

Abstract
The synthesis of 1,2,10,11-tetrahydro-9H-cyclobuta[c]benzo[e]indol-4- one (17, CbBI), containing a deep-seated fundamental structural modification in the CC-1065 and duocarmycin alkylation subunit with the incorporation of a ring expanded fused cyclobutane (vs cyclopropane), its chemical and structural characterization, and its incorporation into a key analogue of the natural products are detailed. The approach to the preparation of CbBI was based on a precedented (Ar-3′ and Ar-5′), but previously unknown Ar-4′ spirocyclization of a phenol onto a tethered alkyl halide to form the desired cyclobutane. The conditions required for the implementation of the Ar-4′ spirocyclization indicate that the entropy of activation substantially impacts the rate of reaction relative to the much more facile Ar-3′ spirocyclization, while the higher enthalpy of activation slows the reaction relative to an Ar-5′ spirocyclization. The characterization of the CbBI-based agents revealed their exceptional stability and exquisite reaction regioselectivity, and a single crystal X-ray structure analysis of N-Boc-CbBI (13) revealed their structural origins. The reaction regioselectivity may be attributed to the stereoelectronic alignment of the two available cyclobutane bonds with the cyclohexadienone π-system, which resides in the bond that extends to the less substituted cyclobutane carbon for 13. The remarkable stability of N-Boc-CbBI (stable even at pH 1) relative to N-Boc-CBI (t1/2 = 133 h at pH 3) containing a cyclopropane may be attributed to a combination of the increased extent of vinylogous amide conjugation, the non-optimal geometric alignment of the cyclobutane with the activating cyclohexadienone, and the intrinsic but modestly lower strain energy (1.8 kcal/mol) of a cyclobutane versus cyclopropane.
Introduction
CC-1065 (1) and duocarmycin SA (2) represent key members of a class of antitumor agents that derive their biological activity from their ability to selectively alkylate duplex DNA (Figure 1).1–3 The study of the natural products, their synthetic unnatural enantiomers,4 derivatives, and key analogues has defined the fundamental features that control the DNA alkylation selectivity, efficiency, and catalysis, providing a detailed understanding of fundamental relationships between structure, reactivity, and biological activity.3
Figure 1.

Natural products
Among the most studied duocarmycin analogues is CBI5 (Figure 2), which is both synthetically more accessible and has been found to enhance both the chemical stability (4×) and the biological potency (4×) of the corresponding derivatives relative to the alkylation subunit found in CC-1065. Thus, it is on this scaffold that new design concepts are often explored, developed, and evaluated.3 Despite the extensive synthetic studies on the duocarmycins, there have been very few examples of modification of the alkylation subunit’s cyclopropane, which is intimately involved in the DNA alkylation reaction responsible for their biological activity.6 For some time, we have been interested in probing the deep-seated change to this cyclopropane with its expansion to a cyclobutane to provide the corresponding 1,2,10,11-tetrahydro-9H-cyclobuta[c]benzo[e]indol-4-one (CbBI) alkylation subunit (Figure 2). We recognized that the modestly smaller strain energy of the cyclobutane in CbBI (1.8 kcal/mol; 27.6 kcal/mol vs 25.8 kcal/mol)7 should make these agents chemically more stable, but the extent of the change in reactivity, reaction regioselectivity, and their impact on the DNA alkylation properties and biological activity of the resulting analogues was not clear. Herein, we report the synthesis of N-Boc-CbBI, its incorporation into a key analogue of the duocarmycins (16, CbBI-TMI), the X-ray structure characterization of 13, and its correlation with the reactivity and reaction regioselectivity impacting the resulting DNA alkylation and biological properties of this class of compounds.
Figure 2.

