

Abstract
Altered regulation of signaling pathways can lead to pathologies including cardiac hypertrophy and heart failure (HF). We report that neonatal and adult cardiomyocytes express chromogranin B (CGB), a calcium (Ca2+) binding protein which modulates Ca2+ release by the inositol 1,4,5-trisphosphate receptor (InsP3R). Using fluorescent Ca2+-indicator dyes, we found that CGB regulates InsP3-dependent Ca2+ release in response to angiotensin-II (ANG-II), an octapeptide hormone that promotes cardiac hypertrophy. ELISA experiments and luciferase reporter assays identified ANG-II as a potent inducer of brain natriuretic peptide (BNP), a hormone that recently emerged as an important biomarker in cardiovascular disease. CGB was found to regulate ANG-II stimulated and basal secretion, expression and promoter activity of BNP that depend on the InsP3R. Moreover, we provide evidence that CGB acts via the transcription factor nuclear factor-kappa B (NF-κB) in an InsP3/Ca2+-dependent manner, but independent of nuclear factor of activated T-cells (NFAT). In-vivo experiments further showed that cardiac hypertrophy induced by ANG-II, a condition characterized by increased ventricular BNP production, is associated with up-regulation of ventricular CGB expression. Over-expression of CGB in cardiomyocytes, in turn, induced the BNP promoter. The evidence presented in this study identifies CGB as a novel regulator of cardiomyocyte InsP3/Ca2+-dependent signaling, NF-κB activity and BNP production.
Keywords: Chromogranin B (CGB); calcium (Ca2+); inositol 1,4,5-trisphosphate receptor (InsP3R); nuclear factor-kappa B (NF-κB); brain natriuretic peptide (BNP)
Introduction
Despite advances in treatment, cardiovascular disease still is the number one cause of morbidity and mortality in the western world.1 A prolonged cardiac hypertrophic state leads to heart failure (HF) and is commonly accompanied by complex changes in gene expression.1, 2 This includes the re-expression of fetal cardiac genes,2 the reciprocal regulation of intracellular Ca2+-release channels3 and an increase in ventricular production of brain natriuretic peptide (BNP), a hallmark of cardiac hypertrophy.4 Importantly, BNP plasma level elevation in patients with HF correlates with disease severity as assessed by New York Heart Association (NYHA) functional class and BNP recently emerged as important cardiac biomarker.4
Myocardial signal-transduction pathways that mediate hypertrophic growth and, eventually, the onset of HF are abundant and complex.1 One pathway involves Ca2+/calmodulin activated calcineurin-nuclear factor of activated T cells (NFAT) signaling.1, 5 In this pathway, cytoplasmic NFAT is dephosphorylated by the serine/threonine protein phosphatase calcineurin and subsequently NFAT is translocated to the nucleus to initiate transcription.1, 5 Recently, the importance of nuclear factor-kappa B (NF-κB) in cardiac hypertrophy was shown and NF-κB was linked to a variety of cardiovascular pathologies.1, 6 In the resting cell, NF-κB dimers reside in the cytoplasm bound to inhibitor proteins, inhibitor-kappa B (IκB).6 Typically, NF-κB signaling is initiated by stimulus-induced phosphorylation of IκB by IκB-kinases (IKKs) that leads to IκB-phosphorylation, polyubiquitination and degradation.6 This unmasks a nuclear translocation sequence resulting in translocation of NF-κB into the nucleus to initiate transcription.6 Even though this NF-κB activation pathway is well described in the immune system7 and its existence in the heart is broadly accepted,6 little is known about NF-κB activation in cardiac cells.
CGB, a Ca2+-binding protein that belongs to the granin-family of acidic proteins,8 resides in the endo-/sarcoplasmic reticulum (ER, SR) and functionally interacts with all three InsP3R-isoforms9 to shape Ca2+ release.10-12 Local InsP3-dependent Ca2+ signaling was only recently linked to cardiac excitation-transcription coupling (ETC) in adult ventricular myocytes.13 Because we found that neonatal and adult ventricular cardiomyocytes express CGB along with all three isoforms of the InsP3R, we hypothesized that CGB would be important in the regulation of cardiomyocyte InsP3-dependent Ca2+ signaling and would modify ETC. In this study we show that CGB regulates cardiomyocyte InsP3/Ca2+-dependent signaling, the activity of the transcription factor NF-κB, and the production of BNP.
Materials and Methods
Expanded Materials and Methods can be found in the Online Data Supplement.
Cell culture
All procedures for animal use were in accordance with guidelines approved by the Yale Animal Care and Use Committee. Primary neonatal rat ventricular cardiomyocytes were prepared as described previously with a purity of at least 95%.14 Cardiac fibroblasts were derived from differential pre-plating.
ANG-II micro-osmotic pump implantation
Micro-osmotic pumps were implanted as described in the Online Data Supplement. Mice were constantly infused with ANG-II for 2 weeks at a rate of 1000 ng/kg/min. Control mice experienced the same surgical procedures without pump implantation and were subsequently treated identically. Cardiac hypertrophy was documented by determination of the LVW/BW ratios at the time of sacrifice.
Plasmids, luciferase reporter vectors, siRNA
Plasmids, luciferase reporter vectors (hBNPLuc, NF-κB-luc, AdNFAT-luc) and siRNA have been described previously or are commercially available.5, 15-18 Luciferase reporter assays were performed as described.14 Intracellular chelation of Ca2+ was accomplished using BAPTA-AM (200 μmol/L).19
Transient transfection, adenoviral infection
Transient transfection and adenoviral infection of cardiomyocytes were previously described.14 Briefly, transfection was performed at day 2 after culture in OptiMEM using Lipofectamine 2000 for 4 hours followed by incubation overnight with complete growth-medium added. Transfection efficiency in cardiomyocytes using the protocol described is at least 30-40% for DNA. Adenoviral infection with NFAT-luciferase reporter vector (AdNFAT-luc)5 was performed for 2 hours.
