infected with F. novicida or the mutant derivatives acpA or ΔABCH. Data are the mean ± SD of triplicate samples from one representative experiment (n = 3).
Colocalization of Francisella with the NADPH oxidase complex component p47phox in human neutrophils and macrophages
Assembly of a functional NADPH oxidase requires the translocation of cytosolic NADPH oxidase components p67phox, p47phox, and p40phox to the membrane component cytochrome b558 (57). However, intracellular F. tularensis LVS did not induce ROS production or colocalize with NADPH complex subunits (36). We examined colocalization of p47phox with bacteria in human neutrophils and MDMs to determine whether the F. novicida, ΔacpA, ΔABCH, and Type A F. tularensis subspecies tularensis associate with NADPH oxidase complex components.
Neutrophils adhered to serum-coated glass coverslips were incubated with F. novicida, ΔacpA, or ΔABCH strains. The colocalization of F. novicida with p47phox was detected using confocal microscopy. Representative confocal micrographs of neutrophils infected with the F. novicida and ΔABCH strains at 30 min postinfection are shown in Fig. 4A. Neutrophils infected with the ΔABCH strain showed a maximum colocalization at 30 min postinfection; ∼90% of the bacteria were colocalized with p47phox (Fig. 4B). The ΔacpA mutant strain colocalized less extensively with p47phox (Fig. 4B). The F. novicida WT strain colocalized with p47phox poorly, reaching a maximum ∼12% at 30 min postinfection (Fig. 4B). Similarly, the virulent Type A and Type B strains colocalized poorly with p47phox in neutrophils (7% and 8.5%, respectively) and MDMs (5.5% and 6.5%, respectively) at 30 min postinfection (data not shown).
FIGURE 4.
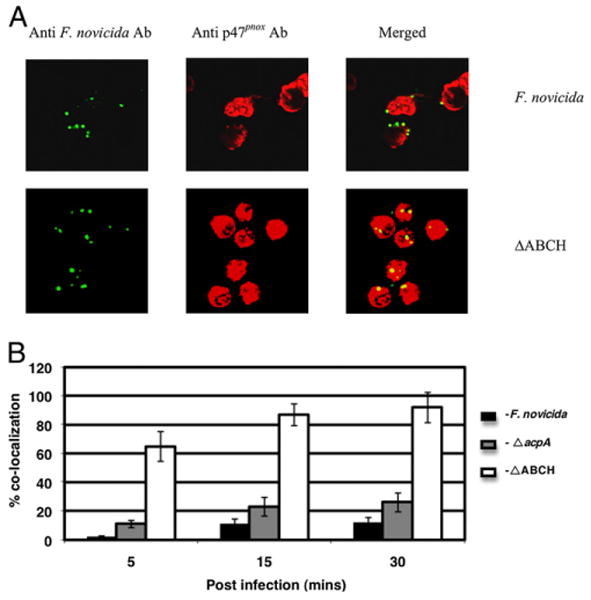
Colocalization of the F. novicida WT strain and mutant strains with p47phox in neutrophils. A, Colocalization of the F. novicida WT and ΔABCH or ΔacpA mutant strains with p47phox was determined at 5, 15, and 30 min postinfection in neutrophils. Francisella was detected following staining with goat anti-mouse Alexa Fluor 488 (green color), and p47phox was detected following staining with donkey anti-rabbit Alexa Fluor 546 (red color). Representative confocal microscopy images of F. novicida and ΔABCH colocalized with p47phox within neutrophils are shown at 30 min postinfection. The images are representative of 1000 infected cells examined from triplicate coverslips in three independent experiments. Original magnification ×63. B, Colocalization of the F. novicida WT, ΔacpA, and ΔABCH mutant strains with p47phox was quantified at 5, 15, and 30 min postinfection. Analyses were based on examination of 1000 infected cells examined from triplicate coverslips in three independent experiments. The results shown are cumulative data of three experiments (mean ± SD of nine samples; triplicate samples were included in each test group in each experiment).
Similar to what was observed in neutrophils, the ΔABCH strain demonstrated marked colocalization with p47phox in MDMs (representative confocal micrographs are shown at 60 min postinfection in Fig. 5A), with a maximum of nearly 48% colocalization at 60 min postinfection (Fig. 5B). The ΔacpA mutant showed an intermediate level of colocalization (Fig. 5B), whereas the WT F. novicida strain showed the least association with p47phox over the time period studied, with a maximum of only ∼8% colocalization observed at 60 min postinfection.
FIGURE 5.
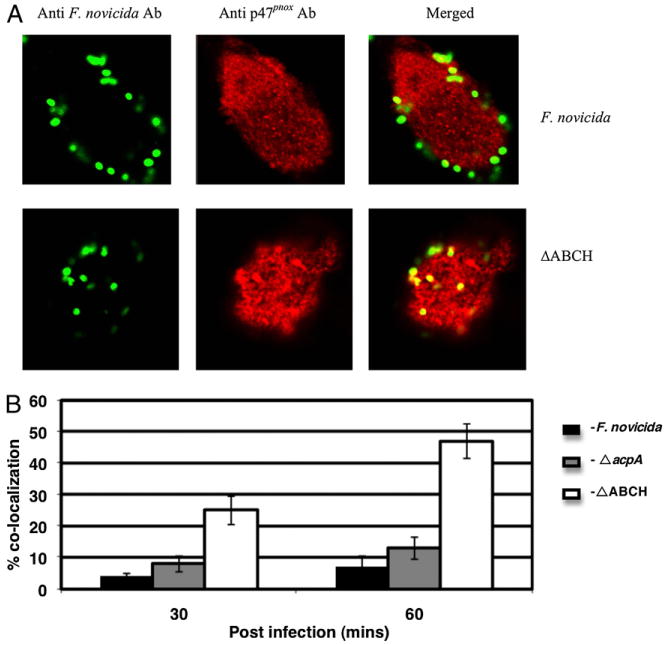
Colocalization of the F. novicida WT strain and mutant strains with p47phox in macrophages. A, Colocalization of the F. novicida WT and ΔABCH or ΔacpA mutant strains with p47phox was determined at 30 and 60 min post-infection in MDMs. Francisella was detected following staining with goat anti-mouse Alexa Fluor 488 (green color), and p47phox was detected following staining with donkey anti-rabbit Alexa Fluor 546 (red color). Representative confocal microscopy images of F. novicida and ΔABCH colocalized with p47phox within MDMs are shown 60 min postinfection. The images are representative of 1000 infected cells examined from triplicate coverslips in three independent experiments. Original magnification ×63. B, Colocalization of the F. novicida WT, ΔacpA, and ΔABCH mutant strains with p47phox was quantified at 30 and 60 min postinfection. Analyses were based on examination of 1000 infected cells examined from triplicate coverslips in three independent experiments. The results shown are cumulative data of three experiments (mean ± SD of nine samples; triplicate samples were included in each test group in each experiment).
