

Abstract
Leber congenital amaurosis (LCA) is a congenital retinal dystrophy characterized by severe visual loss in infancy and nystagmus. Although most often inherited in an autosomal recessive fashion, rare individuals with mutations in the cone-rod homeobox gene, CRX, have dominant disease. CRX is critical for photoreceptor development and acts synergistically with the leucine-zipper transcription factor, NRL. We report on the phenotype of two individuals with LCA due to novel, de novo CRX mutations, c.G264T(p.K74N) and c.413delT(p.I138fs48), that reduce transactivation in vitro to 10% and 30% of control values, respectively. Whereas the c.413delT(p.I138fs48) mutant allows co-expressed NRL to transactivate independently at its normal, baseline level, the c.G264T(p.K74N) mutant reduces co-expressed NRL transactivation and reduces steady state levels of both proteins. Although both mutant proteins predominantly localize normally to the nucleus, they also both show variable cytoplasmic localization. These observations suggest that some CRX-mediated LCA may result from effects beyond haploinsufficiency, such as the mutant protein interefering with other transcription factors’ function. Such patients would therefore not likely benefit from a simple, gene-replacement strategy for their disease.
Keywords: cone-rod homeobox, CRX, neural leucine-zipper protein, NRL, Leber congenital amaurosis, LCA, retinal degeneration
INTRODUCTION
Leber congenital amaurosis (LCA) is a severe, inherited retinal dystrophy of childhood, characterized by severe visual loss at or near birth, nystagmus, and a pigmentary retinopathy (Ahmed and Loewenstein, 2008; den Hollander, et al., 2008). This genetically heterogeneous disorder renders infants with little or no retinal photoreceptor function, resulting in a non- detectable electroretinogram (ERG). LCA is responsible for at least 5% of all retinal dystrophies and is one of the main causes of blindness in children. Although some autosomal dominant families have been described (Freund, et al., 1998), LCA is generally inherited in an autosomal recessive manner.
Cone-rod homeobox (CRX; MIM# 602225) is a paired-like homeodomain transcription factor of the otd/OTX family (Chen, et al., 1997) that is expressed in the photoreceptors (cones and rods) of the retina, regulating vital photoreceptor differentiation and integrity (Furukawa, et al., 1997). CRX acts synergistically with other transcription factors, e.g. NRL and RX, to regulate gene transcription (Kimura, et al., 2000; Mitton, et al., 2000). The protein neural retina leucine zipper (NRL) is a transcription factor, of the v-Maf family, that is primarily expressed in rod photoreceptors (Farjo, et al., 1993; Swaroop, et al., 1992). Its combinatorial and cooperative action with CRX regulates rhodopsin transcription (Kumar, et al., 1996; Rehemtulla, et al., 1996). The deletion of Nrl in mice causes a total loss of rod photoreceptors and super-normal cone function, mediated by S cones(Mears, et al., 2001).
We here report on two, novel, de novo mutations in CRX that lead to presumed autosomal dominant LCA. We describe the clinical presentation of these patients and investigate the ability of the mutant CRX proteins to regulate gene expression via the bovine rhodopsin promoter, both in the presence and absence of NRL. We also characterize the subcellular localization of the mutant proteins and place our findings in the context of what is known about the functional domains of CRX.
MATERIALS AND METHODS
Clinical Examination and Mutation Identification
Both subjects were examined at the National Eye Institute and the University of Maryland by a board-certified ophthalmologist (BPB, MAJ and/or RCC) as part of an IRB-approved protocol for the evaluation and treatment of patients with inherited eye disease. Electroretinograms were recorded under anesthesia according to International Society for Electrophysiology of Vision standards. Research on human subjects was conducted in accordance with the recommendations of the Declaration of Helsinki.
DNA was extracted from whole blood of the two subjects and their parents and the exons and exon-intron boundaries of known LCA genes were amplified and sequenced according to standard protocols. Primers and experimental conditions are available upon request.
Molecular Modeling
Nucleotide numbering reflects cDNA numbering with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence, according to journal guidelines (www.hgvs.org/mutnomen). The initiation codon is codon 1. The structure of wild type and the K88N mutant cone-rod homeobox protein CRX-DNA complex was modeled using the paired homeodomain bound to DNA as a monomer from the RCSB database http://www.rcsb.org/pdb (PDB: 1fjl and 2 dms) as the structural template. The primary sequences of CRX from human, and 1fjl and 2 dms (PDB) were aligned by the method of Needleman & Wunsch (Needleman and Wunsch, 1970), and incorporated in the program Look, version 3.5.2 (Lee, 1994; Lee and Subbiah, 1991) for 3-dimensional structure prediction. The wild-type cone-rod homeobox protein CRX-DNA complex was built by the automatic segment matching method in the program Look (Levitt, 1992) followed by 500 cycles of energy minimization. The same program generated and refined the conformation of the protein with K88N mutation using self-consistent ensemble optimization (500 cycles). Analysis of cone-rod dystrophy causing mutant CRX proteins, R41W and R41Q, were performed identically.