BNP ELISA
Secretion and expression of BNP by cardiomyocytes and cardiac fibroblasts were assayed using the AssayMax Rat BNP-45 ELISA Kit (GENTAUR, Brussels, Belgium) according to the manufacturer’s protocol. Experiments were performed 2 days after siRNA transfection (CGB-siRNA experiments) or at day 4 after culture, respectively, to match time points. Basal and stimulation studies were performed for 4 hours. Pre-incubation with inhibitors (InsP3R inhibitor 2-APB, 25 μmol/L; AT1-R receptor inhibitor telmisartan, 1 μmol/L) was done for 1 hour. Samples were collected and absorbance at 450 nm was measured using a standard microplate reader. Standard points and samples were determined as duplicates or triplicates.
Live cell calcium imaging
Calcium imaging experiments have been described.14 Briefly, cells were loaded with cell-permeant indicator dye (Mag-Fluo-4/AM, 5 μmol/L or Fura Red/AM, 10 μmol/L) and transferred to a Zeiss LSM 510 NLO laser scanning confocal microscope. Fluorescence was measured by defining each cell as one region of interest and quantified in relation to baseline fluorescence (F/F0). The change in whole cell F/F0 was in between the nuclear and cytosolic signals and therefore used as representative read-out in this study (Supplementary Figure 1). The low-affinity (Kd,Ca2+ = 22 μmol/L) Ca2+-indicator Mag-Fluo-4 was used to assess Ca2+ release from internal stores. Differential loading of Mag-Fluo-4 into internal stores was verified (Supplementary Figure 2). In experiments with cells expressing DsRed (co-transfection), the high-affinity Ca2+-indicator Fura Red was used to assess cytosolic Ca2+ changes. This replacement of Mag-Fluo-4 was necessary due to an overlap of Mag-Fluo-4 and DsRed excitation and emission spectra that caused substantial quenching of Mag-Fluo-4 fluorescence by DsRed.
Immunoblotting
Immunoblotting was performed as described.20 Primary antibodies used are: CGB (BD Bioscience); InsP3R-1, InsP3R-2 and InsP3R-3;20 β-actin, GAPDH-HRP (Abcam); SERCA 2a, cardiac RyR (Affinity Bioreagents). Expressions were quantified by scanning densitometry.
Statistical analysis
Data are expressed as mean ± SEM or representative traces. (n/N) describes the number of individual experiments (n) in N independent cultures. Statistical analysis of the differences between multiple groups was performed using one way ANOVA (Student-Newman-Keuls Method), for 2 groups using t-test (SigmaPlot). Statistical significance is indicated: P < 0.05 as *; P < 0.01 as ** and P < 0.001 as ***.
Results
Cardiomyocytes express CGB along with all InsP3R isoforms
Neonatal cardiomyocytes but not cardiac fibroblasts express CGB (Fig. 1a). CGB also is present in adult mouse ventricular myocardium (Fig. 6). Besides CGB, cardiomyocytes express all three InsP3R isoforms (InsP3R-1, InsP3R-2, InsP3R-3) (Fig. 1b). CGB resides in the sarcoplasmic reticulum (SR) and functionally interacts with the InsP3R to shape Ca2+ release.9-12 Therefore, co-existence of CGB and the InsP3R in cardiomyocytes suggests a functional role for CGB in shaping cardiomyocyte InsP3-dependent Ca2+ signaling.
Figure 1. Neonatal cardiomyocytes express CGB and InsP3R isoforms.
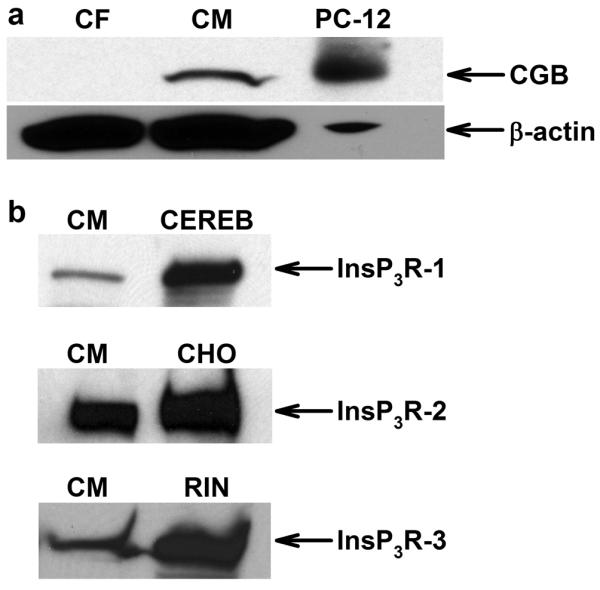
(a) Representative western blot shows that cardiomyocytes (CM) but not cardiac fibroblasts (CF) express CGB. PC-12 cells served as positive control, β-actin as loading control. (b) CM express all InsP3R isoforms; positive controls as indicated: CEREB - cerebellar microsomes, CHO - chinese hamster ovary cells, RIN - rat insulinoma cells.
Figure 6. CGB is up-regulated in ANG-II induced cardiac hypertrophy in-vivo.

(a) Representative western blot shows ventricular expression of CGB and cardiac RyR. GAPDH served as loading control. (b, c) Quantifications by densitometry. (b) ANG-II induces ventricular CGB expression approximately 3-fold. (c) In contrast, cardiac RyR expression is decreased to around 50 %. (d) LVW/BW ratio documents hypertrophy. (e) CGB over-expression in cardiomyocytes induces the BNP promoter.