NADPH oxidase components show increased phosphorylation following infection with the ΔABCH mutant strain relative to F. novicida WT in neutrophils and macrophages
To determine whether the limited generation of NADPH oxidase-mediated ROS in response to WT F. novicida involved the phosphorylation state of NADPH complex subunits by the acid phosphatases, we first examined the phosphorylation of p47phox and p40phox during the course of infection. Neutrophils were infected with F. novicida strains; at different time intervals, cells were lysed, and the phosphorylation of p47phox or p40phox was detected by Western blotting using phospho-p40phox and phospho-p47phox Abs (Fig. 6). In neutrophils, infection by the ΔABCH strain resulted in a marked increase in phosphorylation of p47phox within 15 min of infection compared with the WT strain (Fig. 6). Similarly, phosphorylation of p40phox in neutrophils infected with the ΔABCH strain showed a dramatic increase within 15 min of infection compared with the WT strain. In addition, MDMs infected with the ΔABCH strain showed a steady increase in phosphorylation of p47phox and p40phox over the time course of the experiment, which was not observed with the WT strain (data not shown). The relative increase in p47phox and p40phox phosphorylation during infection of neutrophils and MDMs with the ΔABCH strain versus infection with the WT strain suggests that these NADPH complex components do not become phosphorylated by upstream kinases or are dephosphorylated in human phagocytes when infected with the WT, but not the ΔABCH, strain.
FIGURE 6.
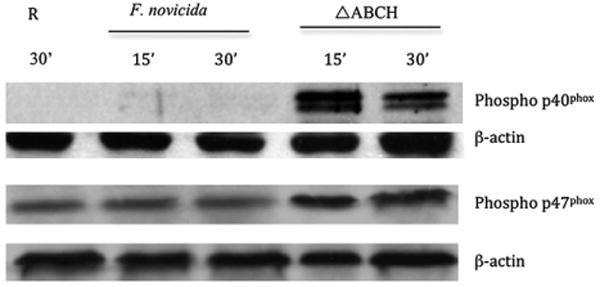
The effect of F. novicida WT and acp mutants on phosphorylation of p47phox and p40phox in neutrophils. Neutrophils were incubated with F. novicida WT or ΔABCH strains for the times shown. Lysates were loaded by protein equivalents, separated by SDS-PAGE, and analyzed by Western blotting with Abs specific for phosphorylated p47phox (rabbit anti-Pp47phox) and phosphorylated p40phox (rabbit anti-Pp40phox). The same membrane was reprobed with β-actin Ab to verify equal protein loading. Resting (R) represents the uninfected PMN cell lysates collected at 30 min postinfection. A representative Western blot image is shown (n = 7).
Direct dephosphorylation of p47phox and p40phox by AcpA
Activation of the NADPH oxidase complex depends upon the phosphorylation and subsequent translocation of the cytosolic components p40phox, p47phox, and p67phox to the phagosomal membrane (58). Several studies reported that phox components are substrates for phosphorylation by PKCs, Akt, and p38 MAPK (46, 59–63). In this study, we used active PKC and Akt to phosphorylate purified p47phox, p40phox, and p67phox proteins in the presence of [32P]-[γ]-ATP (60). The phosphorylated p47phox, p40phox, and p67phox proteins were separated by SDS-PAGE and transferred to a nitrocellulose membrane. The [32P]-labeled proteins were detected by a phosphoimager and autoradiography (Fig. 7A); all three proteins were strongly phosphorylated by the kinases. Membranes containing [32P]-labeled p47phox, p67phox, and p40phox proteins were incubated with purified Francisella AcpA, AcpC, or Hap proteins (purified AcpB was difficult to obtain and was not available). The supernatants containing the released [32Pi] were spotted on thin layer cellulose plates and detected by autoradiography (Fig. 7B). Purified AcpA strongly dephosphorylated p47phox and p40phox but only weakly dephosphorylated p67phox. In the presence of AcpC and Hap, p47phox was dephosphorylated to a small extent, whereas none of the other phosphorylated phox proteins tested were dephosphorylated by these two acid phosphatases (Fig. 7B). To confirm that [32Pi] release was not due to the complete release of the nitrocellulose membrane-bound proteins, the supernatant was subjected to SDS-PAGE and analyzed by autoradiography. These data demonstrated that [32Pi] was released from the proteins (Fig. 7C, 7D). Thus, the results provide evidence that p47phox and p40phox can be directly dephosphorylated by the Francisella acid phosphatases, particularly the AcpA protein.
FIGURE 7.
