

Abstract
Background
Ethanol consumption during pregnancy increases the risk of early pregnancy loss and causes intrauterine growth restriction. We previously showed that chronic gestational exposure to ethanol impairs placentation, and that this effect is associated with inhibition of insulin and insulin growth factor signaling. Since ethanol also causes oxidative stress and DNA damage, we extended our investigations to assess the role of these pathological processes on placentation and placental gene expression.
Methods
Pregnant Long Evans rats were pair-fed liquid diets containing 0% or 24% ethanol by caloric content. Placentas harvested on gestation day 16 were used to examine DNA damage, lipid peroxidation, apoptosis, mitochondrial gene/protein and hormonal gene expression in relation to ethanol exposure.
Results
Gestational exposure to ethanol increased fetal resorption, and trophoblast apoptosis/necrosis, oxidative stress, DNA damage, and lipid peroxidation. These adverse effects of ethanol were associated with increased expression of pro-apoptotic (Bax and Bak) and reduced levels of the anti-apoptotic Bcl-2 protein. In addition, increased trophoblast apoptosis proneness was associated with p53-independent activation of p21, reduced mitochondrial gene and protein expression, and dysregulated expression of prolactin (PRL) family hormones that are required for implantation and pregnancy-related adaptations.
Conclusions
Chronic gestational exposure to ethanol increases fetal demise due to impaired survival and mitochondrial function, increased oxidative stress, DNA damage and lipid peroxidation, and dysregulated expression of prolactin family hormones in placental trophoblasts.
Keywords: Placenta, Fetal Resorption, Fetal Alcohol Spectrum Disorders, Oxidative Stress, Mitochondrial Dysfunction, Prolactin Family Hormones
Ethanol exposure during pregnancy is the most common preventable cause of birth defects and developmental disabilities in the United States. Different degrees, durations, and timing of prenatal ethanol exposure produce a range of structural malformations, and behavioral and neuro-cognitive deficits, that collectively are termed Fetal Alcohol Spectrum Disorders (FASD) (Barr and Streissguth, 2001). Although intrauterine growth restriction (IUGR) is a key feature of FASD, its pathogenesis is still under investigation. Recently, using an established experimental rodent model of FASD, we determined that ethanol-impaired placentation is an important mediator of ethanol-induced IUGR (Gundogan et al., 2008).
Invasion of trophoblast into the uterine wall is an important feature of hemochorial placentation. The motile and invasive subtype of trophoblastic cells, extravillous cytotrophrophoblast (evCTB) in humans and invasive trophoblast cells in rodents, play crucial roles in implantation/placentation. The evCTB invade the endometrium, decidua and upper third of myometrium, and in the process, transform small muscular spiral arteries into flaccid vessels with no muscularis or elastic lamina (Bischof and Irminger-Finger, 2005). This physiologic conversion of maternal spiral arteries into thin-walled vessels that supply the expanded blood flow throughout pregnancy is required for successful establishment of the maternal-fetal circulation (Pijnenborg et al., 1981). Perturbations in this process lead to disease states, e.g., pre-eclampsia (Moffett and Loke, 2006).
Having a hemochorial type placenta, the laboratory rat is a preferred model for studying placental development. Most importantly, in rats the trophoblast invasion involves mesometrial triangle in addition to decidua, which corresponds to placental bed in humans. The role of invasive trophoblast cells in rat spiral artery remodeling has been found to be similar to human pregnancy (Caluwaerts et al., 2005).
Insulin-like growth factors (IGF) regulate placentation due to their stimulatory effects on invasive trophoblast cells in both humans and rodents (DeChiara et al., 1990; Han and Carter, 2000; Laviola et al., 2005). Mechanistically, IGFs stimulate expression of aspartyl-(asparaginyl) β-hydroxylase (AAH), which has an important role in regulating cell motility and invasion regardless of species (Carter et al., 2008; Lahousse et al., 2006; de la Monte et al., 2009), and particularly in human trophoblastic cells (Gundogan et al., 2007). In rats, chronic gestational exposure to ethanol causes IGF resistance, manifested by inhibition of placental IGF signaling through insulin receptor substrate molecules (Gundogan et al., 2008). Consequently, AAH expression is impaired, particularly in the mesometrial triangle (Gundogan et al., 2008). These observations linked ethanol-impaired physiologic conversion of placental spiral arteries to reduced levels of AAH expression in invasive trophoblast, impairing functions required for rat placentation (Gundogan et al., 2008).
A second major factor likely mediating the adverse effects of chronic ethanol exposure is increased oxidative stress, as previously demonstrated for liver and brain (Chu et al., 2007; Hoek and Pastorino, 2004). In this regard, ethanol-induced toxicity and cell death were found to be associated with increased mitochondrial dysfunction, oxidative stress, and DNA damage in brain (Chu et al., 2007; Lee et al., 2005). In addition, in vitro experiments demonstrated that transient ethanol exposure causes oxidative stress in human placental villous tissue (Kay et al., 2000). Indices of oxidative stress were localized in both trophoblasts and villous stroma (Kay et al., 2006).
Although pregnancy is referred to as “a state of oxidative stress” (Wisdom et al., 1991), certain pathological conditions, such as pre-eclampsia, further increase placental oxidative stress and result in early pregnancy loss, IUGR and impaired placentation, similar to the findings in FASD (Myatt and Cui, 2004; Poston and Raijmakers, 2004). Placental mitochondria are a major source of oxidative stress in pre-eclampsia (Wang and Walsh, 1998). We hypothesize that, in addition to IGF resistance, ethanol-impaired rat placentation may be mediated by increased oxidative stress, with attendant increased DNA damage and lipid peroxidation. Herein, using our chronic in utero ethanol exposure model, we characterize ethanol-induced mitochondrial dysfunction, DNA damage and increased apoptosis, and link these abnormalities to dysregulated hormonal gene expression in placenta.