Construct preparation
Vector, pcDNA4TM/HisC, containing wild type human CRX cDNA, as well as the luciferase expression construct, bovine Rho130-luc (position -130 to +72 relative ATG start site) in pGL2-basic (Chen, et al., 2002) and NRL expression plasmid in pcDNA4/HisMaxC(Swaroop, et al., 1992) were sequence verified (ACGT, Inc, Wheeling, IL). Mutation c.G264GT was made using QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) with primers: 5’-GTTTGGTTCAATAACCGGAGGGC-3’ forward and 5’-GCCCTCCGGTTATTGAACCAAAC-3’ reserve. Amplification and transformation of mutant plasmid was per Stratagene protocol and sequence verified. The human CRX deletion mutation, c.413delT, was made by Mutagenex, Inc. (Somerset, NJ) and was sequenced verified. The GenBank reference sequence for human CRX (gene ID1406) was NC_000019.9
Cell Culture and Transactivation Assays
Human ARPE-19 cells (Dunn, et al., 1996) (the kind gift of Dr. Kapil Bharti) were grown in Dulbecco’s Modified Eagles Medium (DMEM) supplemented with 10% fetal bovine serum and 1% antimicrobial agent (GIBCO, Invitrogen, Carlsbad, CA) and maintained at 37°C and 5% CO2. Transfection with Lipofectamine 2000 (Invitrogen) followed the manufacture’s recommendation for cell and DNA preparation. Approximately 19 h before transfection, 1.2 × 105 cells were plated into 12 well plate with growth media. One hour before transfection, growth media was removed and replaced with 0.5 mL plain DMEM. Plasmid quantities: 0.5 μg bRho130-luc ±0.5 μg human CRX ±0.125 μg NRL and 5 ng Renilla with 4 μL Lipofectamine 2000 per well were transfected with a total volume per well after transfection of 0.7 mL plain DMEM. Equimolar amounts of plasmid DNA were transfected throughout all experiments by substituting an equimolar amount of plasmid backbone when expression of CRX or NRL was not desired. Cells were incubated with transfection medium for 6 hours before one milliliter of growth media was added. Cells grew for 48 hours before harvesting for luciferase reporter assay.
Luciferase reporter assay
Both firefly and Renilla expression were assayed in 6 μL of harvested cell lysate, using 30 μL of Dual-Luciferase reporter assay system (Promega, Madison, WI) and Modulus Luminometer (Turner BioSystems, Sunnyvale, CA) for signal quantification. Renilla activity was normalized to the firefly luciferase expression of the same sample as an internal control. Assays were repeated four times in duplicate.
Western Blotting
Cell lysates from the luciferase assay were used for protein analysis. CRX and NRL protein expression was assayed on NuPAGE® Bis-Tris Electrophoresis System: Western Blotting (Invitrogen, Inc.). After blocking the Western membrane with 5% fat-free dry milk in 1X TBST, it was incubated first with 1:200 mouse anti-CRX IgG antibody (GeneTex, Inc., Irvine, CA) or 1:500 rabbit anti-NRL antibody(Rehemtulla, et al., 1996), washed, and then incubated with 1:4000 horseradish peroxidase (HRP)-conjugated goat anti-mouse (Thermo Scientific, Waltham, MA) or anti-rabbit IgG (Thermo Scientific).. Signal was detected by SuperSignal ® West Pico Chemiluminescent Substrate (Thermo Scientific) according to manufactures instructions and exposed to X-ray film.
Immunofluorescence microscopy
2.5 × 104 human ARPE-19 cells were plated onto eight well Lab-Tek II Chamber slides (Nalge Nunc International, Rochester, NY) 20 hours before transfection and maintained in growth chamber at 37°C with 5% CO2. Cells were transfected with 0.2 μg human CRX plasmids (wild-type (WT), c.G264T and c.413delT) using 0.6 μL/well FuGENE HD (Roche) according to manufacture’s recommendations. Transfected cells were allowed to incubate for 24 before they were washed three times with 1X TBS, fixed with 3.7% paraformaldehyde (made with 1X TBS) for 10 min at room temperature (RT). Paraformaldehyde was removed and chilled methanol was applied to cells for three minutes at −20°C, then washed three times with 1X TBS. Chambers were removed from the slide, a hydrophobic barrier was drawn around cellular regions, and cells were then blocked with 10% of goat serum for 30 min at RT. Serum was removed and cells were incubated with 1:500 dilution of mouse anti-CRX IgG antibody (GeneTex, Inc., Irvine, CA) for one hour at RT, then washed three times with 1X TBST for 10 min/wash. Cells were then probed with Alexa Fluor® 488 goat anti-mouse IgG antibody for 45 min at RT at a dilution of 1:1000. Both antibodies were diluted in 1% BSA (Invitrogen) made with 1X TBS. Samples were then washed three times with 1X TBST for 10 min/wash, dipped briefly in ddH2O covered with Vectashield Hard Set mounting medium with DAPI (Vector Laboratories, Inc., Burlingame, CA) and coverslipped. Fluorescence and brightfield images were taken with a Zeiss AxioVert 200 microscope with a digital camera connected to a PC running AxioVision 4.6.3 (Carl Zeiss MicroImaging, Thornwood, NY). When making qualitative comparisons of the intensity of immunofluorescence, care was taken to standardize exposure times.