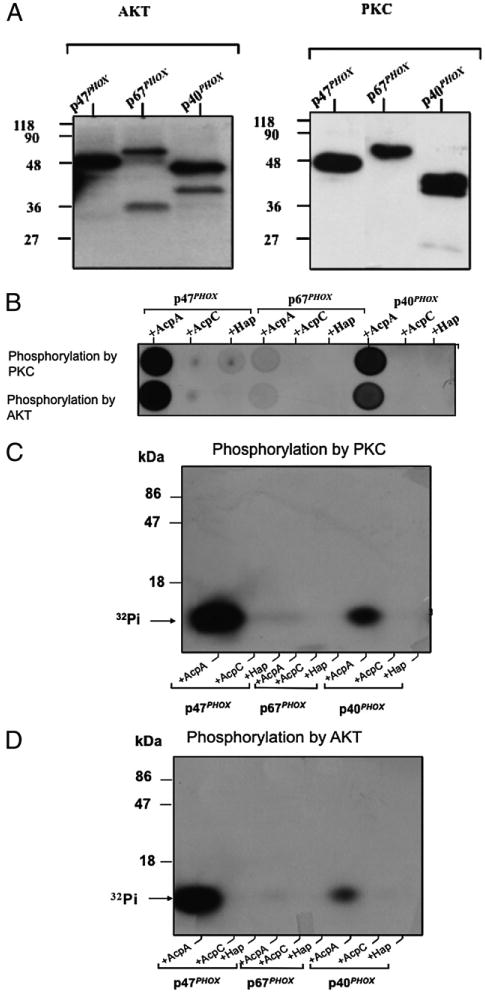
The effect of F. novicida Acps on phosphorylation of p40phox and p47phox in vitro. A, Phosphorylation of p47phox, p67phox, and p40phox by AKT and PKC. Recombinant p47phox, p67phox, and p40phox (5 μg each) were phosphorylated by active PKC or Akt in the presence of 1 μCi of [32P]-[γ]g-ATP for 30 min. The reaction was terminated by adding the same volume of 2× Laemmli sample buffer, and the proteins were separated by SDS-PAGE and transferred to a nitrocellulose membrane. The proteins were stained by ponceau red (data not shown), and [32P]-labeled p47phox, p67phox, and p40phox were detected by phosphoimaging and autoradiography. B, Dot blot of released [32Pi]. The nitrocellulose area containing [32P]-labeled p47phox, p67phox, and p40phox was cut and incubated for 30 min at 37°C with polyvinylpyrrolidone, washed, and then incubated with the different purified acid phosphatases. Supernatants containing released [32P] were spotted onto a thin-layer cellulose plate and detected by autoradiography. C and D, Confirmation of release of [32Pi]. To confirm that the above signal was not due to the release of proteins from the nitrocellulose, the supernatant was subjected to 15% SDS-PAGE and analyzed by autoradiography. C, Phosphorylation by PKC. D, Phosphorylation by Akt.
Discussion
F. tularensis LVS is unable to stimulate the production of an oxidative burst in infected human neutrophils and macrophages and survives within these host cells (36, 51). We previously reported that F. novicida acid phosphatases play a major role in survival within phagocytic cells and in virulence, because the loss of four acid phosphatases (AcpA, AcpB, AcpC, and Hap) in F. novicida (ΔABCH) rendered it unable to survive within and escape from human and murine macrophage phagosomes (19). The ΔABCH strain was also attenuated in the mouse model of tularemia. In addition, purified AcpA was reported to block the generation of an oxidative burst in neutrophils (33). These data led us to investigate the connection between acid phosphatases and ROS production in human neutrophils and macrophages as a potential mechanism for the observed decrease in virulence and intraphagocytic cell survival of the ΔABCH strain.
In this study, we extend the work with F. tularensis LVS (36,51) to show that F. tularensis subspecies tularensis SchuS4, F. tularensis subspecies holarctica, and F. novicida also suppress the production of ROS in human neutrophils, as well as within human macrophages. Interestingly, although the live organisms induced similar low levels of ROS, formalin killed F. novicida induced considerably more ROS than did formalin-killed F. tularensis SchuS4, suggesting that there may be something inherently different about the surfaces of these bacteria that affect the respiratory burst. In addition, we provide evidence that infection with the ΔABCH strain results in a significant induction of ROS, suggesting that the acid phosphatases directly or indirectly interfere with the oxidative burst and that increased ROS production is responsible for the killing of the ΔABCH strain within these phagocytes. Indeed, blocking the assembly and/or function of the NADPH oxidase complex with DPI or mutation of a critical cytosolic component (murine p47phox−/− macrophages) allowed for a WT level of survival of the ΔABCH strain in phagocytes. The loss of only AcpA in F. novicida produced an intermediate amount of ROS production in neutrophils and MDMs and resulted in intermediate survival, suggesting that it was a major, but not the only, acid phosphatase contributing to the observed ΔABCH strain phenotypes.
Assembly of the NADPH oxidase requires that cytosolic phosphorylated p47phox and p40/p67phox heterodimers associate to form p47/p67/p40phox heterotrimers prior to their membrane translocation and subsequent association with flavocytochrome b558 (64–67). Similar to what was observed with the F. tularensis LVS strain, the F. novicida WT strain did not significantly colocalize with the p47phox component of the NADPH oxidase complex in human neutrophils and macrophages. However, the ΔABCH strain, and to a lesser extent, the acpA mutant, significantly colocalized with the p47phox in human neutrophils and human MDMs, correlating with their observed levels of ROS induction.