ANG-II evokes InsP3-dependent Ca2+ release in cardiomyocytes
ANG-II causes the generation of phosphoinositides in cardiomyocytes through its interaction with the G-protein coupled ANG-II type 1 receptor (AT1-R).21, 22 We monitored changes in SR and nuclear envelope Ca2+ content in response to ANG-II (1 μmol/L) using the low-affinity fluorescent Ca2+-indicator Mag-Fluo-4/AM. Experiments were performed in Ca2+ free (0 Ca2+, EGTA 1 mmol/L) extracellular solution. ANG-II caused a marked decrease in Mag-Fluo-4 fluorescence intensity that recovered over time to baseline (Fig. 2a, 2d), as would be expected for Ca2+ release and subsequent re-uptake into internal stores. The magnitude of the ANG-II evoked Ca2+ release was 1.3 ± 0.04 (30/2) (Fig. 2c). Pre-treatment of cardiomyocytes with the InsP3R inhibitors 2-aminoethoxydiphenylborate (2-APB, 25 μmol/L) or xestospongin D (XeD, 5 μmol/L) prevents Ca2+ release upon ANG-II stimulation (Fig. 2b, 2c). Depolarization served as cell viability control and provided the means to distinguish between excitable myocytes and contaminating fibroblasts (“D”; Fig. 2a, 2b). These results show that ANG-II evokes InsP3-dependent Ca2+ release from internal stores in cardiomyocytes.
Figure 2. ANG-II evokes InsP3-dependent Ca2+ release in cardiomyocytes.

Ca2+ release from internal stores was monitored using the low-affinity Ca2+-indicator Mag-Fluo-4/AM. (a, b) Representative traces. (a) ANG-II causes Ca2+ release that recovers to baseline. (b) No significant Ca2+ release was observed in cardiomyocytes pre-treated with either one of the InsP3R inhibitors 2-APB or XeD. (c) Quantification of released Ca2+. ANG-II causes Ca2+ release only in the absence of InsP3R inhibitors. (d) Representative pseudo-colored time course of cardiomyocytes challenged with ANG-II. Letters indicate respective time points in panel a. Scale bar: 10 μm.
CGB shapes ANG-II evoked Ca2+ release
CGB is known to modulate Ca2+ release by the InsP3R at the single channel level and in intact cells.10-12, 17 Because we found that cardiomyocytes express CGB along with the InsP3R (Fig. 1), we hypothesized that CGB would be important in cardiomyocytes in shaping InsP3-dependent Ca2+ signaling. Therefore, CGB expression in cardiomyocytes was silenced by small interfering RNA (siRNA) (Fig. 3a). Densitometry revealed a knock-down to 34 ± 7 % compared to mock treatment (3/1) (p<0.001, Fig. 3b). To determine whether this down-regulation impacts InsP3-dependent Ca2+ release, cells were co-transfected with CGB-siRNA and DsRed and changes in cytosolic Ca2+ upon ANG-II stimulation (1 μmol/L) monitored using the fluorescent Ca2+-indicator Fura Red/AM. DsRed-only transfected cells served as control. The rise in cytosolic Ca2+ was blunted after CGB knock-down (Fig. 3c). Note that an increase in cytosolic Ca2+ causes a decrease in Fura Red fluorescence when excited at 488 nm. CGB knock-down diminished peak Ca2+ release to 84 ± 2 % (17/1) compared to control (100 ± 6 %; (17/1)) (p<0.05, Fig. 3d) and decreased the velocity of Ca2+ release to 32 ± 13 % (12/1) compared to control (100 ± 28.5 %; (13/1)) (p<0.05, Fig. 3e). These effects could also be explained by store depletion following CGB knock-down. To address this question, we depleted ER stores by adding 10 μmol/L of the sarcoplasmic-endoplasmic reticulum calcium-ATPase (SERCA) inhibitor thapsigargin in Ca2+-free medium and calculated the ER-Ca2+ content as area under the release curve. Both CGB-siRNA transfected and control cells had similar amounts of Ca2+ stored in the ER (Fig. 3f). We conclude that CGB shapes InsP3-dependent Ca2+ signaling in cardiomyocytes.
Figure 3. CGB shapes ANG-II evoked Ca2+ release.
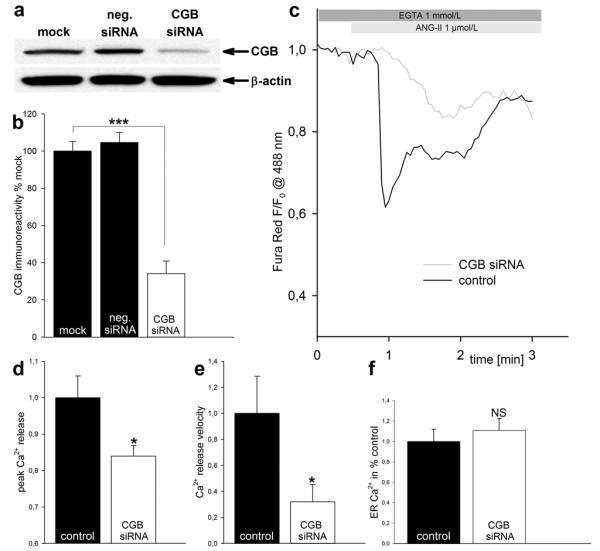
(a, b) Documentation of CGB knock-down. (a) Representative western blot. Note the decrease in CGB immunoreactivity in CGB knock-down cells only. (b) Quantification of CGB immunoreactivity by densitometry. (c) Representative traces. CGB knock-down decreases peak Ca2+ release and velocity of Ca2+ release upon ANG-II stimulation. (d, e) Quantification of peak Ca2+ release (d) and velocity of Ca2+ release (e). (f) CGB knock-down does not deplete ER-Ca2+ stores.