METHODS
In Vivo Ethanol Exposure Model
Pregnant Long Evans rats were pair-fed liquid diets (BioServ, Frenchtown, NJ) in which ethanol comprised 0% (N = 5) or 24% (N = 9) of the caloric content (Xu et al., 2003). The diets were initiated on gestation day (GD) 6, and continued throughout pregnancy. Gestation day 0 (GD0) was designated as the day in which sperm was first detected in vaginal smears (Vercruysse et al., 2006). The 24% ethanol-containing diet typically produces blood alcohol concentrations of 31.3 ± 8.3 mM (Soscia et al., 2006). The rats were monitored daily to ensure equivalent food consumption and maintenance of body weight. Based on preliminary experiments, we determined that the oxidative-stress related changes are more prominent within the placenta, especially junctional zone, relative to mesometrial triangle. Therefore, we decided to analyze the chorioallantoic placenta considering that the junctional zone is well-established during the rat pregnancy days 13 to 21. Rats were killed on GD16 to harvest placentas and fetuses. We chose day 16 because at this point in gestation, the placenta is well-established and trophoblast cell invasion into the decidua and spiral arteries has already initiated. The implantation sites were dissected and placental tissue with underlying mesometrial triangle were weighed and immersion fixed in Histochoice (Amresco Corp., Solon, OH), embedded in paraffin, and used to generate histological sections (Gundogan et al., 2008). Maintaining tissue integrity at the maternal–fetal interface enabled better analysis of the junctional zone and decidua basalis. Alternatively, placental tissue separated from the mesometrial triangle (Gundogan et al., 2008), was snap frozen in a dry ice-methanol bath and stored at −80°C for later mRNA and protein studies. The resorbed placental sites were excluded from histochemical and molecular analyses due to extensive necrosis. Experiments were repeated 3 times.
Immunohistochemical Staining
Histological sections of placenta (5 μm thick) were immunostained with monoclonal antibodies to 8-hydroxy-deoxyguanosine (8-OHdG) or 4-hydroxy-2,3-nonenal (HNE) to detect DNA damage or lipid peroxidation as previously described (Chu et al., 2007). In brief, deparaffinized, rehydrated tissue sections were permeabilized by a 20-minute incubation in 0.1 mg/mL saponin in phosphate-buffered saline (10 mM sodium phosphate, 0.9% NaCl, pH 7.4; PBS). Endogenous peroxidase activity was quenched with 0.6% hydrogen peroxide in methanol (30 minutes). Nonspecific binding sites were blocked with SuperBlock-TBS (Pierce Chemical Co., Rockford, IL). The sections were incubated overnight at 4°C with 1 to 2 μg/ml of primary antibody. Immunoreactivity was detected using biotinylated secondary antibody, avidin–biotin horseradish peroxidase complex (ABC) reagents, and diaminobenzidine as the chromogen (Vector Laboratories, Burlingame, CA) (Soscia et al., 2006). The sections were lightly counterstained with hematoxylin. The slides were examined under code to assess the presence, distribution and intensity of immunoreactivity by light microscopy.
Enzyme-Linked Immunosorbent Assay
We measured immunoreactivity in placental tissue homogenates by direct binding ELISA. The objectives were to assess levels of oxidative stress, DNA damage, and apoptosis proneness. The proteins assayed and their functions are listed in Table 1. Fresh frozen tissues were homogenized in radio-immunoprecipitation assay (RIPA) buffer (50 mM Tris–HCl, pH 7.5, 1%NP-40, 0.25%Na-deoxycholate, 150 mM NaCl, 1 mM EDTA, 2 mM EGTA) containing protease (1 mM PMSF, 0.1 mM TPCK, 1 μg/ml pepstatin A, 0.5 μg/ml leupeptin, 1 mM NaF, 1 mM Na4P2O7) and phosphatase (2 mM Na3VO4) inhibitors. The supernatant fractions obtained after centrifuging the samples at 12,000×g for 15 minutes at 4°C were used for ELISAs. Protein concentrations were determined with the bicinchoninic acid (BCA) assay (Pierce, Rockford, IL).
Table 1.
Summary of the Genes/Proteins Assayed in the Study and Their Functions
| Protein/gene abbreviation | Full name | Function |
|---|---|---|
| HNE | 4-Hydroxy-2, 3-nonenal | Caspase activation Cell death Induction of TGF-β1 Disruption of neuronal calcium homeostasis |
| 8-OHdG | 8-Hydroxy-deoxyguanosine | Cell cycle arrest Cell death |
| p53 | Protein 53 Tumor suppressor protein |
Cell cycle arrest Apoptosis Senescence DNA repair |
| p21 | Cyclin-dependent kinase inhibitor | Cell cycle arrest |
| Bcl-2 | B-cell CLL/Lymphoma 2 | Inhibits apoptosis |
| Bax | Bcl-2 associated X-protein | Promotes apoptosis |
| Bak | Bcl-2 antogonist killer | Promotes apoptosis |
| Mdm2 | Murine double minute 2 | Negative regulator of p53 tumor suppressor |
| Complex I | NADH ubiquinol dehydrogenase | Accepts electrons from NADH, transfers them to ubiquinone (Coenzyme Q10), and uses the energy released to pump protons across the mitochondria inner membrane |
| Complex II | Succinate dehydrogenase | Catalyzes the electron transport from succinate to ubiquinone |
| Complex III | Ubiquinol cytochrome c oxidoreductase | Catalyzes the transfer of electrons from ubiquinol (reduced Coenzyme Q10) to cytochrome c and utilizes the energy to translocate protons from inside the mitochondrial inner membrane to outside |
| Complex IV | Cytochrome c oxidoreductase | Collects electrons from reduced cytochrome c and transfers them to oxygen to produce water; the energy released is used to transport protons across the mitochondrial inner membrane |
| Complex V | ATP synthase | Catalyzes the synthesis of adenosine triphosphate (ATP) with energy derived from electrochemical proton gradients |
| Prl3d1 (PL-I) | Prolactin family 3, subfamily d, member 1 (placental lactogen 1) | Mammary gland development Maintenance of corpus luteum integrity Progesterone and relaxin biosynthesis |
| Prl3b1 (PL-II) | Prolactin family 3, subfamily b, member 1 (placental lactogen 2) | Mammary gland development Maintanence of corpus luteum integrity Progesterone and relaxin biosynthesis |
| PRL-R | Prolactin receptor | Mammary gland development Progesterone production Lactation Fertility Implantation Activation of Jak kinase, STAT1, −3, −5, Ras/Raf/MAP kinase pathways |
Samples containing ~50 ng of protein in 100 μl Tris-buffered saline (TBS) were adsorbed to the flat surfaces of opaque white polystyrene 96-well plates (Packard Optiplate-96, Perkin-Elmer, Boston, MA) by overnight incubation at 4°C. Nonspecific binding sites were blocked by a 3-hour, room temperature incubation with 1% bovine serum albumin dissolved in TBS + 0.05% Tween 20 + 2% bovine serum albumin (TBST-BSA). Samples were incubated with primary antibody (0.5 to 1.0 μg/ml) for 1 hour at 37°C. Antibody binding was detected with horseradish peroxidase-conjugated secondary anti-body (1:10,000; Pierce, Rockford, IL) and the Amplex UltraRed soluble fluorophore (Molecular Probes, Eugene, OR) (Cohen et al., 2007). Immunoreactivity was measured (Ex 530/Em 590) in a SpectraMax M5 microplate reader (Molecular Devices Corp., Sunnyvale, CA). The results were normalized to protein content in each well (quantified using the NanoOrange® Protein Quantitation Kit (Molecular Probes, Eugene, OR). Negative control reactions included omission of specific protein, primary antibody, secondary antibody, or both antibodies. Between steps, the wells were washed 3 times with TBST using a Nunc Immunowash apparatus (Nunc, Rochester, NY).