Several other bacteria and parasites were shown to mediate exclusion or disruption of NADPH oxidase assembly or ROS production during phagocytosis (52–55, 68–72). For example, Helicobacter pylori disrupts phagosomal NADPH oxidase targeting, because superoxide anions are released extracellularly instead of within the phagosomes, and these phagosomes do not contain p47phox or p67phox (73–75). Leishmania exclude NADPH oxidase cytosolic components p47phox and p67phox from phagosomes in a lipophosphoglycan-dependent fashion (68, 69). In contrast, Salmonella excludes flavocytochrome b558 from the phagosomal membrane, thus preventing NADPH oxidase assembly (54, 72). Finally, Coxiella burnetii produces an acid phosphatase that, similar to Francisella, inhibits ROS production from activated human neutrophils by an unknown mechanism (55).
Recently published work suggested that AcpA, AcpB, and AcpC do not play a role in the pathogenesis of the F. tularensis SchuS4 strain (using a triple mutant) (76). This is contrary to our previous finding with F. novicida, which demonstrated increased virulence defects upon the accumulation of acp deletions, culminating with a strong virulence defect for the ΔABCH strain (quadruple mutant) (19). Our ongoing work with the acid phosphatases (Acps) in F. tularensis SchuS4 (data not shown), coupled with the data presented in this study, suggests that the acid phosphatases play a role in F. tularensis SchuS4 pathogenesis but perhaps not to the same extent as observed in F. novicida.
The hypothesis we developed based on the data presented in this work states that the acid phosphatases collectively directly participate in the dephosphorylation of phox components and/or of their kinases, which inhibits NADPH oxidase assembly and ROS production. In support of an effect on the upstream kinases, human MDMs infected with the ΔABCH strain have an increased level of p38 MAPK phosphorylation, but not Akt and Erk1/2, compared with the MDMs infected with F. novicida WT strain (data not shown). Further studies are required to fully understand the phosphorylation patterns of the upstream kinases, including p38 MAPK, during ΔABCH strain infection. To address the phox components directly, phosphorylation of p47phox was weak and phospho-p40phox was undetectable at early time points post-infection of human neutrophils with the F. novicida WT strain. However, ΔABCH strain infection resulted in a marked increase in the phosphorylation of these phox components. To determine whether the Acps had the capacity to directly dephosphorylate the phox components, phosphorylated p40phox, p47phox, and p67phox proteins were treated individually in vitro with three purified Francisella Acps. AcpA strongly dephosphorylated the p40phox and p47phox proteins, but dephosphorylated p67phox weakly, demonstrating specificity in this reaction. As opposed to the actions of AcpA, AcpC and Hap weakly dephosphorylated p47phox but not p40phox or p67phox. Although AcpA demonstrated the most activity toward the phox components in the in vivo assay, the virulence data (19) and the data presented in this article (including the colocalization data with p47phox) suggest that a combined effort of the phosphatases is required for a maximal effect toward ROS suppression. Thus, there are likely targets of these phosphatases beyond the phox components, such as upstream kinases mentioned above, that are necessary for proper NADPH oxidase activation.
The rapid kinetics of NADPH oxidase inactivation by Francisella spp., coupled with the location of the targeted phox components, provide a conceptual challenge to these findings. Thus, how might AcpA or the other acid phosphatases be able to mediate these effects? Several pieces of data support a role for the Francisella acid phosphatases in this process. We previously demonstrated that two of the acid phosphatases, AcpA and Hap, were induced 219- and 10-fold, respectively, at 2 h postinfection with THP-1 macrophages (19). This induction decreased over time, suggesting that the peak activation may be prior to the 2 h time point. In addition, AcpA is outer membrane-associated in logarithmically growing cells (19), and it has been described as a Francisella-secreted protein, although this has not been observed in all studies that have screened for secreted factors. An acid phosphatase of Legionella pneumophila, a pathogen that resembles F. tularensis in a number of ways, is also secreted (77). None of the other Francisella Acps were reported to be secreted in vitro, but nothing is known about the potential secretion of these Francisella Acps in vivo. Thus, we hypothesize that Francisella affects the phagocyte (e.g., kinase dephosphorylation) very soon after contact, which may be Acp dependent and independent. Early secretion/release of acid phosphatases within the phagosome, or after phagosomal escape, which was described to occur rapidly after phagocytosis (17, 78), would result in a further reduction of the phosphorylation of phox components normally necessary for proper NADPH oxidase assembly and function. A less likely possibility is that the ΔABCH mutant uses an unknown receptor for phagocyte entry that is associated with a more robust oxidative burst than normally occurs following entry via CR3 and the mannose receptor, known receptors for Francisella WT strains (12). Additional molecular and biochemical characterization, including localization, of the Francisella acid phosphatases within host phagocytes will further define the mechanisms by which this bacterial pathogen evades an early innate immune response, allowing for bacterial colonization and disease progression. Targeted inhibition of these phosphatases could be a therapeutic strategy against Francisella infection.
Supplementary Material
Acknowledgments
We thank Dr. Chandan Sen for providing p47phox knockout mice for this study. The authors also thank Dr. Francis Nano for guidance in the initial work on Francisella acid phosphatases.
This work was supported by the National Institutes of Health, National Institute of Allergy and Infectious Diseases Regional Center of Excellence for Biodefense and Emerging Infectious Diseases Research Program. We acknowledge membership within and support from the Region V Great Lakes Regional Center of Excellence (National Institutes of Health Award 2-U54-AI-057153).
Abbreviations used in this paper
- ΔABCH
deletion of acid phosphatases AcpA, AcpB, AcpC, and Hap
- Acp
acid phosphatase
- BMDM
bone marrow-derived macrophage
- DPI
diphenylene iodonium
- FN
Francisella novicida
- FKFN
formalin-killed Francisella novicida
- FKFT
formalin-killed Francisella tularensis
- FPI
Francisella pathogenicity island
- LVS
live vaccine strain
- MDM
monocyte-derived macrophage
- MOI
multiplicity of infection
- OPZ
serum-opsonized zymosan
- PKC
protein kinase C
- PMN
polymorphonuclear leukocyte
- ROS
reactive oxygen species
- SchuS4
F. tularensis subspecies tularensis SchuS4
- WT
wild-type
Footnotes
The online version of this article contains supplemental material.