ANG-II stimulated and basal BNP secretion depend on the InsP3R
BNP secretion is regulated at the transcriptional level.4 ANG-II increases cardiomyocyte BNP mRNA levels by its interaction with the G-protein coupled AT1-R that causes the formation of phosphoinositides.21-25 Therefore, we hypothesized that secretion of BNP would be stimulated by ANG-II and would depend on the InsP3 signaling pathway. To address this question, we first studied the time-course of BNP secretion in cardiomyocytes. Supernatant samples were collected after 1 hour without any treatment (baseline) followed by repetitive collections from cells incubated with ANG-II (1 μmol/L) or vehicle for 2, 4, 8 and 12 hours and assayed by ELISA. Cells incubated with vehicle only showed an increase in BNP secretion over time due to tonic basal secretion (Fig. 4a). However, cells incubated with ANG-II showed a markedly different kinetics (Fig. 4a). The ANG-II dependent portion of BNP secretion was maximal after 4 hours of incubation (Fig. 4b). This time period was used in any further ELISA experiment. Next, we studied the requirement of the AT1-R and Ca2+ release by the InsP3R in ANG-II stimulated BNP secretion. Both the AT1-R inhibitor telmisartan (Telm, 1 μmol/L) and the InsP3R inhibitor 2-APB (25 μmol/L) prevented the stimulating effect of ANG-II (Fig. 4c). Inhibition of the InsP3R also diminished basal BNP secretion (Fig. 4d). These results show that ANG-II stimulates BNP secretion in cardiomyocytes and that InsP3-mediated Ca2+ release is a component of the pathway for ANG-II stimulated and basal BNP secretion.
Figure 4. Basal and ANG-II stimulated BNP production depend on the InsP3R and are regulated by CGB.

(a) Time course of BNP secretion. Note different kinetics of vehicle and ANG-II. (b) ANG-II stimulated secretion reaches a relative maximum after 4 hours. (c) ANG-II stimulated BNP secretion depends on the AT1-R and the InsP3R. (d) Inhibition of the InsP3R diminishes basal BNP secretion. (e, f) CGB knock-down decreases basal BNP secretion and expression and abrogates the stimulating effect of ANG-II. (g) Linear regression analysis suggests that CGB regulates BNP production at the transcriptional level.
CGB knock-down diminishes basal and abrogates ANG-II stimulated BNP production
Basal and ANG-II stimulated BNP secretion depend on the activation of the InsP3R (Fig. 4d, 4c). We hypothesized that CGB which shapes InsP3-dependent Ca2+ release (Fig. 3) would also modify cardiomyocyte BNP production. In order to address this question, cardiomyocytes were transiently transfected with CGB-siRNA, negative-siRNA or mock treated. Basal BNP secretion was reduced to 65 ± 7 % (12/3) after CGB knock-down compared to mock treatment (100 ± 3 %; (12/3)) (p<0.001, Fig. 4e). CGB knock-down also prevented the stimulating effect of ANG-II (56 ± 8 %; (12/3)) (Fig. 4e). To investigate whether CGB knock-down inhibits BNP secretion by preventing its release or by decreasing its production, we also measured BNP expression. CGB knock-down significantly decreased basal BNP expression to 63 ± 5 % (11/3) compared to mock treatment (100 ± 3%; (10/3)) (p<0.01, Fig. 4f). As for secretion, ANG-II failed to exert positive effects on BNP expression after CGB knock-down (56 ± 5 %; (10/2)) (Fig. 4f). Negative (non-targeting) siRNA transfected cells showed slightly increased BNP production (Fig. 4e, 4f) which we attribute to cell stress during the transfection. Values measured for BNP expression and secretion of each individual experiment were matched and subjected to bi-directional analysis and linear regression. These data revealed a strong correlation of BNP expression and secretion (r2 = 0.98) (Fig. 4g). This suggests that CGB impacts BNP production at the transcriptional level rather than by preventing the formation of secretory granules or their secretion; either of which would lead to a build up of BNP inside the cell.
CGB regulates the BNP promoter
To study the potential role of CGB on BNP production at the transcriptional level, we used a human BNP promoter luciferase reporter (hBNPLuc).15 To investigate the effect of CGB on basal BNP promoter luciferase activity, cardiomyocytes were transiently transfected with hBNPLuc alone (“mock”) or co-transfected with hBNPLuc and negative-siRNA or CGB-siRNA. Basal luciferase activity after CGB knock-down was significantly diminished to 37 ± 6 % (7/2) compared to mock (100 ± 5 %; (6/2)) (p<0.001, Fig. 5a). Cells transiently transfected with negative-siRNA showed a non-significant reduction in hBNPLuc activity (87 ± 6 %; (7/2)) (Fig. 5a). Next, we asked whether ANG-II would induce BNP and whether this induction would be modified by CGB. Cardiomyocytes were incubated with vehicle (control) or 1 μmol/L ANG-II for 4 hours. ANG-II increased luciferase activity to 210 ± 9 % (3/1) compared to control (100 ± 5 %; (3/1)) (p<0.001, Fig. 5b). In contrast, ANG-II failed to exert stimulating effects in CGB knock-down cells (Fig. 5b). Remarkably, CGB knock-down kept luciferase activity below basal levels even with ANG-II present (27 ± 3 % of control; (3/1)) (p<0.001, Fig 5b). These results show that CGB regulates the BNP promoter under both basal and ANG-II stimulated conditions.
Figure 5. CGB regulates the BNP promoter and NF-κB.

(a, b) BNP promoter luciferase activities. CGB knock-down decreases basal promoter activity and prevents its induction by ANG-II. (c) Basal NFAT activity is not affected by CGB knock-down. Cyclosporine A (CsA) served as control. (d, e) NF-κB luciferase activities. CGB knock-down decreases basal NF-κB activity and prevents its activation by ANG-II. (f) Basal NF-κB activity depends on Ca2+.