Quantitative Reverse Transcriptase Polymerase Chain Reaction (qRT-PCR) Analyses
We used qRT-PCR to measure mRNA levels of genes encoding the 5 major mitochondrial complex proteins, apoptosis/cell survival mechanisms, and prolactin (PRL) family polypeptides and receptors. Total RNA was isolated from frozen tissue using TRIzol (Invitrogen, Carlsbad, CA). RNA concentration and purity were determined from the absorbances measured at 260 and 280 nm. Samples containing 2 μg RNA were reverse transcribed with the AMV First Strand cDNA synthesis kit (Roche, Basel, Switzerland) and random oligodeoxynucleotide primers. PCR amplifications were performed in 20 μl reactions containing cDNA generated from 2.5 ng of original RNA template, 300 nM each of gene specific forward and reverse primer pairs (Table 2), and 10 μl of 2× QuantiTect SYBR Green PCR Mix (Qiagen Inc, Valencia, CA). The amplified signals were detected continuously with the Mastercycler ep realplex detection system (Eppendorf North America, Westbury, NY). The amplification protocol used was as follows: initial 15-minutes denaturation and enzyme activation at 95°C, 45 cycles of 95°C × 30 seconds, 60°C × 30 seconds, and 72°C × 30 seconds. Annealing temperatures were optimized using the temperature gradient program provided with the Mastercycler ep realplex software. Experiments were performed in triplicate.
Table 2.
Primer Pair Sequences for Real-Time Quantitative RT-PCR*
| Primer | Sequence (5′ → 3′) | Position (mRNA) | Amplicon size (bp) | |
|---|---|---|---|---|
| 18S | For Rev |
GGA CAC GGA CAG GAT TGA CA ACC CAC GGA ATC GAG AAA GA |
1278 1327 |
50 |
| Complex I | For Rev |
CCT GGA ACG GTC ACA CAT TTG CAT ACA GAA CTC GGT CCC TGA TTC |
198 282 |
85 |
| Complex II-A | For Rev |
CAG CAG AAG AAG CCA TTT GCG AGC ACA GTC AGC CTC ATT CAA GG |
1854 1982 |
129 |
| Complex II-C | For Rev |
GGA CAC TTC TGT TCA TTG GTC AGC CCA TTT CTC TTC TCT CAG TCA CGC |
843 913 |
71 |
| Complex II-D | For Rev |
GCA CTT TGA TTG TAT GCC TCC TTG C GCC CAC CAA CGG ATT TAT TCT TC |
507 596 |
90 |
| Compex III | For Rev |
CAA AGA ACC GTC CTG GCA ATG CCT TGA TGA GGT AAG CAG TGT GC |
306 407 |
102 |
| Complex IV | For Rev |
GAG AGC CAT TTC TAC TTC GGT GTG CCA CTC ATT GGT GCC CTT GTT C |
125 389 |
265 |
| Complex V | For Rev |
AGA CTG GTT TTG GGA GGT AGC CC ATT TTG ATT GGT GCC CCC G |
301 423 |
123 |
| Prolactin 3d1 | For Rev |
ACA CTT GGT CGT CTG CAG TG GAC CAG GCA GGG TAG TCA AA |
408 587 |
180 |
| Prolactin 3b1 | For Rev |
CAT AGT GGC AGC AGT GGC TA CAT GCA CCG ATA CAG GAC AC |
418 649 |
232 |
| Prolactin receptor | For Rev |
TCG CAA CAA ATG CTG AGA AC AGG GAT CAA GAT GGC AGA TG |
1180 1365 |
186 |
| P53 | For Rev |
CCA TCC TTA CCA TCA TCA CGC TG GGC ACA AAC ACG AAC CTC AAA G |
766 845 |
80 |
| P21 | For Rev |
CAC TTG GAC TCG CTG AGC AAA G CGC TGA TAA TCC CAC TTG AGG TC |
1177 1235 |
58 |
For: forward primer; Rev: reverse primer; Position: initial nucleotide for primer binding; bp: base pair size of amplicon.
PCR primers were designed using Mac Vector 7.0 software (Accelrys Inc., Oxford Molecular Ltd., Oxford, England) and primer-target specificity was checked using NCBI-BLASTn (Basic Local Alignment Search Tool–nucleotide) searcHES. Ribosomal 18S RNA measured in parallel reactions was used to calculate relative abundance of each mRNA transcript (Xu et al., 2003; Yeon et al., 2003). Results were normalized to 18S because 18S is highly abundant and the levels were essentially invariant among samples, whereas housekeeping genes were modulated by disease state. Control studies included analysis of: (1) template-free reactions; (2) RNA that had not been reverse transcribed; (3) RNA samples that were pre-treated with DNAse I; (4) samples treated with RNAse A prior to reverse transcriptase reaction; and (5) genomic DNA.
Statistical Analyses
Data are depicted as mean ± SEM. in the graphs. Inter-group comparisons were made using Student’s t-tests or Mann–Whitney tests. Statistical analyses were performed using GraphPad Prism 5 software (GraphPad Software, Inc, La Jolla, CA). Significant p-values are indicated over the graphs.