RESULTS
Clinical Descriptions and Genotyping
Patient #1 (c.G264T, p.K88N)
Patient 1, a 2 month old female, presented with nystagmus mimicking opsoclonus, which prompted a work-up for neuroblastoma (negative) and a brain MRI (normal). At age 3 months, she would blink to light, but not fix and follow with either eye. At age 5 months, she underwent an electroretinogram under sedation, which showed absent scotopic and photopic responses. At age 8 months, her nystagmus had dampened slightly. Pupils were sluggishly reactive without paradoxical response. Anterior segment exam was normal. Dilated fundus exam showed bilateral pigment granularity, particular in the maculae (Figure 1A). Her cycloplegic refraction was +6.50 OU. At 2.5 years of age, her visual acuity was 20/1000 using single HOTV optotypes. Fundus exam and refraction were stable. DNA sequencing of the CEP290, AIPL1, CRB1, GUCY2D, RPE65, RDH12 and RPGRIP1 exons and exon-intron boundaries revealed no likely pathological sequence changes. Sequencing of the CRX gene showed a c.G264T mutation, predicted to change lysine 88 to an asparagine in the third helix of the homeodomain (Figure 2). Neither parent possessed this sequence change in peripheral blood.
Figure 1.
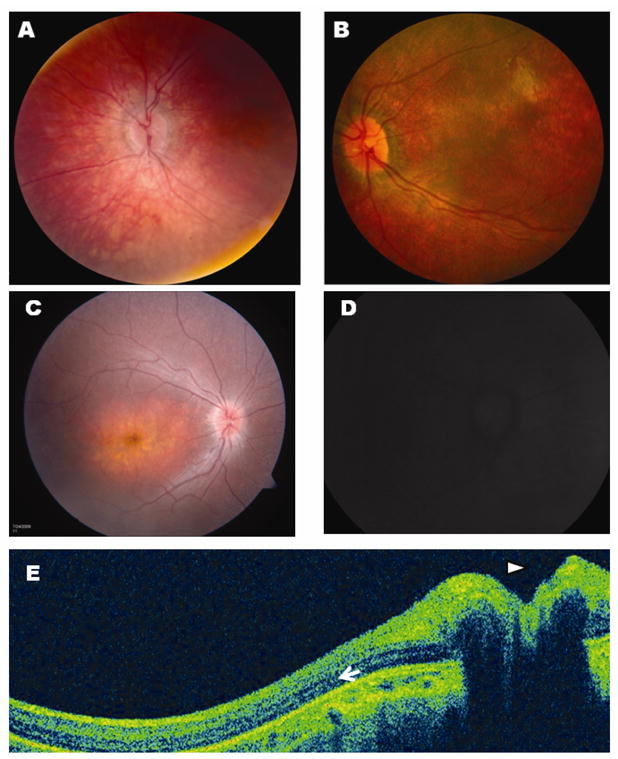
Fundus appearance of Patient #1 (A) and Patient #2 (B) at the time of diagnosis. Both patients had bilateral pigment mottling and unrecordable full-field scotopic and photopic ERGs. Fundus photos were obtainable on Patient #2 at 2.5 years (C), showing progression of macular atrophy and diffuse pigment mottling and retinal arteriorle attenuation. Mild blurring of the optic disc margins, consistent with pseudopapilledema, was noted on exam (C) and on ocular coherence tomography (OCT, E, arrowhead). Fundus autofluorescence (D) showed no abnormal lipofuscin accumulation. OCT also demonstrated significant retinal thinning (approximately 200μm), although a reflectance band consistent with some preservation of outer nuclear layer cells is present (arrow).
Figure 2.

Schematic of CRX protein, indicating the position of the two mutations from this report (boxed), as well as other mutations reported to caused Leber congenital amaurosis (LCA, above the diagram) or cone-rod dystrophy (CORD, below the diagram). Functional domains are noted by shading. Numbers refer to amino acid position.