CGB regulates NF-κB activity but does not affect NFAT
We aimed to identify the transcription factor that mediates the regulatory role of CGB on the BNP promoter. We first focused on NFAT, a Ca2+-dependent transcription factor that plays a central role in cardiac hypertrophy.1, 5 Cardiomyocytes were infected with adenoviral NFAT-luciferase reporter (AdNFAT-luc)5 alone (“mock”) and after transient transfection with CGB-siRNA or negative-siRNA. We did not observe any significant change in basal NFAT activity after CGB knock-down (mock 100 ± 6 % (7/2); negative-siRNA 96 ± 7 % (7/2); CGB-siRNA 102 ± 6 % (15/2)) (Fig. 5c). However, in control experiments using the calcineurin inhibitor cyclosporine A1 (CsA, 1 μmol/L) basal NFAT activity was significantly decreased with no significant differences among the three groups (mock/CsA 25 ± 2 % (7/2); negative-siRNA/CsA 29 ± 3 % (7/2); CGB-siRNA/CsA 30 ± 3 % (7/2)) (p<0.001, Fig. 5c). These results suggest a NFAT-independent mechanism.
Recently, the NF-κB signaling pathway has been recognized as an important mediator of cardiac hypertrophic signaling.1, 6 Little is known about Ca2+-dependent activation of NF-κB beyond its well characterized Ca2+-dependent function in the immune system and recent reports showing Ca2+-dependent activation of NF-κB in hippocampal neurons and skeletal muscle.7, 19, 26 To study the effect of CGB on NF-κB activity in cardiomyocytes, we utilized a NF-κB luciferase reporter (NF-κB-luc).16 Cardiomyocytes were transiently transfected with NF-κB-luc alone (“mock”) or co-transfected with NF-κB-luc and CGB-siRNA or negative-siRNA. Following CGB knock-down, basal NF-κB luciferase-activity was significantly decreased to 50 ± 6 % (8/2) compared to mock (100 ± 3 %; (8/2)) (p<0.001, Fig. 5d). The non-significant increase in NF-κB activity following negative-siRNA transfection is most likely due to cell stress during the transfection and consistent with our observations on BNP production (Fig. 4e, 4f). Next, we investigated whether NF-κB would be induced by ANG-II and whether this induction was regulated by CGB as we found for the BNP promoter (Fig. 5b). In fact, ANG-II (1 μmol/L) increased NF-κB luciferase activity to 121 ± 10 % (3/1) compared to control (100 ± 7 %; (3/1)) (p=0.093, Fig. 5e) and CGB knock-down kept NF-κB luciferase activity below basal levels even with ANG-II present (17 ± 2 % of control; (3/1)) (p<0.001, Fig. 5e). The Ca2+-dependence of basal NF-κB activity was tested by chelation of intracellular Ca2+ with BAPTA. Basal luciferase activity following Ca2+ chelation was reduced to 63 ± 16 % (5/1) compared to control (100 ± 2 %; (6/1)) (p<0.05, Fig. 5f). These results provide evidence for a novel role of CGB in the regulation of basal and ANG-II stimulated BNP promoter and NF-κB activities in a Ca2+-dependent manner that is independent of NFAT signaling.
Cardiac hypertrophy induces ventricular CGB in-vivo
Cardiac hypertrophy is commonly associated with increased ventricular BNP production and elevated BNP plasma levels in patients.4 Because we found that CGB is an important modulator of BNP transcription, expression and secretion in cardiomyocytes (Fig. 4, 5), we studied ventricular CGB expression in-vivo during cardiac hypertrophy. Cardiac hypertrophy was induced in adult mice by chronic ANG-II treatment using micro-osmotic pumps. Hypertrophy was documented by determination of the left ventricular weight/body weight (LVW/BW) ratios (Fig. 6d). Chronic ANG-II treatment increased LVW/BW ratio 1.7 fold (4.7 ± 0.3 in ANG-II treated mice compared to 2.7 ± 0.1 in sham treated matched littermates) (p<0.05, Fig. 6d). Hypertrophy was associated with a marked up-regulation of ventricular CGB expression (Fig. 6a). Densitometry revealed an increase in CGB expression to 322 ± 46 % (5/1) in hypertrophied ventricles compared to control (100 ± 14 %; (9/1)) (p<0.001, Fig. 6b). Moreover, consistent with previous reports on decreased cardiac ryanodine receptor (RyR) mRNA levels in failing human hearts,3 we found the expression of cardiac RyR in the same samples significantly decreased to 52 ± 16 % (5/1) compared to control (100 ± 9 %; (9/1)) (p<0.05, Fig. 6a, 6c). No significant change in the expression of any of the InsP3R isoforms or SERCA 2a was observed (data not shown).
CGB over-expression increases BNP promoter activity in cardiomyocytes
Because we found CGB induced in-vivo in adult mice ventricular myocardium following the induction of cardiac hypertrophy (Fig. 6a, 6b), we next asked whether elevated CGB expression would, in turn, increase BNP promoter activity. To address this question, we over-expressed CGB. Cardiomyocytes were co-transfected with the hBNPLuc reporter plasmid15 and full length CGB18 or empty vector (control). In fact, CGB over-expression increased BNP promoter luciferase activity to 172 ± 16 % (4/1) compared to control (100 ± 24 %; (3/1)) (p<0.05, Fig. 6e).
Discussion
In the present study we provide evidence for a novel role of the Ca2+ binding protein CGB in the regulation of cardiomyocyte signaling. We show that CGB shapes InsP3-dependent Ca2+ release in cardiomyocytes and that CGB also regulates NF-κB activity and BNP production. Our in-vitro results are supported by in-vivo experiments that show the involvement of CGB in ANG-II mediated cardiac hypertrophy.