Source of Reagents
Antibodies to 8-OHdG and HNE were purchased from Chemicon Inc. (Temecula, CA). Antibodies to p53, p21, mdm2, Bcl-2, Bax and Bak were obtained from Santa Cruz (Santa Cruz, CA). Antibodies to complex I, II, III, IV, and V were from Molecular Probes, Invitrogen (Eugene, OR). All the antibodies were commercially characterized and demonstrated to specifically detect the corresponding rat antigens. Reagents for immunohistochemical analysis were purchased from Vector Laboratories (Burlingame, CA). QuantiTect SYBR Green PCR Mix was obtained from Qiagen Inc. (Valencia, CA). All other fine chemical and reagents were purchased from Calbiochem-EMD Biosciences (La Jolla, CA), Pierce Biotechnology Inc. (Rockford, IL), or Sigma-Aldrich (St. Louis, MO).
RESULTS
Chronic Gestational Exposure to Ethanol Increases Fetal Resorption
We used an established model of FASD in which pregnant Long-Evans rats were fed isocaloric liquid control or ethanol-containing (24% by caloric content) diets from GD6 through termination (GD16) (Xu et al., 2003). Chow-fed controls were studied simultaneously. In the chow-fed control group (N = 2) litter sizes were 12 and 15, consistent with our findings. In the liquid diet control group (N = 5), litter sizes ranged from 10 to 16 (median: 14; mean ± SD: 13.8 ± 2.3), whereas in the ethanol-exposed group (N = 9), litter sizes ranged from 5-to-15 (median: 10; mean ± SD: 10.2 ± 3.0; p = .05). In addition, in 5 of the ethanol-fed dams, between 1 and 7 additional implantation sites that lacked fetal tissue and therefore represented fetal resorption sites were identified. Similar sites were not detected in either control group.
Chronic Gestational Exposure to Ethanol Causes Placental Oxidative Stress
We examined H&E stained sections of full-thickness placentas from dams fed with chow (N = 4), control liquid diet (N = 4), or ethanol-containing liquid diet (N = 5). Placentas from chow and control liquid diet fed dams had similar mean weights, including underlying mesometrial triangle (Chow: 0.43 ± 0.02 g; Control liquid diet: 0.42 ± 0.01 g), and they were histologically indistinguishable. In contrast, the mean weight of placentas from ethanol-exposed dams (0.55 ± 0.02 g; p = 4.2E−09) was significantly higher than in the pair-fed liquid diet control group. Histological studies revealed that the excess weight was due to marked congestion within the labyrinthine region of ethanol-exposed placentas (Fig. 2C,D) compared with control (Fig. 1A,B). In addition, ethanol-exposed placentas had conspicuous foci of necrosis in the spongiotrophoblast layer and decidua basalis (Fig. 2C), primarily in the most central regions of placentas, and both intracellular and extracellular amorphous hyaline deposits within the decidua basalis (Fig. 2D). The control placentas were devoid of these lesions (Fig. 2A,B).
Fig. 2.
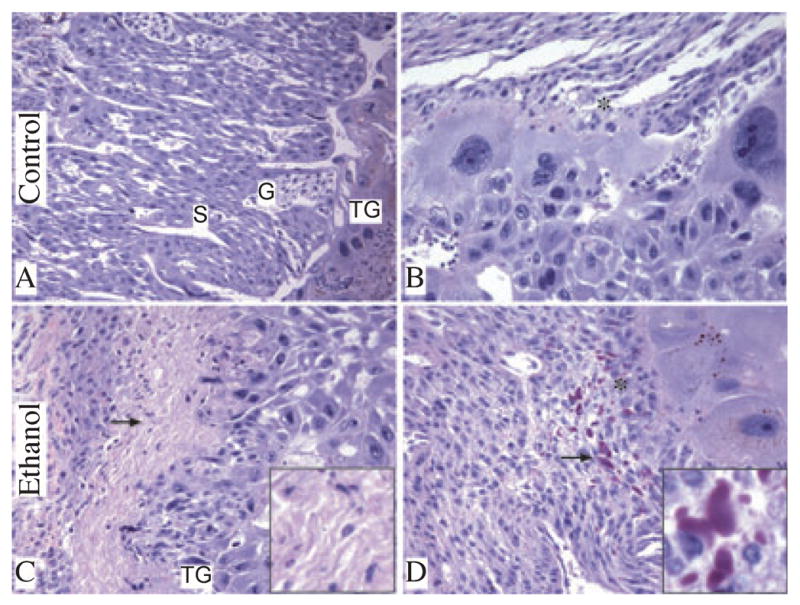
Chronic gestational exposure to ethanol alters placental morphology. Junctional zone composed of spongiotrophoblast [S], glycogen cells [G], trophoblast giant cells [TG] (A) and decidua [*] (B) is demonstrated in control placenta. Foci of necrosis in junctional zone (arrow, C) and intracellular and extracellular amorphous hyaline deposits (arrow, D) were noted within decidua basalis of placentas from ethanol-exposed dams. Insets in panels C and D show higher magnification images of necrosis and amorphous hyaline deposits, respectively (original magnification X800). (H&E stained sections, original magnification X20 [A, C] and X400 [B, D]).
Fig. 1.
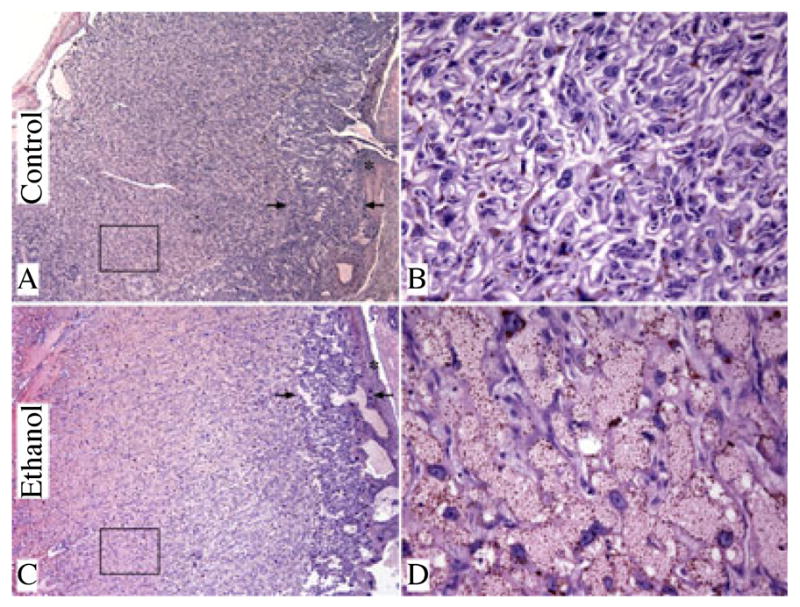
Prominent congestion in the labyrinthine region of placenta in association with chronic gestational ethanol exposure. Full-thickness images of placentas from control (A, B) and ethanol-exposed (C, D) dams (H&E stained sections, original magnification X4 [A, C] and X400 [B, D]). The arrows delineate the borders of the junctional zone. The boxed areas in A and C are the labyrinthine regions depicted in high magnification images (B, D). The decidua is marked with (*) in low magnification images.