Disclosures: The authors have no financial conflicts of interest.
References
- 1.Oyston PC. Francisella tularensis: unravelling the secrets of an intracellular pathogen. J Med Microbiol. 2008;57:921–930. doi: 10.1099/jmm.0.2008/000653-0. [DOI] [PubMed] [Google Scholar]
- 2.Anthony LD, Burke RD, Nano FE. Growth of Francisella spp. in rodent macrophages. Infect Immun. 1991;59:3291–3296. doi: 10.1128/iai.59.9.3291-3296.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ben Nasr A, Haithcoat J, Masterson JE, Gunn JS, Eaves-Pyles T, Klimpel GR. Critical role for serum opsonins and complement receptors CR3 (CD11b/CD18) and CR4 (CD11c/CD18) in phagocytosis of Francisella tularensis by human dendritic cells (DC): uptake of Francisella leads to activation of immature DC and intracellular survival of the bacteria. J Leukoc Biol. 2006;80:774–786. doi: 10.1189/jlb.1205755. [DOI] [PubMed] [Google Scholar]
- 4.Lindemann SR, McLendon MK, Apicella MA, Jones BD. An in vitro model system used to study adherence and invasion of Francisella tularensis live vaccine strain in nonphagocytic cells. Infect Immun. 2007;75:3178–3182. doi: 10.1128/IAI.01811-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shepard CC, Ribi E, Larson C. Electron microscopically revealed structural elements of Bacterium tularense and their in vitro and in vivo role in immunologic reactions. J Immunol. 1955;75:7–14. [PubMed] [Google Scholar]
- 6.Shepard CC. Nonacid-fast bacteria and HeLa cells: their uptake and subsequent intracellular growth. J Bacteriol. 1959;77:701–714. doi: 10.1128/jb.77.6.701-714.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Councilman WTS, Strong RP. Plague-like infections in rodents. Trans Assoc Am Physicians. 1921;36:135–143. [Google Scholar]
- 8.Buddingh GJW, Womack FC. Observations on the infection of chick embryos with Bacterium tularense, Brucella, and Pasteurella pestis. J Exp Med. 1941;74:213–222. doi: 10.1084/jem.74.3.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Francis E. Microscopic changes of tularemia in the tick Dermacentor andersoni and the bedbug Cimex lectularius. Public Health Rep. 1927;42:2763–2772. [Google Scholar]
- 10.Fortier AH, Green SJ, Polsinelli T, Jones TR, Crawford RM, Leiby DA, Elkins KL, Meltzer MS, Nacy CA. Life and death of an intracellular pathogen: Francisella tularensis and the macrophage. Immunol Ser. 1994;60:349–361. [PubMed] [Google Scholar]
- 11.Clemens DL, Lee BY, Horwitz MA. Francisella tularensis enters macrophages via a novel process involving pseudopod loops. Infect Immun. 2005;73:5892–5902. doi: 10.1128/IAI.73.9.5892-5902.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balagopal A, MacFarlane AS, Mohapatra N, Soni S, Gunn JS, Schlesinger LS. Characterization of the receptor-ligand pathways important for entry and survival of Francisella tularensis in human macrophages. Infect Immun. 2006;74:5114–5125. doi: 10.1128/IAI.00795-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schulert GS, Allen LA. Differential infection of mononuclear phagocytes by Francisella tularensis: role of the macrophage mannose receptor. J Leukoc Biol. 2006;80:563–571. doi: 10.1189/jlb.0306219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Santic M, Asare R, Skrobonja I, Jones S, Abu Kwaik Y. Acquisition of the vacuolar ATPase proton pump and phagosome acidification are essential for escape of Francisella tularensis into the macrophage cytosol. Infect Immun. 2008;76:2671–2677. doi: 10.1128/IAI.00185-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chong A, Wehrly TD, Nair V, Fischer ER, Barker JR, Klose KE, Celli J. The early phagosomal stage of Francisella tularensis determines optimal phagosomal escape and Francisella pathogenicity island protein expression. Infect Immun. 2008;76:5488–5499. doi: 10.1128/IAI.00682-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clemens DL, Lee BY, Horwitz MA. Francisella tularensis phagosomal escape does not require acidification of the phagosome. Infect Immun. 2009;77:1757–1773. doi: 10.1128/IAI.01485-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Checroun C, Wehrly TD, Fischer ER, Hayes SF, Celli J. Autophagy-mediated reentry of Francisella tularensis into the endocytic compartment after cytoplasmic replication. Proc Natl Acad Sci USA. 2006;103:14578–14583. doi: 10.1073/pnas.0601838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Santic M, Molmeret M, Barker JR, Klose KE, Dekanic A, Doric M, Abu Kwaik Y. A Francisella tularensis pathogenicity island protein essential for bacterial proliferation within the host cell cytosol. Cell Microbiol. 2007;9:2391–2403. doi: 10.1111/j.1462-5822.2007.00968.x. [DOI] [PubMed] [Google Scholar]