Local InsP3-dependent Ca2+ signaling was only recently linked to cardiac ETC in adult ventricular myocytes and it was also shown that these local Ca2+ signals act in a manner that is segregated from the global Ca2+ transients that mediate ECC.13 InsP3Rs are ubiquitous intracellular Ca2+ release channels that are present in the heart, although at lower levels than the related RyRs which mediate ECC.27, 28 The InsP3R-2 is the most InsP3-sensitive and the predominant isoform in cardiomyocytes.27, 29, 30 In atrial cardiomyocytes the InsP3R-2 is believed to co-localize with “junctional” RyRs at the subsarcolemmal space, possibly modifying ECC and thereby triggering arrhythmias27 whereas in ventricular cardiomyocytes the InsP3R-2 was found throughout the cell, but elevated within or in close proximity to the nuclear envelope31 and an important role in cardiac ETC was reported.13
CGB, which is a member of the granin-family of acidic proteins,8 interacts with all three InsP3R-isoforms9 and modulates InsP3-dependent Ca2+ release.10-12 Here we show that CGB is expressed in neonatal cardiomyocytes (Fig. 1) as well as adult mice ventricular myocardium (Fig. 6) and impacts InsP3-dependent Ca2+ release upon ANG-II stimulation that could not be explained by store depletion following CGB knock-down (Fig. 2, 3). Signaling specificity of the multifunctional second-messenger Ca2+ is achieved by variations in and combinations of magnitude, frequency and duration of Ca2+ signals.32 Our results that CGB simultaneously modifies two of these modalities (i.e. magnitude and velocity of Ca2+ release; Fig. 3) suggests a crucial role for CGB in fine-tuning of the local InsP3-dependent Ca2+ signals that target gene transcription in cardiomyocyte ETC. In fact, the decrease in basal and the prevention of ANG-II stimulated BNP secretion and expression after CGB knock-down (Fig. 4) is also seen at the transcriptional level in similarly impaired basal and stimulated BNP promoter activities (Fig. 5). Notably, both basal and ANG-II stimulated BNP production depend on Ca2+ release by the InsP3R (Fig. 4) that is modified by CGB (Fig. 3). Although CGB impacts InsP3-dependent Ca2+ release (Fig. 3) and the calcineurin-NFAT signaling pathway is considered one of the central players in integrating cardiac InsP3/Ca2+-mediated hypertrophic inputs,1, 5 we did not observe any significant impairment in basal NFAT activity after CGB knock-down (Fig. 5) that would explain the consistent decrease in basal BNP production (Fig. 4) and promoter activity (Fig. 5). However, basal - Ca2+-dependent (Fig. 5f) - and ANG-II stimulated activity of another transcription factor, NF-κB, were markedly decreased following CGB gene-silencing (Fig. 5), similar to the observed impairments in basal and ANG-II stimulated BNP production and promoter activity (Fig. 4, 5). Therefore, linkage of NF-κB signaling and BNP production in cardiomyocytes seems very likely. Specific NF-κB binding sites identified on the human BNP promoter further suggest that both are linked directly.33 CGB knock-down consistently kept BNP secretion, expression, promoter activity and NF-κB activity below basal/control levels even with the inducer ANG-II present (Fig. 4, 5). This suggests that ANG-II recruits the same CGB-regulated pathway that is responsible for basal BNP production to exert its positive effects, most likely the InsP3R, InsP3 and Ca2+.
Ca2+-dependent NF-κB activation was recently shown in neurons and skeletal muscle.19, 26 Here we provide evidence for the existence of a similar Ca2+-dependent NF-κB activation in cardiomyocytes that is independent of the Ca2+/calcineurin-activated NFAT pathway; even though both transcription factors utilize the same second messenger – Ca2+. Therefore, our results provide another example for how precisely Ca2+ signals can activate different signaling pathways in cardiomyocytes, possibly similar to the differential activation of transcription factors by Ca2+ described in lymphocytes.32
In neurons, Ca2+-dependent activation of NF-κB was shown to require Ca2+/calmodulin-dependent kinase II (CaMKII).19 However, using the general CaMKs-inhibitor KN-62 (10 μmol/L), we did not observe any significant suppression in basal NF-κB activity that could explain the suppressive effect of CGB knock-down (data not shown). This suggests that CGB impacts NF-κB signaling independent of CaMKs in cardiomyocytes and that NF-κB activation is regulated differently in various cell types.
In-vivo experiments showed that ANG-II induced cardiac hypertrophy, a condition characterized by increased ventricular BNP production, is associated with an impressive up-regulation of CGB in ventricular myocardium (Fig. 6). Because we also show in this study that CGB is an important regulator of basal and ANG-II stimulated BNP transcription, expression and secretion in cardiomyocytes in-vitro (Fig. 4, 5), this result suggests that CGB might be of similar importance in the regulation of BNP production in-vivo, possibly contributing to the induction of ventricular BNP in cardiac hypertrophy. This hypothesis becomes even more likely since CGB over-expression in cardiomyocytes significantly induces the BNP promoter (Fig. 6e).
Based on the evidence presented in this study we propose that CGB is of crucial importance in the InsP3/Ca2+-dependent regulation of hypertrophic signaling in cardiomyocytes (Fig. 7a-c). Physiologically, CGB resides in the SR and shapes Ca2+ release from internal stores by modulating the activity of the InsP3R. The generation of InsP3 is triggered by various hypertrophic agonists including ANG-II. Ca2+ released from the SR by the InsP3R triggers the activation of Ca2+-dependent transcription factors including NF-κB that initiate BNP transcription (ETC) (Fig. 7a). Decreased CGB expression levels in cardiomyocytes lead to impaired CGB-InsP3R coupling that results in decreased InsP3-dependent Ca2+ release, NF-κB activity and BNP production (Fig. 7b). Because we found that CGB expression is markedly up-regulated in ANG-II induced cardiac hypertrophy in-vivo and that CGB over-expression in cardiomyocytes leads to enhanced activity of the human BNP promoter, we hypothesize that CGB contributes to the increase in ventricular BNP production commonly associated with cardiac hypertrophy (Fig. 7c). In this scenario, the up-regulation of CGB expression in hypertrophic ventricles improves the CGB-InsP3R coupling leading to increased InsP3-dependent Ca2+ release, NF-κB activity and BNP production.
Figure 7. Proposed model for the role of CGB in cardiac hypertrophic signaling.