Patient #2 (c.413delT, p.I138fs48)
Patient 2, a 10 month-old girl, presented with a history of nystagmus since 2–3 months of age. Her father had noted reduced visual behavior in dimly-lit surroundings; in normal lighting, she seemed to see up close, but not at a distance. The child was otherwise healthy and was on- or above-target for developmental milestones. Her parents were both of Ashkenazi Jewish descent and there was no family history of relevant eye disease. On exam, she fixed and followed well with either eye and had no ocular preference. Her binocular Teller acuity was 20/400. She had a high frequency, moderate amplitude nystagmus, but no anomalous head posture or strabismus. Anterior segment exam was normal. Dilated fundoscopic examination showed bilateral attenuation of her retinal arterioles, diffuse macular and extramacular pigment granularity, and early waxy pallor to the optic nerves bilaterally (Figure 1B). An electroretinogram performed under anesthesia showed unrecordable photopic and scotopic responses. Cycloplegic refraction showed mild myopic astigmatism bilaterally. At 16 months of age, her exam was approximately stable, save that she saw 20/600 on binocular Teller acuity testing. At 2.5 years, her binocular visual acuity was 20/700 with crowded, single HOTV optotypes. Fundus examination showed some progression of pigment changes and significant macular atrophy with thinning of the temporal nerve fiber layer (Figure 1C). The optic nerves exhibited mild pseudopapilledema. Fundus autofluorescence shows little retinal signal, consistent with little to no lipofuscin accumulation; the optic disc exhibited mild fluorescence, although the exact reason for this finding is unclear (Figure 1D). Cirrus ocular coherence tomography (OCT) performed as a 5 line raster through the nerve and temporal retina—although limited due to the patient’s age and nystagmus—confirmed elevation of the optic nerve (arrowhead) and diffuse retinal thinning (thickness 200μm)(Figure 1E), including thinning of the temporal nerve fiber layer. However, an outer band of reflectivity seen in all images suggests the presence of some residual cells in the outer nuclear layer (arrow). DNA sequencing of the CEP290, AIPL1, CRB1, GUCY2D, RPE65, RDH12 and RPGRIP1 exons and exon-intron boundaries revealed no likely pathological sequence changes. Sequencing of the CRX gene showed a c.413delT mutation, predicted to frameshift the CRX protein sequence after p.I138, followed by 48 amino acids of unique sequence, shortening the OTX tail of the protein (Figure 2). Neither parent possessed this sequence change.
In silico modeling of wild-type and p.K88N mutant protein
We first sought to understand the potential effects these mutations had on DNA binding using atomic modeling of the wild-type and p.K88N mutant proteins bound to a paired domain DNA sequence. Because of the complexity created by the “random” incorporation of amino acids in the p.I138fs48 mutant after the predicted frameshift and the lack of a good crystallographic template, we were unable to reliably model this protein.
The atomic model of the CRX paired homeodomain include 3 α-helices, H1 (residues 41-61), H2 (residues 67–77), and H3 (residues 80–100) (Figure 3A).
Figure 3.

Molecular model of wild-type (red) and K88N mutant (pink) CRX proteins bound to DNA. The three alpha helices of the CRX homeodomain (H1, H2, and H3) bind to the major groove of DNA via several hydrogen bonds (A). Mutation of lysine 88 destroys one such critical hydrogen bond to DNA (B, arrow), making it likely that the mutant protein has a lower affinity for binding to DNA than wild-type protein. The conservation of lysine 88 across multiple vertebrate species also argues for the functional significance of this residue (C).
Our model predicts that p.K88N CRX mutation affects the binding of the recognition α 3-helix (H3) (Figure 3B) by changing a postively-charged lysine is to a smaller, polar asparagine residue. This change is predicted to break a hydrogen bond necessary to stabilize binding of CRX to the major groove of the duplex DNA consensus oligonucleotide. Indeed, in wild type protein this 3.5 Å H-bond is connecting the side chain nitrogen of lysine 88 and oxygen O4’ of T6 from the duplex DNA nucleotide. Thus, the mutation of lysine 88 to aspargine is predicted to affect the ability of CRX to bind DNA. The functional importance of this residue is supported by its absolute conservation across several vertebrate species (Figure 3C).
Previous reports have implicated mutations in the homeodomain of CRX in cone-rod dystrophies (Figure 2). We therefore sought to determine if our model could predict why these mutations produced a less severe phenotype than p.K88N, which causes LCA. The p.R41W and p.R41Q mutant proteins are altered in the N-terminal fragment of the homeodomain and are structurally less defined compared to other regions. However, the p.E80A and p.R90W are predicted to disturb electrostatic interactions between charged residues, which are energetically weaker than the hydrogen bond disrupted by the p.K88N sequence change. Specifically, the replacement of glutamic acid in position 80 to a small hydrophobic alanine could disturb the predicted electrostatic interaction between side chains of p.E80 and p.R69, residing at a distance of 5.6 Å in the native homeodomain. Located in the third helix of the homeodomain, arginine 90 stabilizes an electrostatic interaction between CRX and the negatively-charged DNA backbone, forming a salt bridge at a distance of 4.68 Å. Replacement of p.R90 to a hydrophobic tryptophan will disrupt this salt bridge and introduce a strong hydrophobic interaction in a cavity created by hydrophobic residues W86, F46, F87, and F58. Our model thus predicts a rough phenotype-genotype correlation in that disruption of a strong, hydrogen bond by the p.K88N sequence change results in a more severe phenotype than disruption of weaker, electrostatic interactions in p.E80A and p.R90W, which cause a less severe phenotype.