- 19.Mohapatra NP, Soni S, Reilly TJ, Liu J, Klose KE, Gunn JS. Combined deletion of four Francisella novicida acid phosphatases attenuates virulence and macrophage vacuolar escape. Infect Immun. 2008;76:3690–3699. doi: 10.1128/IAI.00262-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lai XH, Golovliov I, Sjöstedt A. Francisella tularensis induces cytopathogenicity and apoptosis in murine macrophages via a mechanism that requires intracellular bacterial multiplication. Infect Immun. 2001;69:4691–4694. doi: 10.1128/IAI.69.7.4691-4694.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mariathasan S, Weiss DS, Dixit VM, Monack DM. Innate immunity against Francisella tularensis is dependent on the ASC/caspase-1 axis. J Exp Med. 2005;202:1043–1049. doi: 10.1084/jem.20050977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rajaram MV, Butchar JP, Parsa KV, Cremer TJ, Amer A, Schlesinger LS, Tridandapani S. Akt and SHIP modulate Francisella escape from the phagosome and induction of the Fas-mediated death pathway. PLoS One. 2009;4:e7919. doi: 10.1371/journal.pone.0007919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mougous JD, Cuff ME, Raunser S, Shen A, Zhou M, Gifford CA, Goodman AL, Joachimiak G, Ordoñez CL, Lory S, et al. A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science. 2006;312:1526–1530. doi: 10.1126/science.1128393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pukatzki S, Ma AT, Sturtevant D, Krastins B, Sarracino D, Nelson WC, Heidelberg JF, Mekalanos JJ. Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc Natl Acad Sci USA. 2006;103:1528–1533. doi: 10.1073/pnas.0510322103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meibom KL, Forslund AL, Kuoppa K, Alkhuder K, Dubail I, Dupuis M, Forsberg A, Charbit A. Hfq, a novel pleiotropic regulator of virulence-associated genes in Francisella tularensis. Infect Immun. 2009;77:1866–1880. doi: 10.1128/IAI.01496-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baron GS, Nano FE. MglA and MglB are required for the intramacrophage growth of Francisella novicida. Mol Microbiol. 1998;29:247–259. doi: 10.1046/j.1365-2958.1998.00926.x. [DOI] [PubMed] [Google Scholar]
- 27.Brotcke A, Monack DM. Identification of fevR, a novel regulator of virulence gene expression in Francisella novicida. Infect Immun. 2008;76:3473–3480. doi: 10.1128/IAI.00430-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brotcke A, Weiss DS, Kim CC, Chain P, Malfatti S, Garcia E, Monack DM. Identification of MglA-regulated genes reveals novel virulence factors in Francisella tularensis. Infect Immun. 2006;74:6642–6655. doi: 10.1128/IAI.01250-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Charity JC, Costante-Hamm MM, Balon EL, Boyd DH, Rubin EJ, Dove SL. Twin RNA polymerase-associated proteins control virulence gene expression in Francisella tularensis. PLoS Pathog. 2007;3:e84. doi: 10.1371/journal.ppat.0030084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mohapatra NP, Soni S, Bell BL, Warren R, Ernst RK, Muszynski A, Carlson RW, Gunn JS. Identification of an orphan response regulator required for the virulence of Francisella spp. and transcription of pathogenicity island genes. Infect Immun. 2007;75:3305–3314. doi: 10.1128/IAI.00351-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buchan BW, McCaffrey RL, Lindemann SR, Allen LA, Jones BD. Identification of migR, a regulatory element of the Francisella tularensis live vaccine strain iglABCD virulence operon required for normal replication and trafficking in macrophages. Infect Immun. 2009;77:2517–2529. doi: 10.1128/IAI.00229-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mohapatra NP, Balagopal A, Soni S, Schlesinger LS, Gunn JS. AcpA is a Francisella acid phosphatase that affects intramacrophage survival and virulence. Infect Immun. 2007;75:390–396. doi: 10.1128/IAI.01226-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reilly TJ, Baron GS, Nano FE, Kuhlenschmidt MS. Characterization and sequencing of a respiratory burst-inhibiting acid phosphatase from Francisella tularensis. J Biol Chem. 1996;271:10973–10983. doi: 10.1074/jbc.271.18.10973. [DOI] [PubMed] [Google Scholar]
- 34.Appelberg R. Neutrophils and intracellular pathogens: beyond phagocytosis and killing. Trends Microbiol. 2007;15:87–92. doi: 10.1016/j.tim.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 35.Nauseef WM. How human neutrophils kill and degrade microbes: an integrated view. Immunol Rev. 2007;219:88–102. doi: 10.1111/j.1600-065X.2007.00550.x. [DOI] [PubMed] [Google Scholar]
- 36.McCaffrey RL, Allen LA. Francisella tularensis LVS evades killing by human neutrophils via inhibition of the respiratory burst and phagosome escape. J Leukoc Biol. 2006;80:1224–1230. doi: 10.1189/jlb.0406287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Olakanmi O, Britigan BE, Schlesinger LS. Gallium disrupts iron metabolism of mycobacteria residing within human macrophages. Infect Immun. 2000;68:5619–5627. doi: 10.1128/iai.68.10.5619-5627.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fortier AH, Leiby DA, Narayanan RB, Asafoadjei E, Crawford RM, Nacy CA, Meltzer MS. Growth of Francisella tularensis LVS i n macrophages: the acidic intracellular compartment provides essential iron required for growth. Infect Immun. 1995;63:1478–1483. doi: 10.1128/iai.63.4.1478-1483.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clay CD, Soni S, Gunn JS, Schlesinger LS. Evasion of complement-mediated lysis and complement C3 deposition are regulated by Francisella tularensis lipopolysaccharide O antigen. J Immunol. 2008;181:5568–5578. doi: 10.4049/jimmunol.181.8.5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pierini LM, Eddy RJ, Fuortes M, Seveau S, Casulo C, Maxfield FR. Membrane lipid organization is critical for human neutrophil polarization. J Biol Chem. 2003;278:10831–10841. doi: 10.1074/jbc.M212386200. [DOI] [PubMed] [Google Scholar]