(a) Physiologically, CGB resides inside the SR and shapes Ca2+ release from internal stores through its interaction with the InsP3R. Released Ca2+ activates transcription factors including NF-κB that initiate BNP transcription. Nuclear Ca2+ release by the InsP3R might exert direct effects. (b) Decreased CGB expression impairs CGB – InsP3R coupling and leads to decreased InsP3-dependent Ca2+ release, NF-κB activity and BNP production. (c) Up-regulation of CGB, as observed in ANG-II induced ventricular hypertrophy, improves CGB – InsP3R coupling and leads to increased InsP3-dependent Ca2+ release, NF-κB activity and BNP production.
In the present study we provide insights into how CGB might regulate InsP3-dependent Ca2+-release, NF-κB activity and BNP production at the cellular level in neonatal cardiomyocytes. Our results also show that up-regulation of CGB occurs in-vivo in adult hypertrophic cardiomyocytes, suggesting that CGB plays a role in the complex cellular processes involved in cardiac hypertrophy.
Supplementary Material
Acknowledgments
We are grateful to Wolfgang Boehmerle, Craig J. Gibson, Andjelka Celic and Brenda DeGray for invaluable advice regarding the design of the experiments and thoughtful discussions and comments on the manuscript. We thank Peter Burch and Dr. Anton Bennett for their help with the adenoviral infection and Dr. Richard Wojcikiewicz for generously providing the InsP3R-2 antibody. We thank Dr. Margot C. LaPointe (hBNPLuc), Dr. Gabriel Nùñez (NF-κB-luc) and Dr. Jeffery D. Molkentin (AdNFAT-luc) for kindly providing reporter constructs as well as Dr. Jean-Pierre Julien and Dr. Makoto Urushitani for providing the full length CGB plasmid.
Sources of Funding Supported by grants from the NIH (BEE, FJG), scholarships from the German National Merit Foundation (FMH, KZ), Roland Ernst Foundation (FMH) and FONDECYT 1060077 (ME).
Footnotes
Disclosures None.
References
- 1.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 2.Swynghedauw B. Phenotypic plasticity of adult myocardium: molecular mechanisms. J Exp Biol. 2006;209:2320–2327. doi: 10.1242/jeb.02084. [DOI] [PubMed] [Google Scholar]
- 3.Go LO, Moschella MC, Watras J, Handa KK, Fyfe BS, Marks AR. Differential regulation of two types of intracellular calcium release channels during end-stage heart failure. J Clin Invest. 1995;95:888–894. doi: 10.1172/JCI117739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Lemos JA, McGuire DK, Drazner MH. B-type natriuretic peptide in cardiovascular disease. Lancet. 2003;362:316–322. doi: 10.1016/S0140-6736(03)13976-1. [DOI] [PubMed] [Google Scholar]
- 5.Wilkins BJ, Dai YS, Bueno OF, Parsons SA, Xu J, Plank DM, Jones F, Kimball TR, Molkentin JD. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. 2004;94:110–118. doi: 10.1161/01.RES.0000109415.17511.18. [DOI] [PubMed] [Google Scholar]
- 6.Hall G, Hasday JD, Rogers TB. Regulating the regulator: NF-kappaB signaling in heart. J Mol Cell Cardiol. 2006;41:580–591. doi: 10.1016/j.yjmcc.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 7.Baeuerle PA, Henkel T. Function and activation of NF-kappa B in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 8.Helle KB. The granin family of uniquely acidic proteins of the diffuse neuroendocrine system: comparative and functional aspects. Biol Rev Camb Philos Soc. 2004;79:769–794. doi: 10.1017/s146479310400644x. [DOI] [PubMed] [Google Scholar]
- 9.Yoo SH, Oh YS, Kang MK, Huh YH, So SH, Park HS, Park HY. Localization of three types of the inositol 1,4,5-trisphosphate receptor/Ca(2+) channel in the secretory granules and coupling with the Ca(2+) storage proteins chromogranins A and B. J Biol Chem. 2001;276:45806–45812. doi: 10.1074/jbc.M107532200. [DOI] [PubMed] [Google Scholar]
- 10.Thrower EC, Choe CU, So SH, Jeon SH, Ehrlich BE, Yoo SH. A functional interaction between chromogranin B and the inositol 1,4,5-trisphosphate receptor/Ca2+ channel. J Biol Chem. 2003;278:49699–49706. doi: 10.1074/jbc.M309307200. Epub 42003 Sep 49623. [DOI] [PubMed] [Google Scholar]
- 11.Choe CU, Ehrlich BE. The inositol 1,4,5-trisphosphate receptor (IP3R) and its regulators: sometimes good and sometimes bad teamwork. Sci STKE. 2006;2006:re15. doi: 10.1126/stke.3632006re15. [DOI] [PubMed] [Google Scholar]
- 12.Jacob SN, Choe CU, Uhlen P, DeGray B, Yeckel MF, Ehrlich BE. Signaling microdomains regulate inositol 1,4,5-trisphosphate-mediated intracellular calcium transients in cultured neurons. J Neurosci. 2005;25:2853–2864. doi: 10.1523/JNEUROSCI.4313-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu X, Zhang T, Bossuyt J, Li X, McKinsey TA, Dedman JR, Olson EN, Chen J, Brown JH, Bers DM. Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. J Clin Invest. 2006;116:675–682. doi: 10.1172/JCI27374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uhlen P, Burch PM, Zito CI, Estrada M, Ehrlich BE, Bennett AM. Gain-of-function/Noonan syndrome SHP-2/Ptpn11 mutants enhance calcium oscillations and impair NFAT signaling. Proc Natl Acad Sci U S A. 2006;103:2160–2165. doi: 10.1073/pnas.0510876103. Epub 2006 Feb 2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.LaPointe MC, Wu G, Garami M, Yang XP, Gardner DG. Tissue-specific expression of the human brain natriuretic peptide gene in cardiac myocytes. Hypertension. 1996;27:715–722. doi: 10.1161/01.hyp.27.3.715. [DOI] [PubMed] [Google Scholar]