Immunohistochemical staining revealed low levels and sparsely distributed HNE immunoreactivity only in labyrinthine trophoblasts. There was no identifiable HNE (Fig. 3A) or 8-OHdG (Fig. 3C) immunoreactivity in the junctional zone of control placentas. In contrast, ethanol-exposed placentas had strikingly increased HNE (Fig. 3B) and 8-OHdG (Fig. 3D) immunoreactivity in trophoblastic cells within the labyrinthine and spongiotrophoblast cells in the junctional zone. HNE and 8-OHdG immunoreactivities were mainly distributed in the perinuclear zones of trophoblastic cells. ELISA studies confirmed these in situ observations, demonstrating significantly higher levels of HNE immunoreactivity in ethanol-exposed (582.8 ± 21.7 FLU/μg protein; N = 6) relative to control (510.4 ± 6.5 FLU/μg protein, p = .002; N = 6) placentas.
Fig. 3.

Chronic gestational exposure to ethanol causes oxidative stress and DNA damage. Paraffin-embedded histological sections were immunostained with monoclonal antibodies to 4-hydroxy-2,3-nonenal [HNE (A, B)] and 8-hydroxydeoxyguanosine [8-OHdG (C, D)]. Immunoreactivity was revealed by the ABC method with DAB (brown) as the chromogen. High magnification (original, X400) images depict relative levels of HNE and 8-OHdG immunoreactivity in junctional zone of control (A, C) and ethanol-exposed (B, D) placentas.
Ethanol-Induced Oxidative Stress is Associated With Elevated Pro-Apoptotic Protein Expression
We next examined whether ethanol-induced oxidative stress and DNA damage were associated with increased expression of p53-associated pro-apoptosis genes by measuring p53 and cyclin-dependent kinase inhibitor 1A (p21) by qRT-PCR, and p53, p21, and mdm2 (p53’s regulatory protein) by ELISA. Those studies revealed that chronic gestational exposure to ethanol significantly increased the mean placental levels of p21 and mdm2 protein (Fig. 4B,C). In contrast, p53 protein (Fig. 4A) and mRNA (0.8 ± 0.03 × 10−6, control 1.01 ± 0.08 × 10−6; p = .02) levels were significantly reduced in ethanol-exposed placentas. The studies were extended to explore the roles of Bcl family member proteins as mediators of apoptosis and potential cross-talk with p53 signaling mechanisms (Toledo and Wahl, 2006) by using ELISAs to measure Bax (pro-apoptosis), Bak (pro-apoptosis), Bcl-2 (pro-survival). Those investigations revealed significantly higher mean levels of Bak and Bax, and reduced levels of Bcl-2 in ethanol-exposed relative to control placentas (Fig. 4D–F).
Fig. 4.

Effects of chronic gestational ethanol exposure on apoptosis associated proteins. Placental expression levels of p53 (A), cyclin-dependent kinase inhibitor 1A (p21, B), p53’s regulatory protein mdm2 (C), pro-apoptotic proteins Bak (D) and Bax(E) and anti-apoptotic protein Bcl-2 (F) were determined by enzyme-linked immunosorbent assay (ELISA) in placentas from control and ethanol-exposed dams. The results were normalized to protein content. The graphs depict the mean ± SEM levels. Inter-group comparisons were made using Student t-tests. Significant p-values are indicated above the graphs.
Ethanol Impairs Placental Mitochondrial Function
QRT-PCR studies detected expression of genes encoding Complexes I through V in both control and ethanol-exposed placentas. In both groups, Complex II-D was most abundant, followed by Complex IV, Complex II-A, Complex III, Complex V, Complex I, and finally Complex II-C (Fig. 5). Chronic gestational ethanol exposure significantly reduced expression of all mitochondrial complex genes, except for II-A and II-C (Fig. 5). In fact, the mean level of Complex II-C mRNA was significantly higher in ethanol-exposed relative to control placentas (Fig. 5C). The mean levels of 18S rRNA were similar between the groups (Fig. 5H). ELISAs also detected expression of all 5 mitochondrial protein complexes in control and ethanol-exposed placentas. Corresponding with the qRT-PCR analyses, Complex II was most abundantly expressed. Otherwise, Complexes I, III, IV, and V proteins were expressed at similar levels in control placentas (Fig. 5). Chronic ethanol exposure significantly reduced the mean placental levels of Complexes I and II, while significantly increasing the mean levels of Complexes III, IV, and V (Fig. 6A,B). Since Complexes I and II regulate entry into the mitochondrial transport chain, ethanol exposure impaired oxidative phosphorylation at the initial steps. The significant increases in later Complexes could represent compensatory efforts to generate ATP.
Fig. 5.

Gestational ethanol exposure impairs mitochondrial gene expression. The mRNA levels of genes encoding the 5 major mitochondrial complex proteins (A–G) were measured by quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) in control and ethanol-exposed placentas and normalized to ribosomal 18S RNA (H) measured in parallel reactions. The graphs depict the mean ± SEM levels. Inter-group comparisons were made using Student t-tests. Significant p-values are indicated above the graphs.
Fig. 6.

Gestational ethanol exposure alters mitochondrial complex protein expression. The protein levels of the 5 major mitochondrial complex proteins (A–E) were measured by ELISA in placentas from control and ethanol-exposed dams. The results were normalized to protein content. The graphs depict the mean ± SEM levels. Inter-group comparisons were made using Student’s t-tests. Significant p-values are indicated above the graphs.