Functional Characterization of Mutations
In order to test how these two mutations affected CRX function in a model system, we transiently transfected a human retinal pigment epithelial-like cell like (h-ARPE-19 cells) with a expression constructs of human wild-type, c.G264T, or c.413delT CRX cDNA, along with a reporter construct consisting of the bovine rhodopsin promoter (pBR130-luc) coupled to the luciferase gene. ARPE-19 cells were chosen as a model, as they do not express detectable levels of CRX or NRL protein on Western (thus reducing any complication from endogenous protein), are human in origin, and may have more “neuro-ectoderm-like” properties than peripherally-derived cell lines. The results presented are typical of those obtained in eight independent experiments. Expression of the wild-type CRX protein resulted in a 5-fold activation of the bovine rhodopsin promoter. When compared to the wild-type protein, the c.G264T and c.413delT constructs resulted in 10% (p= 1.5 × 10−11) and 30% (p=3.7 × 10−11) of normal CRX activity (Figure 4A).
Figure 4.

Functional analysis of c.G264T (p.K88N) and c.413delT (p.I138fs48) mutant CRX genes compared to the wild-type gene, both in the presence and the absence of the retinal transcription factor, NRL. (A) Transactivation of wild-type and mutant CRX proteins in the presence and absence of NRL, as measured using a bovine rhodopsin promoter-luciferase construct. (B) Western blot of CRX protein compared to GAPDH, a housekeeping gene, in corresponding transactivation experiments. Presence of NRL is noted in brackets. (C) Western blot of NRL protein compared to GAPDH in corresponding transactivation experiments.
Since the retinal leucine zipper transcription factor, NRL, cooperates with CRX to facilitate rhodopsin gene activation (Chen, et al., 1997), we investigated the effect of co-expression of NRL in our experimental paradigm. As previously reported, expression of NRL by itself results in approximately a 5-fold activation of the bovine rhodopsin promoter and this effect is increased to 30-fold in the presence of CRX (Figure 4A). Co-expression of NRL with the c.G264T mutant CRX resulted in a 2-fold increase in transactivation. However, co-expression of the c.413delT mutation (predicted to affect the WSP and OTX domains) with NRL resulted in 9-fold activation of the rhodopsin promoter, slightly more than we observed with NRL expression alone.
In order to evaluate the effect that these mutations had on CRX and NRL expression in our cell culture system, we performed Western blot analysis on protein lysates from our transactivation experiments. While expression levels of wild-type and Lys88Asn CRX were comparable relative to the housekeeping gene GAPDH, the p.I138fs48 mutant had a higher steady-state level of protein expression (Figure 4B). Co-expression of the wild-type NRL protein did not affect wild-type CRX protein expression, although levels of NRL protein were slightly lower in this co-expression paradigm than when NRL was expressed in the absence of CRX (Figure 4C). In contrast, the steady-state levels of CRX and NRL were both drastically reduced in the presence of the K88N missense mutation. Co-expression of p.I138fs48 CRX with wild-type NRL resulted in a slight reduction of the steady-state levels of the truncated CRX protein and had no significant effect on the level of NRL expression (when compared to the expression of NRL in the absence of CRX). Similar transactivation and protein expression results were obtained independently using HEK293 cells (human endothelial cell line) in more than three experiments.
We next compared the sub-cellular localization of wild-type and mutant CRX proteins in human ARPE-19 cells. As previously reported in other cell types, wild-type CRX is predominantly a nuclear protein (Figure 5) (Fei and Hughes, 2000). The p.K88N protein remains mostly nuclear, but some faint cytoplasmic localization was detected. The p.Ifs48 mutant also was predominantly nuclear, although the percentage of cytoplasmic localization was qualitatively greater than in the p.K88N mutation.
Figure 5.

Immunolocalization of wild-type and mutant CRX proteins in ARPE-19 cells. While wild-type protein is almost exclusively nuclear, both mutants show varying degrees of cytoplasmic localization in addition to a predominant, nuclear pattern.
DISCUSSION
LCA is a severe, inherited retinal dystrophy of childhood characterized by severe visual loss at or near birth, nystagmus, and a pigmentary retinopathy. This genetically heterogeneous disorder renders infants with little or no retinal photoreceptor function. In this report, we characterize two novel, de novo CRX mutations in two unrelated patients that lead to presumed autosomal dominant LCA.
CRX is a homeodomain protein expressed in photoreceptor cells, regulating vital photoreceptor differentiation and integrity (Chen, et al., 1997; Furukawa, et al., 2002; Furukawa, et al., 1997). Mutations have been described in patients with a variety of presentations including autosomal dominant LCA, dominant cone-rod dsytrophy, and retinitis pigmentosa (Freund, et al., 1997; Freund, et al., 1998; Sohocki, et al., 1998). As with all transcription factors, CRX is thought to interact with other proteins such as the neural leucine zipper transcription factor NRL in vivo to activate tissue-specific gene transcription. We demonstrate that different mutations of CRX can not only differentially affect CRX function alone, but that they may also have different effects on transactivation in the presence of a co-activating factor such as NRL. Specifically, the K88N mutant protein not only has little intrinsic transactivation capability, but it also interferes with the independent ability of NRL to activate transcription—an effect akin to a classic “dominant negative effect”, where a mutant protein interferes with the normal function of a co-expressed wild-type protein. Because co-expression of K88N with wild-type NRL results in a reduction in steady state levels of both proteins, we posit that the mutant protein is capable of binding NRL, but is recognized as misfolded by cells and targeted for degradation. In contrast, co-expression of Ile138fs48 CRX with wild-type NRL does not drastically alter the steady state level of either protein. While the ability of Ile138fs48 CRX to transactivate at the rhodopsin promoter is reduced compared to wild-type CRX, this mutant protein does not appear to drastically interfere with the ability of NRL to independently activate transcription. These results agree with previous observations by Mitton et al., who found that CRX-NRL interactions were mostly localized to the homeodomain and the immediate surrounding sequences (Mitton, et al., 2000).