- 41.Crowther JE, Schlesinger LS. Endocytic pathway for surfactant protein A in human macrophages: binding, clathrin-mediated uptake, and trafficking through the endolysosomal pathway. Am J Physiol Lung Cell Mol Physiol. 2006;290:L334–L342. doi: 10.1152/ajplung.00267.2005. [DOI] [PubMed] [Google Scholar]
- 42.Jackson SH, Gallin JI, Holland SM. The p47phox mouse knockout model of chronic granulomatous disease. J Exp Med. 1995;182:751–758. doi: 10.1084/jem.182.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parsa KV, Ganesan LP, Rajaram MV, Gavrilin MA, Balagopal A, Mohapatra NP, Wewers MD, Schlesinger LS, Gunn JS, Tridandapani S. Macrophage pro-inflammatory response to Francisella novicida infection is regulated by SHIP. PLoS Pathog. 2006;2:e71. doi: 10.1371/journal.ppat.0020071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dang PM, Stensballe A, Boussetta T, Raad H, Dewas C, Kroviarski Y, Hayem G, Jensen ON, Gougerot-Pocidalo MA, El-Benna J. A specific p47phox-serine phosphorylated by convergent MAPKs mediates neutrophil NADPH oxidase priming at inflammatory sites. J Clin Invest. 2006;116:2033–2043. doi: 10.1172/JCI27544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rajaram MV, Ganesan LP, Parsa KV, Butchar JP, Gunn JS, Tridandapani S. Akt/Protein kinase B modulates macrophage inflammatory response to Francisella infection and confers a survival advantage in mice. J Immunol. 2006;177:6317–6324. doi: 10.4049/jimmunol.177.9.6317. [DOI] [PubMed] [Google Scholar]
- 46.Dang PM, Fontayne A, Hakim J, El Benna J, Périanin A. Protein kinase C zeta phosphorylates a subset of selective sites of the NADPH oxidase component p47phox and participates in formyl peptide-mediated neutrophil respiratory burst. J Immunol. 2001;166:1206–1213. doi: 10.4049/jimmunol.166.2.1206. [DOI] [PubMed] [Google Scholar]
- 47.Felts RL, Reilly TJ, Calcutt MJ, Tanner JJ. Crystallization of a newly discovered histidine acid phosphatase from Francisella tularensis. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006;62:32–35. doi: 10.1107/S1744309105039813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Felts RL, Reilly TJ, Tanner JJ. Crystallization of AcpA, a respiratory burst-inhibiting acid phosphatase from Francisella tularensis. Biochim Biophys Acta. 2005;1752:107–110. doi: 10.1016/j.bbapap.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 49.Felts RL, Reilly TJ, Tanner JJ. Structure of Francisella tularensis AcpA: prototype of a unique superfamily of acid phosphatases and phospholipases C. J Biol Chem. 2006;281:30289–30298. doi: 10.1074/jbc.M606391200. [DOI] [PubMed] [Google Scholar]
- 50.Reilly TJ, Felts RL, Henzl MT, Calcutt MJ, Tanner JJ. Characterization of recombinant Francisella tularensis acid phosphatase A. Protein Expr Purif. 2006;45:132–141. doi: 10.1016/j.pep.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 51.Schulert GS, McCaffrey RL, Buchan BW, Lindemann SR, Hollenback C, Jones BD, Allen LA. Francisella tularensis genes required for inhibition of the neutrophil respiratory burst and intramacrophage growth identified by random transposon mutagenesis of strain LVS. Infect Immun. 2009;77:1324–1326. doi: 10.1128/IAI.01318-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aguirre-García MM, Okhuysen PC. Cryptosporidium parvum: identification and characterization of an acid phosphatase. Parasitol Res. 2007;101:85–89. doi: 10.1007/s00436-006-0457-8. [DOI] [PubMed] [Google Scholar]
- 53.Hartland EL, Green SP, Phillips WA, Robins-Browne RM. Essential role of YopD in inhibition of the respiratory burst of macrophages by Yersinia enterocolitica. Infect Immun. 1994;62:4445–4453. doi: 10.1128/iai.62.10.4445-4453.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gallois A, Klein JR, Allen LA, Jones BD, Nauseef WM. Salmonella pathogenicity island 2-encoded type III secretion system mediates exclusion of NADPH oxidase assembly from the phagosomal membrane. J Immunol. 2001;166:5741–5748. doi: 10.4049/jimmunol.166.9.5741. [DOI] [PubMed] [Google Scholar]
- 55.Baca OG, Roman MJ, Glew RH, Christner RF, Buhler JE, Aragon AS. Acid phosphatase activity in Coxiella burnetii: a possible virulence factor. Infect Immun. 1993;61:4232–4239. doi: 10.1128/iai.61.10.4232-4239.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lindgren H, Stenmark S, Chen W, Tärnvik A, Sjöstedt A. Distinct roles of reactive nitrogen and oxygen species to control infection with the facultative intracellular bacterium Francisella tularensis. Infect Immun. 2004;72:7172–7182. doi: 10.1128/IAI.72.12.7172-7182.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heyworth PG, Curnutte JT, Nauseef WM, Volpp BD, Pearson DW, Rosen H, Clark RA. Neutrophil nicotinamide adenine dinucleotide phosphate oxidase assembly. Translocation of p47-phox and p67-phox requires interaction between p47-phox and cytochrome b558. J Clin Invest. 1991;87:352–356. doi: 10.1172/JCI114993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Volpp BD, Nauseef WM, Clark RA. Two cytosolic neutrophil oxidase components absent in autosomal chronic granulomatous disease. Science. 1988;242:1295–1297. doi: 10.1126/science.2848318. [DOI] [PubMed] [Google Scholar]