Chronic Gestational Ethanol Exposure Inhibits the Prolactin-Related Gene Expressions
Spongiotrophoblast are the major source of most PRL family member molecules needed to maintain pregnancy throughout the latter half of the gestation (Soares, 2004). To further explore the mechanisms and consequences of ethanol-impaired placentation, we used qRT-PCR analysis to measure mRNA levels of PRL family 3, subfamily d, member 1, which is also referred to as placental lactogen-1 (Prl3d1), PRL family 3, subfamily b, member 1, or placental lactogen-2 (Prl3b1), and PRL receptor (PRLR). Corresponding with the degenerative histopathological changes with increased oxidative stress, DNA damage, and mitochondrial dysfunction, ethanol-exposed placentas had significantly reduced mean levels of Prl3b1 and PRLR mRNA transcripts relative to control (Fig. 7B,C). Although the mean levels of Prl3d1 mRNA transcripts were reduced in ethanol-exposed placentas, the difference from control did not reach statistical significance (Fig. 7A).
Fig. 7.
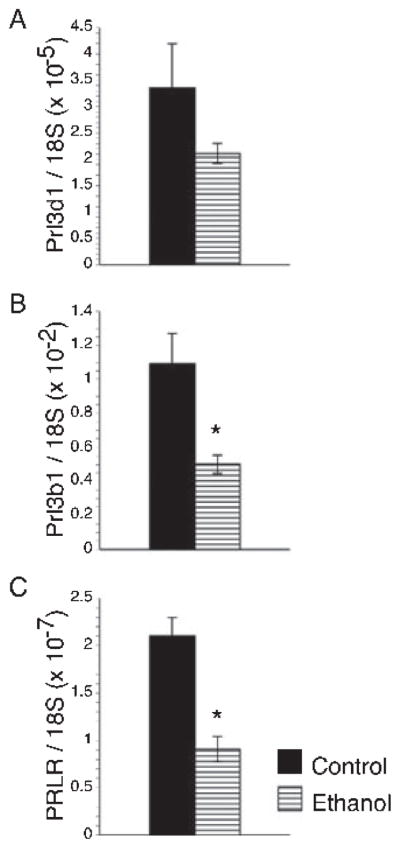
Chronic gestational exposure to ethanol inhibits the prolactin-related gene expressions. The mRNA levels of Prl3d1 (prolactin family 3, subfamily d, member 1; also called placental lactogen-1; (A) Prl3b1 (prolactin family 3, subfamily b, member 1; also called placental lactogen-2; (B) and prolactin receptor (PRLR, C) were measured by qRT-PCR in control and ethanol-exposed placentas and normalized to ribosomal 18S RNA measured in parallel reactions. The graphs depict the mean ± SEM levels. Inter-group comparisons were made using Student t-tests. Significant p-values are indicated above the graphs.
DISCUSSION
This study demonstrates underlying mechanisms and functional consequences of ethanol-induced oxidative stress in rat placenta. One of the major adverse effects of maternal ethanol consumption is early pregnancy loss (Henriksen et al., 2004). Previous studies demonstrated increased pregnancy loss following in utero ethanol exposure in which the mean blood alcohol concentration achieved was 51.1 ± 11.9 mM (Gundogan et al., 2008). Herein, we found that reducing ethanol exposure from 37 to 24% of the caloric content still resulted in substantial fetal demise with a significant trend toward reduced litter size. Histological studies revealed complete necrosis of placental and fetal tissue vis-à-vis intactness of the implantation site (data not shown) within the regions of fetal resorption. Moreover, similar less frequent and smaller foci of necrosis were detected in the spongiotrophoblast layer and decidua basalis of ethanol-exposed placentas. In addition, DNA damage and lipid peroxidation were more prominent in these zones compared with other regions of ethanol-exposed placentas. These findings suggest that besides IGF resistance (Gundogan et al., 2008), extensive placental oxidative damage contributes to pregnancy loss after chronic gestational exposure to ethanol.
Further analysis of gene expression directed toward uncovering mechanisms of pregnancy loss revealed that chronic gestational exposure to ethanol significantly increased pro-apoptosis and inhibited pro-survival mechanisms in placentas. In addition we demonstrated that the pro-apoptosis mechanisms involved p21, which is a downstream target of p53 (Gartel and Tyner, 2002), with increased levels of mdm2 but not p53. Sincemdm2 binds to p53 and inhibits its transactivation of target genes (Barak et al., 1994), the increased levels of mdm2 most likely reflect inhibition of p53 activity and signaling. Therefore, the data suggest that ethanol-induced oxidative stress and associated apoptosis/necrosis are mediated through p21, Bax, and Bak via a p53-independent pathway.
Mitochondria are the main organelles responsible for the oxidative phosphorylation needed to generate ATP. We used direct binding ELISAs to measure expression levels of the 5 membrane-bound protein complexes (Complex I–V) that catalyze oxidative phosphorylation. The major finding was that Complexes I and II were significantly reduced by chronic in utero exposure to ethanol, corresponding with previous observations in ethanol-exposed developing brains (Chu et al., 2007). This finding is of particular interest because Complexes I and II represent the principal entry sites into the electron transport chain (Cecchini, 2003; Hirst, 2005), and halting oxidative phosphorylation at the initial steps of electron transport could have a major impact on the generation of ATP.
Successful implantation, placental development and maintenance of pregnancy depend on functioning trophoblastic cells. Examples of trophoblast functions include, establishment of the maternal–fetal interface, nutrient-waste exchange, and hormone production. Prolactin (PRL) family hormones, including prolactin, placental lactogen I (Prl3d1), and placental lactogen II (Prl3b1), regulate pregnancy-dependent adaptations to physiological stress (Soares et al., 2007). Prolactin, which is produced by the maternal anterior pituitary gland, is the dominant lactogenic hormone of early pregnancy (GD 0 to 10). Trophoblast giant cells are the main source of Prl3d1 and Prl3b1 in developing rat placenta (Faria et al., 1990). Prl3d1 expression begins to increase on GD6, and by mid-gestation (GD11 to 12), it becomes the dominant lactogenic hormone. Prl3b1 production starts mid-gestation, and is the dominant lactogenic hormone throughout the second half of gestation (Soares, 2004).