It is difficult to make a clear correlation between genotype and phenotype in our two patients. Although Patient#2 initially had better acuity than Patient#1—at least by preferential looking tests—her subsequent macular atrophy seems to have reduced her acuity by the time a formal measurement could be made. It is theoretically possible that this mutant protein, by allowing NRL to independently regulate retinal gene expression, would lead to a less severe phenotype, at least initially.
Consistent with this hypothesis, Koenekoop et al. have described visual improvement in a child with a similar frameshift mutation in CRX (Koenekoop, et al., 2002). To date, nearly all the reported CRX mutations that cause LCA are expected to frameshift the CRX protein, usually downstream of the homeodomain protein. Missense mutations in the homeodomain and elsewhere have been largely associated with cone-rod dystrophy. As suggested by our molecular modeling, the disruption of a critical hydrogen bond in the p.K88N mutant protein—which is energetically stronger than the electrostatic interactions disrupted by two CORD-causing mutations—may help explain why Patient #1 had more severe phenotype. Consistent with the observations of Fei and Hughes, our point mutation at amino acid 88 causes some of the CRX protein to localize to the cytoplasm, as well as the normal, nuclear localization (Fei and Hughes, 2000). Somewhat surprisingly, the frameshift mutation we describe has similar cytoplasmic and nuclear localization, which differs from these authors’ findings using CRX deletion constructs in HEK-290 cells. The reason for this difference may be a difference in the cell type studied and/or differences caused by the novel C-terminal protein sequence created by the Ile138fs48 mutation.
Recent gene therapy trials of one genetic form of LCA caused by mutations in the RPE65 gene have been very encouraging (Bainbridge, et al., 2008; Cideciyan, et al., 2008; Hauswirth, et al., 2008; Maguire, et al., 2009; Maguire, et al., 2008). Because RPE65 causes autosomal recessive LCA that is likely due to loss of protein function and some outer nuclear layer is preserved anatomically (Jacobson, et al., 2008), it is an excellent choice for a gene replacement strategy in humans. The clinical situation in patients with CRX mutations may be more complex. First, adult patients with cone-rod dystrophy due to CRX mutation have dramatic thinning of the outer nuclear layer, suggesting that the target cells may rapidly degenerate (Jacobson, et al., 1998). Our OCT results confirm that this thinning is present early in life. However, it is somewhat encouraging that a band of reflectivity consistent with some preservation of an outer nuclear layer (although not photoreceptor outer segments) was observed in Patient #2. Pronounced nystagmus and the age of the child limit our ability to make more detailed observations using OCT at this time. A second consideration in designing a therapy for dominant, CRX-mediated LCA is that it is not clear that the mechanism of degeneration is purely due to haploinsufficiency/loss-of-protein function. Our data suggest that some mutations in CRX may have an effect akin to a dominant negative effect on other retinal transcription factor genes, such as NRL. Chen et al., in their analysis of multiple CRX point mutations and frameshift mutations found no specific correlation between residual activity of CRX and disease severity (Chen, et al., 2002). Swaroop et al. have reported a family with a homeodomain missense mutation, R90W, that causes a relatively mild, adult-onset cone dysfunction syndrome when heterozygous and a severe, LCA phenotype when homozygous(Swaroop, et al., 1999). In fact, Silva et al. have reported a frameshift mutation in CRX upstream of the homeodomain region that is found in both normal subjects and patients with LCA, suggesting that haploinsufficiency for CRX function isn’t necessarily pathogenic (Silva, et al., 2000). In order to clarify the pathogenesis of dominant CRX mutations in vivo, “knock-in” of specific CRX mutations in mouse models may need to be compared with the CRX knockout mouse. If dominant-negative-like effects or other, complex mechanisms are responsible for disease in humans, specific knock-down of the mutant CRX allele may be more beneficial than replacing CRX function.
Acknowledgments
We would like to thank NEI ophthalmic photographers, Denise Cunningham, Mel Palmer, Mike Bono, and Alicia Zetina for their expert assistance. This research was sponsored by the intramural program of the National Eye Institute, National Institutes of Health, U.S. Department of Health and Human Services. Contract grant sponsor: Intramural Program, NEI. We would like to thank the staff of the Carver Molecular Diagnostic Laboratory at the University of Iowa for excellent technical service.