- 59.Chen Q, Powell DW, Rane MJ, Singh S, Butt W, Klein JB, McLeish KR. Akt phosphorylates p47phox and mediates respiratory burst activity in human neutrophils. J Immunol. 2003;170:5302–5308. doi: 10.4049/jimmunol.170.10.5302. [DOI] [PubMed] [Google Scholar]
- 60.Fontayne A, Dang PM, Gougerot-Pocidalo MA, El-Benna J. Phosphorylation of p47phox sites by PKC alpha, beta II, delta, and zeta: effect on binding to p22phox and on NADPH oxidase activation. Biochemistry. 2002;41:7743–7750. doi: 10.1021/bi011953s. [DOI] [PubMed] [Google Scholar]
- 61.Torres M. Mitogen-activated protein kinase pathways in redox signaling. Front Biosci. 2003;8:d369–d391. doi: 10.2741/999. [DOI] [PubMed] [Google Scholar]
- 62.Torres M, Forman HJ. Redox signaling and the MAP kinase pathways. Biofactors. 2003;17:287–296. doi: 10.1002/biof.5520170128. [DOI] [PubMed] [Google Scholar]
- 63.Wang Y, Zeigler MM, Lam GK, Hunter MG, Eubank TD, Khramtsov VV, Tridandapani S, Sen CK, Marsh CB. The role of the NADPH oxidase complex, p38 MAPK, and Akt in regulating human monocyte/macrophage survival. Am J Respir Cell Mol Biol. 2007;36:68–77. doi: 10.1165/rcmb.2006-0165OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Babior BM. The leukocyte NADPH oxidase. Isr Med Assoc J. 2002;4:1023–1024. [PubMed] [Google Scholar]
- 65.Brown GE, Stewart MQ, Liu H, Ha VL, Yaffe MB. A novel assay system implicates PtdIns(3,4)P(2), PtdIns(3)P, and PKC delta in intracellular production of reactive oxygen species by the NADPH oxidase. Mol Cell. 2003;11:35–47. doi: 10.1016/s1097-2765(03)00005-4. [DOI] [PubMed] [Google Scholar]
- 66.El-Benna J, Dang PM, Gougerot-Pocidalo MA. Priming of the neutrophil NADPH oxidase activation: role of p47phox phosphorylation and NOX2 mobilization to the plasma membrane. Semin Immunopathol. 2008;30:279–289. doi: 10.1007/s00281-008-0118-3. [DOI] [PubMed] [Google Scholar]
- 67.Bokoch GM, Diebold BA. Current molecular models for NADPH oxidase regulation by Rac GTPase. Blood. 2002;100:2692–2696. doi: 10.1182/blood-2002-04-1149. [DOI] [PubMed] [Google Scholar]
- 68.Lodge R, Descoteaux A. Phagocytosis of Leishmania donovani amastigotes is Rac1 dependent and occurs in the absence of NADPH oxidase activation. Eur J Immunol. 2006;36:2735–2744. doi: 10.1002/eji.200636089. [DOI] [PubMed] [Google Scholar]
- 69.Lodge R, Diallo TO, Descoteaux A. Leishmania donovani lipophosphoglycan blocks NADPH oxidase assembly at the phagosome membrane. Cell Microbiol. 2006;8:1922–1931. doi: 10.1111/j.1462-5822.2006.00758.x. [DOI] [PubMed] [Google Scholar]
- 70.Vázquez-Torres A, Fantuzzi G, Edwards CK, 3rd, Dinarello CA, Fang FC. Defective localization of the NADPH phagocyte oxidase to Salmonella-containing phagosomes in tumor necrosis factor p55 receptor-deficient macrophages. Proc Natl Acad Sci USA. 2001;98:2561–2565. doi: 10.1073/pnas.041618998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vazquez-Torres A, Jones-Carson J, Mastroeni P, Ischiropoulos H, Fang FC. Antimicrobial actions of the NADPH phagocyte oxidase and inducible nitric oxide synthase in experimental salmonellosis. I. Effects on microbial killing by activated peritoneal macrophages in vitro. J Exp Med. 2000;192:227–236. doi: 10.1084/jem.192.2.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vazquez-Torres A, Xu Y, Jones-Carson J, Holden DW, Lucia SM, Dinauer MC, Mastroeni P, Fang FC. Salmonella pathogenicity island 2-dependent evasion of the phagocyte NADPH oxidase. Science. 2000;287:1655–1658. doi: 10.1126/science.287.5458.1655. [DOI] [PubMed] [Google Scholar]
- 73.Allen LA, Allgood JA, Han X, Wittine LM. Phosphoinositide3-kinase regulates actin polymerization during delayed phagocytosis of Helicobacter pylori. J Leukoc Biol. 2005;78:220–230. doi: 10.1189/jlb.0205091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Allen LA, Beecher BR, Lynch JT, Rohner OV, Wittine LM. Helicobacter pylori disrupts NADPH oxidase targeting in human neutrophils to induce extracellular superoxide release. J Immunol. 2005;174:3658–3667. doi: 10.4049/jimmunol.174.6.3658. [DOI] [PubMed] [Google Scholar]
- 75.DeLeo FR, Allen LA, Apicella M, Nauseef WM. NADPH oxidase activation and assembly during phagocytosis. J Immunol. 1999;163:6732–6740. [PubMed] [Google Scholar]
- 76.Child R, Wehrly TD, Rockx-Brouwer D, Dorward DW, Celli J. Acid phosphatases do not contribute to the pathogenesis of type A Francisella tularensis. Infect Immun. 2010;78:59–67. doi: 10.1128/IAI.00965-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Aragon V, Kurtz S, Flieger A, Neumeister B, Cianciotto NP. Secreted enzymatic activities of wild-type and pilD-deficient Legionella pneumophila. Infect Immun. 2000;68:1855–1863. doi: 10.1128/iai.68.4.1855-1863.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Santic M, Molmeret M, Abu Kwaik Y. Modulation of biogenesis of the Francisella tularensis subsp. novicida-containing phagosome in quiescent human macrophages and its maturation into a phagolysosome upon activation by IFN-gamma. Cell Microbiol. 2005;7:957–967. doi: 10.1111/j.1462-5822.2005.00529.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.