To better understand the impact of ethanol-mediated oxidative stress on placental trophoblast function and the potential impact on pregnancy loss, we measured gene expression corresponding to PRLR, Prl3d1, and Prl3b1 by qRT-PCR analysis. Prolactin receptor interacts with PRL, Prl3d1, and Prl3b1 to transmit signals that mediate trophoblastic cell functions needed to maintain pregnancy (Soares, 2004). The PRLR is expressed by the uterine decidual cells (Gu et al., 1996) and its interaction with Prl3d1 and Prl3b1 was shown to play a role in decidual cell survival through PI3K/protein kinase B-mediated inhibition of caspase-3 activity (Tessier et al., 2001). The finding that all 3 mRNA transcripts were reduced by chronic gestational exposure to ethanol suggests that ethanol mediates its adverse effects on pregnancy maintenance via inhibition of PRL family genes and their interaction with decidua. Considering the inhibitory effects of ethanol on insulin signaling pathway including PI3-kinase, it is suggestive of a double-hit on decidual function. In this regard, it is noteworthy that we initiated the ethanol feedings on GD6 when Prl3d1 expression begins, and continued them through midgestation when Prl3b1 production takes over to become the dominant lactogenic hormone. Since prominent 8-OHdG and 4-HNE immunoreactivities were observed in Prl3d1-and Prl3b1-producing zones of ethanol-exposed placentas, ethanol-mediated DNA damage and oxidative stress may contribute to the impairments in placental hormone expression and function, and thereby promote pregnancy loss.
In conclusion, chronic gestational exposure to ethanol causes early pregnancy loss associated with increased DNA damage, lipid peroxidation, mitochondrial dysfunction, and activation of pro-apoptosis/anti-survival mechanisms. Mechanistically, these adverse effects of ethanol may contribute to early pregnancy loss and IUGR by impairing gene expression, survival, and function of lactogenic hormone-producing and PRL receptor bearing cells, which are needed to maintain pregnancy.
Acknowledgments
Supported by K08AA-016783 and K24-16126 from the National Institutes of Health.
References
- Barak Y, Gottlieb E, Juven-Gershon T, Oren M. Regulation of mdm2 expression by p53: alternative promoters produce transcripts with nonidentical translation potential. Genes Dev. 1994;8(15):1739–1749. doi: 10.1101/gad.8.15.1739. [DOI] [PubMed] [Google Scholar]
- Barr HM, Streissguth AP. Identifying maternal self-reported alcohol use associated with fetal alcohol spectrum disorders. Alcohol Clin Exp Res. 2001;25(2):283–287. [PubMed] [Google Scholar]
- Bischof P, Irminger-Finger I. The human cytotrophoblastic cell, a mononuclear chameleon. Int J Biochem Cell Biol. 2005;37(1):1–16. doi: 10.1016/j.biocel.2004.05.014. [DOI] [PubMed] [Google Scholar]
- Caluwaerts S, Vercruysse L, Luyten C, Pijnenborg R. Endovascular trophoblast invasion and associated structural changes in uterine spiral arteries of the pregnant rat. Placenta. 2005;26(7):574–584. doi: 10.1016/j.placenta.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Carter JJ, Tong M, Silbermann E, Lahousse SA, Ding FF, Longato L, Roper N, Wands JR, de laMonte SM. Ethanol impaired neuronal migration is associated with reduced aspartyl-asparaginyl-beta-hydroxylase expression. Acta Neuropathol. 2008;116(3):303–315. doi: 10.1007/s00401-008-0377-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecchini G. Function and structure of complex II of the respiratory chain. Annu Rev Biochem. 2003;72:77–109. doi: 10.1146/annurev.biochem.72.121801.161700. [DOI] [PubMed] [Google Scholar]
- Chu J, Tong M, de la Monte SM. Chronic ethanol exposure causes mitochondrial dysfunction and oxidative stress in immature central nervous system neurons. Acta Neuropathol. 2007;113(6):659–673. doi: 10.1007/s00401-007-0199-4. [DOI] [PubMed] [Google Scholar]
- Cohen AC, Tong M, Wands JR, de la Monte SM. Insulin and insulin-like growth factor resistance with neurodegeneration in an adult chronic ethanol exposure model. Alcohol Clin Exp Res. 2007;31(9):1558–1573. doi: 10.1111/j.1530-0277.2007.00450.x. [DOI] [PubMed] [Google Scholar]
- DeChiara TM, Efstratiadis A, Robertson EJ. A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature. 1990;345(6270):78–80. doi: 10.1038/345078a0. [DOI] [PubMed] [Google Scholar]
- Faria TN, Deb S, Kwok SC, Talamantes F, Soares MJ. Ontogeny of placental lactogen-I and placental lactogen-II expression in the developing rat placenta. Dev Biol. 1990;141(2):279–291. doi: 10.1016/0012-1606(90)90384-u. [DOI] [PubMed] [Google Scholar]
- Gartel AL, Tyner AL. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther. 2002;1(8):639–649. [PubMed] [Google Scholar]
- Gu Y, Srivastava RK, Clarke DL, Linzer DI, Gibori G. The decidual prolactin receptor and its regulation by decidua-derived factors. Endocrinology. 1996;137(11):4878–4885. doi: 10.1210/endo.137.11.8895360. [DOI] [PubMed] [Google Scholar]
- Gundogan F, Elwood G, Greco D, Rubin LP, Pinar H, Carlson RI, Wands JR, de la Monte SM. Role of aspartyl-(asparaginyl) beta-hydroxylase in placental implantation: relevance to early pregnancy loss. Hum Pathol. 2007;38(1):50–59. doi: 10.1016/j.humpath.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Gundogan F, Elwood G, Longato L, Tong M, Feijoo A, Carlson RI, Wands JR, de la Monte SM. Impaired placentation in fetal alcohol syndrome. Placenta. 2008;29(2):148–157. doi: 10.1016/j.placenta.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han VK, Carter AM. Spatial and temporal patterns of expression of messenger RNA for insulin-like growth factors and their binding proteins in the placenta of man and laboratory animals. Placenta. 2000;21(4):289–305. doi: 10.1053/plac.1999.0498. [DOI] [PubMed] [Google Scholar]