Footnotes
Communicated by Henrik Dahl
References
- Ahmed E, Loewenstein J. Leber congenital amaurosis: disease, genetics and therapy. Semin Ophthalmol. 2008;23(1):39–43. doi: 10.1080/08820530701745215. [DOI] [PubMed] [Google Scholar]
- Bainbridge JW, Smith AJ, Barker SS, Robbie S, Henderson R, Balaggan K, Viswanathan A, Holder GE, Stockman A, Tyler N, et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med. 2008;358(21):2231–9. doi: 10.1056/NEJMoa0802268. [DOI] [PubMed] [Google Scholar]
- Chen S, Wang QL, Nie Z, Sun H, Lennon G, Copeland NG, Gilbert DJ, Jenkins NA, Zack DJ. Crx, a novel Otx-like paired-homeodomain protein, binds to and transactivates photoreceptor cell-specific genes. Neuron. 1997;19(5):1017–30. doi: 10.1016/s0896-6273(00)80394-3. [DOI] [PubMed] [Google Scholar]
- Chen S, Wang QL, Xu S, Liu I, Li LY, Wang Y, Zack DJ. Functional analysis of cone-rod homeobox (CRX) mutations associated with retinal dystrophy. Hum Mol Genet. 2002;11(8):873–84. doi: 10.1093/hmg/11.8.873. [DOI] [PubMed] [Google Scholar]
- Cideciyan AV, Aleman TS, Boye SL, Schwartz SB, Kaushal S, Roman AJ, Pang JJ, Sumaroka A, Windsor EA, Wilson JM, et al. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc Natl Acad Sci U S A. 2008;105(39):15112–7. doi: 10.1073/pnas.0807027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Hollander AI, Roepman R, Koenekoop RK, Cremers FP. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res. 2008;27(4):391–419. doi: 10.1016/j.preteyeres.2008.05.003. [DOI] [PubMed] [Google Scholar]
- Dunn KC, Aotaki-Keen AE, Putkey FR, Hjelmeland LM. ARPE-19, a human retinal pigment epithelial cell line with differentiated properties. Exp Eye Res. 1996;62(2):155–69. doi: 10.1006/exer.1996.0020. [DOI] [PubMed] [Google Scholar]
- Farjo Q, Jackson AU, Xu J, Gryzenia M, Skolnick C, Agarwal N, Swaroop A. Molecular characterization of the murine neural retina leucine zipper gene, Nrl. Genomics. 1993;18(2):216–22. doi: 10.1006/geno.1993.1458. [DOI] [PubMed] [Google Scholar]
- Fei Y, Hughes TE. Nuclear trafficking of photoreceptor protein crx: the targeting sequence and pathologic implications. Invest Ophthalmol Vis Sci. 2000;41(10):2849–56. [PubMed] [Google Scholar]
- Freund CL, Gregory-Evans CY, Furukawa T, Papaioannou M, Looser J, Ploder L, Bellingham J, Ng D, Herbrick JA, Duncan A, et al. Cone-rod dystrophy due to mutations in a novel photoreceptor-specific homeobox gene (CRX) essential for maintenance of the photoreceptor. Cell. 1997;91(4):543–53. doi: 10.1016/s0092-8674(00)80440-7. [DOI] [PubMed] [Google Scholar]
- Freund CL, Wang QL, Chen S, Muskat BL, Wiles CD, Sheffield VC, Jacobson SG, McInnes RR, Zack DJ, Stone EM. De novo mutations in the CRX homeobox gene associated with Leber congenital amaurosis. Nat Genet. 1998;18(4):311–2. doi: 10.1038/ng0498-311. [DOI] [PubMed] [Google Scholar]
- Furukawa A, Koike C, Lippincott P, Cepko CL, Furukawa T. The mouse Crx 5’-upstream transgene sequence directs cell-specific and developmentally regulated expression in retinal photoreceptor cells. J Neurosci. 2002;22(5):1640–7. doi: 10.1523/JNEUROSCI.22-05-01640.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa T, Morrow EM, Cepko CL. Crx, a novel otx-like homeobox gene, shows photoreceptor-specific expression and regulates photoreceptor differentiation. Cell. 1997;91(4):531–41. doi: 10.1016/s0092-8674(00)80439-0. [DOI] [PubMed] [Google Scholar]
- Hauswirth WW, Aleman TS, Kaushal S, Cideciyan AV, Schwartz SB, Wang L, Conlon TJ, Boye SL, Flotte TR, Byrne BJ, et al. Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum Gene Ther. 2008;19(10):979–90. doi: 10.1089/hum.2008.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson SG, Cideciyan AV, Aleman TS, Sumaroka A, Windsor EA, Schwartz SB, Heon E, Stone EM. Photoreceptor layer topography in children with leber congenital amaurosis caused by RPE65 mutations. Invest Ophthalmol Vis Sci. 2008;49(10):4573–7. doi: 10.1167/iovs.08-2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson SG, Cideciyan AV, Huang Y, Hanna DB, Freund CL, Affatigato LM, Carr RE, Zack DJ, Stone EM, McInnes RR. Retinal degenerations with truncation mutations in the cone-rod homeobox (CRX) gene. Invest Ophthalmol Vis Sci. 1998;39(12):2417–26. [PubMed] [Google Scholar]