- 16.Inohara N, Koseki T, Lin J, del Peso L, Lucas PC, Chen FF, Ogura Y, Nunez G. An induced proximity model for NF-kappa B activation in the Nod1/RICK and RIP signaling pathways. J Biol Chem. 2000;275:27823–27831. doi: 10.1074/jbc.M003415200. [DOI] [PubMed] [Google Scholar]
- 17.Huh YH, Jeon SH, Yoo JA, Park SY, Yoo SH. Effects of chromogranin expression on inositol 1,4,5-trisphosphate-induced intracellular Ca2+ mobilization. Biochemistry. 2005;44:6122–6132. doi: 10.1021/bi048070w. [DOI] [PubMed] [Google Scholar]
- 18.Urushitani M, Sik A, Sakurai T, Nukina N, Takahashi R, Julien JP. Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat Neurosci. 2006;9:108–118. doi: 10.1038/nn1603. Epub 2005 Dec 2020. [DOI] [PubMed] [Google Scholar]
- 19.Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D. NF-kappa B functions in synaptic signaling and behavior. Nat Neurosci. 2003;6:1072–1078. doi: 10.1038/nn1110. [DOI] [PubMed] [Google Scholar]
- 20.Johenning FW, Zochowski M, Conway SJ, Holmes AB, Koulen P, Ehrlich BE. Distinct intracellular calcium transients in neurites and somata integrate neuronal signals. J Neurosci. 2002;22:5344–5353. doi: 10.1523/JNEUROSCI.22-13-05344.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dostal DE, Hunt RA, Kule CE, Bhat GJ, Karoor V, McWhinney CD, Baker KM. Molecular mechanisms of angiotensin II in modulating cardiac function: intracardiac effects and signal transduction pathways. J Mol Cell Cardiol. 1997;29:2893–2902. doi: 10.1006/jmcc.1997.0524. [DOI] [PubMed] [Google Scholar]
- 22.Sadoshima J, Izumo S. Signal transduction pathways of angiotensin II--induced c-fos gene expression in cardiac myocytes in vitro. Roles of phospholipid-derived second messengers. Circ Res. 1993;73:424–438. doi: 10.1161/01.res.73.3.424. [DOI] [PubMed] [Google Scholar]
- 23.Wiese S, Breyer T, Dragu A, Wakili R, Burkard T, Schmidt-Schweda S, Fuchtbauer EM, Dohrmann U, Beyersdorf F, Radicke D, Holubarsch CJ. Gene expression of brain natriuretic peptide in isolated atrial and ventricular human myocardium: influence of angiotensin II and diastolic fiber length. Circulation. 2000;102:3074–3079. doi: 10.1161/01.cir.102.25.3074. [DOI] [PubMed] [Google Scholar]
- 24.Nakagawa O, Ogawa Y, Itoh H, Suga S, Komatsu Y, Kishimoto I, Nishino K, Yoshimasa T, Nakao K. Rapid transcriptional activation and early mRNA turnover of brain natriuretic peptide in cardiocyte hypertrophy. Evidence for brain natriuretic peptide as an “emergency” cardiac hormone against ventricular overload. J Clin Invest. 1995;96:1280–1287. doi: 10.1172/JCI118162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lokuta AJ, Cooper C, Gaa ST, Wang HE, Rogers TB. Angiotensin II stimulates the release of phospholipid-derived second messengers through multiple receptor subtypes in heart cells. J Biol Chem. 1994;269:4832–4838. [PubMed] [Google Scholar]
- 26.Valdes JA, Hidalgo J, Galaz JL, Puentes N, Silva M, Jaimovich E, Carrasco MA. Nf-Kb Activation by Depolarization of Skeletal Muscle Cells Depends on Ryanodine and Ip3 Receptors-Mediated Calcium Signals. Am J Physiol Cell Physiol. 2007 doi: 10.1152/ajpcell.00320.2006. [DOI] [PubMed] [Google Scholar]
- 27.Lipp P, Laine M, Tovey SC, Burrell KM, Berridge MJ, Li W, Bootman MD. Functional InsP3 receptors that may modulate excitation-contraction coupling in the heart. Curr Biol. 2000;10:939–942. doi: 10.1016/s0960-9822(00)00624-2. [DOI] [PubMed] [Google Scholar]
- 28.Mackenzie L, Roderick HL, Proven A, Conway SJ, Bootman MD. Inositol 1,4,5-trisphosphate receptors in the heart. Biol Res. 2004;37:553–557. doi: 10.4067/s0716-97602004000400008. [DOI] [PubMed] [Google Scholar]
- 29.Thrower EC, Hagar RE, Ehrlich BE. Regulation of Ins(1,4,5)P3 receptor isoforms by endogenous modulators. Trends Pharmacol Sci. 2001;22:580–586. doi: 10.1016/s0165-6147(00)01809-5. [DOI] [PubMed] [Google Scholar]
- 30.Perez PJ, Ramos-Franco J, Fill M, Mignery GA. Identification and functional reconstitution of the type 2 inositol 1,4,5-trisphosphate receptor from ventricular cardiac myocytes. J Biol Chem. 1997;272:23961–23969. doi: 10.1074/jbc.272.38.23961. [DOI] [PubMed] [Google Scholar]
- 31.Bare DJ, Kettlun CS, Liang M, Bers DM, Mignery GA. Cardiac type 2 inositol 1,4,5-trisphosphate receptor: interaction and modulation by calcium/calmodulin-dependent protein kinase II. J Biol Chem. 2005;280:15912–15920. doi: 10.1074/jbc.M414212200. Epub 12005 Feb 15913. [DOI] [PubMed] [Google Scholar]
- 32.Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature. 1997;386:855–858. doi: 10.1038/386855a0. [DOI] [PubMed] [Google Scholar]
- 33.Liang F, Gardner DG. Mechanical strain activates BNP gene transcription through a p38/NF-kappaB-dependent mechanism. J Clin Invest. 1999;104:1603–1612. doi: 10.1172/JCI7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.