- Henriksen TB, Hjollund NH, Jensen TK, Bonde JP, Andersson AM, Kolstad H, Ernst E, Giwercman A, Skakkebaek NE, Olsen J. Alcohol consumption at the time of conception and spontaneous abortion. Am J Epidemiol. 2004;160(7):661–667. doi: 10.1093/aje/kwh259. [DOI] [PubMed] [Google Scholar]
- Hirst J. Energy transduction by respiratory complex I–an evaluation of current knowledge. Biochem Soc Trans. 2005;33(Pt 3):525–529. doi: 10.1042/BST0330525. [DOI] [PubMed] [Google Scholar]
- Hoek JB, Pastorino JG. Cellular signaling mechanisms in alcohol-induced liver damage. Semin Liver Dis. 2004;24(3):257–272. doi: 10.1055/s-2004-832939. [DOI] [PubMed] [Google Scholar]
- Kay HH, Grindle KM, Magness RR. Ethanol exposure induces oxidative stress and impairs nitric oxide availability in the human placental villi: a possible mechanism of toxicity. Am J Obstet Gynecol. 2000;182(3):682–688. doi: 10.1067/mob.2000.104201. [DOI] [PubMed] [Google Scholar]
- Kay HH, Tsoi S, Grindle K, Magness RR. Markers of oxidative stress in placental villi exposed to ethanol. J Soc Gynecol Investig. 2006;13(2):118–121. doi: 10.1016/j.jsgi.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Lahousse SA, Carter JJ, Xu XJ, Wands JR, de la Monte SM. Differential growth factor regulation of aspartyl-(asparaginyl)-beta-hydroxylase family genes in SH-Sy5y human neuroblastoma cells. BMC Cell Biol. 2006;7:41. doi: 10.1186/1471-2121-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laviola L, Perrini S, Belsanti G, Natalicchio A, Montrone C, Leonardini A, Vimercati A, Scioscia M, Selvaggi L, Giorgino R, Greco P, Giorgino F. Intrauterine growth restriction in humans is associated with abnormalities in placental insulin-like growth factor signaling. Endocrinology. 2005;146(3):1498–1505. doi: 10.1210/en.2004-1332. [DOI] [PubMed] [Google Scholar]
- Lee RD, An SM, Kim SS, Rhee GS, Kwack SJ, Seok JH, Chae SY, Park CH, Choi YW, Kim HS, Cho HY, Lee BM, Park KL. Neurotoxic effects of alcohol and acetaldehyde during embryonic development. J Toxicol Environ Health A. 2005;68(23–24):2147–2162. doi: 10.1080/15287390500177255. [DOI] [PubMed] [Google Scholar]
- Moffett A, Loke C. Immunology of placentation in eutherian mammals. Nat Rev Immunol. 2006;6(8):584–594. doi: 10.1038/nri1897. [DOI] [PubMed] [Google Scholar]
- de la Monte SM, Tong M, Carlson RI, Carter JJ, Longato L, Silbermann E, Wands JR. Ethanol inhibition of aspartyl-asparaginyl-beta-hydroxylase in fetal alcohol spectrum disorder: potential link to the impairments in central nervous system neuronal migration. Alcohol. 2009;43(3):225–240. doi: 10.1016/j.alcohol.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myatt L, Cui X. Oxidative stress in the placenta. Histochem Cell Biol. 2004;122(4):369–382. doi: 10.1007/s00418-004-0677-x. [DOI] [PubMed] [Google Scholar]
- Pijnenborg RBJ, Robertson WB, Dixon G, Brosens I. The pattern of interstitial trophoblastic invasion of the myometrium in early human pregnancy. Placenta. 1981;2(4):303–316. doi: 10.1016/s0143-4004(81)80027-6. [DOI] [PubMed] [Google Scholar]
- Poston L, Raijmakers MT. Trophoblast oxidative stress, antioxidants and pregnancy outcome–a review. Placenta. 2004;25(Suppl A):S72–S78. doi: 10.1016/j.placenta.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Soares MJ. The prolactin and growth hormone families: pregnancy-specific hormones/cytokines at the maternal-fetal interface. Reprod Biol Endocrinol. 2004;2:51. doi: 10.1186/1477-7827-2-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares MJ, Konno T, Alam SM. The prolactin family: effectors of pregnancy-dependent adaptations. Trends Endocrinol Metab. 2007;18(3):114–121. doi: 10.1016/j.tem.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Soscia SJ, Tong M, Xu XJ, Cohen AC, Chu J, Wands JR, de la Monte SM. Chronic gestational exposure to ethanol causes insulin and IGF resistance and impairs acetylcholine homeostasis in the brain. Cell Mol Life Sci. 2006;63(17):2039–2056. doi: 10.1007/s00018-006-6208-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier C, Prigent-Tessier A, Ferguson-Gottschall S, Gu Y, Gibori G. PRL antiapoptotic effect in the rat decidua involves the PI3K/protein kinase B-mediated inhibition of caspase-3 activity. Endocrinology. 2001;142(9):4086–4094. doi: 10.1210/endo.142.9.8381. [DOI] [PubMed] [Google Scholar]
- Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer. 2006;6(12):909–923. doi: 10.1038/nrc2012. [DOI] [PubMed] [Google Scholar]
- Vercruysse L, Caluwaerts S, Luyten C, Pijnenborg R. Interstitial trophoblast invasion in the decidua and mesometrial triangle during the last third of pregnancy in the rat. Placenta. 2006;27(1):22–33. doi: 10.1016/j.placenta.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Wang Y, Walsh SW. Placental mitochondria as a source of oxidative stress in pre-eclampsia. Placenta. 1998;19(8):581–586. doi: 10.1016/s0143-4004(98)90018-2. [DOI] [PubMed] [Google Scholar]
- Wisdom SJ, Wilson R, McKillop JH, Walker JJ. Antioxidant systems in normal pregnancy and in pregnancy-induced hypertension. Am J Obstet Gynecol. 1991;165(6 Pt 1):1701–1704. doi: 10.1016/0002-9378(91)90018-m. [DOI] [PubMed] [Google Scholar]
- Xu J, Yeon JE, Chang H, Tison G, Chen GJ, Wands J, de la Monte S. Ethanol impairs insulin-stimulated neuronal survival in the developing brain: role of PTEN phosphatase. J Biol Chem. 2003;278(29):26929–26937. doi: 10.1074/jbc.M300401200. [DOI] [PubMed] [Google Scholar]
- Yeon JE, Califano S, Xu J, Wands JR, De LaMonte SM. Potential role of PTEN phosphatase in ethanol-impaired survival signaling in the liver. Hepatology. 2003;38(3):703–714. doi: 10.1053/jhep.2003.50368. [DOI] [PubMed] [Google Scholar]