- Kimura A, Singh D, Wawrousek EF, Kikuchi M, Nakamura M, Shinohara T. Both PCE-1/RX and OTX/CRX interactions are necessary for photoreceptor-specific gene expression. J Biol Chem. 2000;275(2):1152–60. doi: 10.1074/jbc.275.2.1152. [DOI] [PubMed] [Google Scholar]
- Koenekoop RK, Loyer M, Dembinska O, Beneish R. Visual improvement in Leber congenital amaurosis and the CRX genotype. Ophthalmic Genet. 2002;23(1):49–59. doi: 10.1076/opge.23.1.49.2200. [DOI] [PubMed] [Google Scholar]
- Kumar R, Chen S, Scheurer D, Wang QL, Duh E, Sung CH, Rehemtulla A, Swaroop A, Adler R, Zack DJ. The bZIP transcription factor Nrl stimulates rhodopsin promoter activity in primary retinal cell cultures. J Biol Chem. 1996;271(47):29612–8. doi: 10.1074/jbc.271.47.29612. [DOI] [PubMed] [Google Scholar]
- Lee C. Predicting protein mutant energetics by self-consistent ensemble optimization. J Mol Biol. 1994;236(3):918–39. doi: 10.1006/jmbi.1994.1198. [DOI] [PubMed] [Google Scholar]
- Lee C, Subbiah S. Prediction of protein side-chain conformation by packing optimization. J Mol Biol. 1991;217(2):373–88. doi: 10.1016/0022-2836(91)90550-p. [DOI] [PubMed] [Google Scholar]
- Levitt M. Accurate modeling of protein conformation by automatic segment matching. J Mol Biol. 1992;226(2):507–33. doi: 10.1016/0022-2836(92)90964-l. [DOI] [PubMed] [Google Scholar]
- Maguire AM, High KA, Auricchio A, Wright JF, Pierce EA, Testa F, Mingozzi F, Bennicelli JL, Ying GS, Rossi S, et al. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: a phase 1 dose-escalation trial. Lancet. 2009 doi: 10.1016/S0140-6736(09)61836-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire AM, Simonelli F, Pierce EA, Pugh EN, Jr, Mingozzi F, Bennicelli J, Banfi S, Marshall KA, Testa F, Surace EM, et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med. 2008;358(21):2240–8. doi: 10.1056/NEJMoa0802315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mears AJ, Kondo M, Swain PK, Takada Y, Bush RA, Saunders TL, Sieving PA, Swaroop A. Nrl is required for rod photoreceptor development. Nat Genet. 2001;29(4):447–52. doi: 10.1038/ng774. [DOI] [PubMed] [Google Scholar]
- Mitton KP, Swain PK, Chen S, Xu S, Zack DJ, Swaroop A. The leucine zipper of NRL interacts with the CRX homeodomain. A possible mechanism of transcriptional synergy in rhodopsin regulation. J Biol Chem. 2000;275(38):29794–9. doi: 10.1074/jbc.M003658200. [DOI] [PubMed] [Google Scholar]
- Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 1970;48(3):443–53. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- Rehemtulla A, Warwar R, Kumar R, Ji X, Zack DJ, Swaroop A. The basic motif-leucine zipper transcription factor Nrl can positively regulate rhodopsin gene expression. Proc Natl Acad Sci U S A. 1996;93(1):191–5. doi: 10.1073/pnas.93.1.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva E, Yang JM, Li Y, Dharmaraj S, Sundin OH, Maumenee IH. A CRX null mutation is associated with both Leber congenital amaurosis and a normal ocular phenotype. Invest Ophthalmol Vis Sci. 2000;41(8):2076–9. [PubMed] [Google Scholar]
- Sohocki MM, Sullivan LS, Mintz-Hittner HA, Birch D, Heckenlively JR, Freund CL, McInnes RR, Daiger SP. A range of clinical phenotypes associated with mutations in CRX, a photoreceptor transcription-factor gene. Am J Hum Genet. 1998;63(5):1307–15. doi: 10.1086/302101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaroop A, Wang QL, Wu W, Cook J, Coats C, Xu S, Chen S, Zack DJ, Sieving PA. Leber congenital amaurosis caused by a homozygous mutation (R90W) in the homeodomain of the retinal transcription factor CRX: direct evidence for the involvement of CRX in the development of photoreceptor function. Hum Mol Genet. 1999;8(2):299–305. doi: 10.1093/hmg/8.2.299. [DOI] [PubMed] [Google Scholar]
- Swaroop A, Xu JZ, Pawar H, Jackson A, Skolnick C, Agarwal N. A conserved retina-specific gene encodes a basic motif/leucine zipper domain. Proc Natl Acad Sci U S A. 1992;89(1):266–70. doi: 10.1073/pnas.89.1.266. [DOI] [PMC free article] [PubMed] [Google Scholar]