CBI versus CbBI and retrosynthetic analysis
In addition and perhaps the origin that such derivatives have not yet been examined, the parent spirocyclobutylcyclohexadienone ring system embedded in CbBI is unknown and many well established methods for forming 4-membered rings (i.e., 2+2 cycloaddition) cannot be easily adapted to its preparation. The approach pursued and ultimately implemented entailed a previously unreported Ar-4′ alkylative spirocyclization of the corresponding phenol analogous to the Ar-3′ spirocyclization used for the synthesis of CBI (Figure 2). Pioneering efforts by Winstein and Baird first demonstrated the spirocyclization of such phenol precursors to form reactive cyclopropanes (Ar-3′ alkylation) and the more stable cyclopentanes (Ar-5′ alkylation), but no reports of its extension to an Ar-4′ spirocyclization for preparation of cyclobutanes have been disclosed.8
Results and Discussion
Synthesis
The synthesis of CbBI began with the N-alkylation of 59,10 with 6, which proceeded in 83% yield (Scheme 1). Key 5-exo-trig free radical cyclization11 of 7 upon treatment with tributyltin hydride gave the desired ethyl ester 8, and reduction of 8 with lithium borohydride provided 9 in quantitative yield. Treatment of the resulting alcohol 9 with methanesulfonyl chloride in pyridine, followed by the addition of LiCl cleanly provided the desired chloride 10. Removal of the benzyl protecting group was achieved by hydrogenolysis with 10% Pd/C and ammonium formate, providing the first key spirocyclization substrate 11. However, all attempts to spirocyclize 11 to provide 13 were unsuccessful, providing only recovered starting material in most cases and chloride elimination and/or Boc deprotected products upon heating at high reaction temperatures (150–160 °C). An extensive range of bases, solvents, and reaction temperatures were examined without success. Thus, in spite of the relative release of ring strain, the Ar-4′ spirocyclization of 11 is kinetically much slower than the analogous Ar-3′ spirocyclization used to provide N-Boc-CBI; a reaction that occurs at room temperature upon exposure to even aqueous NaHCO3 (2 h). The reactivity of the cyclization substrate was increased by conversion of 11 to the corresponding iodide using NaI in 2-butanone in nearly quantitative yield. Although spirocyclization of 12 was not observed at or near room temperature under a wide range of reaction conditions, warming a solution of this substrate in saturated aqueous NaHCO3/THF (1:1) at 110 °C for 3 h provided the desired spirocycle 13 (confirmed by X-ray,12 Figure 3). Further optimization of the spirocyclization to minimize competitive Boc deprotection and iodide solvolysis (130 °C for 1 h) provided N-Boc-CbBI in 55% yield. It is remarkable, but not unprecedented, that the 4-membered ring closure to provide 13 is kinetically so much slower than the corresponding 3-membered ring spirocyclization used for the preparation of N-Boc-CBI. This may be attributed largely to the increased entropy of activation required for the Ar-4′ spirocyclization and is not reflective of the relative stabilities of the resulting products. In contrast, the slow rate of ring closure relative to an Ar-5′ spirocyclization8 may be related to the corresponding enthalpy of activation and is likely reflective of the relative stabilities of the resulting products.
Scheme 1.

Figure 3.
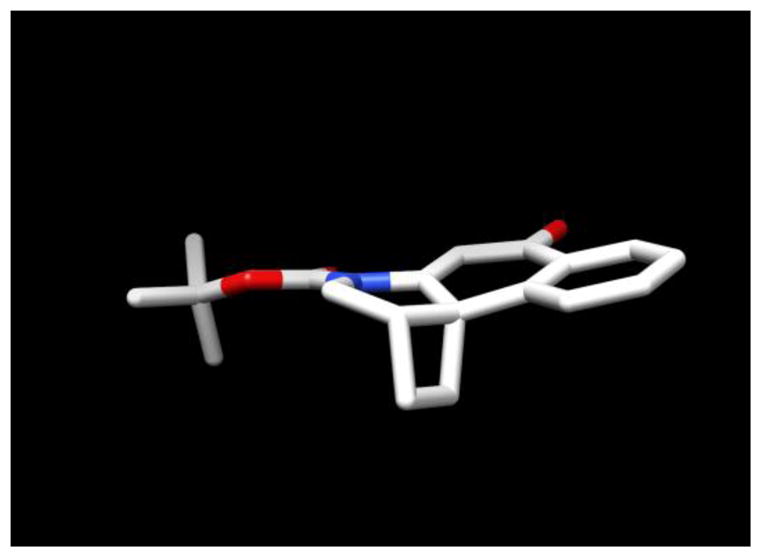
X-ray structure of 13 (N-Boc-CbBI)
In order to accurately assess the biological properties of such cyclobutane derivatives, the corresponding analogue 16 (CbBI-TMI) of duocarmycin SA (2) was prepared. Thus, Boc deprotection of 11 (4 N HCl in EtOAc, 23 °C, 30 min) followed by direct coupling of the resulting indoline hydrochloride salt with 5,6,7-trimethoxyindole-2-carboxylic acid13 (1.1 equiv, 3 equiv of 1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide (EDCI), DMF, 23 °C, 16 h, 65%) afforded 14 (Scheme 2). Conversion of the chloride to the iodide 15 (5.0 equiv of NaI, 2-butanone, 65 °C, 3 h, 82%) provided the cyclization substrate that was subjected to the optimized spirocyclization conditions. However, the hydrolytic lability of the linking amide bond proved significant, generating only small amounts of 16 (<5%) and providing predominantly the free amine of CbBI, resulting from spirocyclization and subsequent hydrolysis of the resulting labile amide. By running the spirocyclization reaction at a lower temperature for a longer reaction time (110 °C for 5 h), amide bond hydrolysis was minimized and the yield of 16 was improved. As discussed below, the linking amide bond in 16 is weak and diagnostic of the preferential nitrogen lone pair vinylogous amide (vs amide) conjugation with the cyclohexadienone system of CbBI. Since this vinylogous amide is cross-conjugated with the cyclobutylcyclohexadienone, its dominance serves to stabilize the otherwise reactive cyclobutane.
Scheme 2.

Reactivity and Reaction Regioselectivity
The study of both the rate of acid-catalyzed solvolysis and the regioselectivity of addition to the activated cyclopropane has proven key to understanding structural features that contribute to the biological properties of the CC-1065 and duocarmycin alkylation subunits. The rates of solvolysis have provided insights into the structural features that stabilize or activate the cyclopropane for nucleophilic attack as well as into the source of catalysis for the DNA alkylation reaction,14 and their study defined a fundamental parabolic relationship between the alkylation subunit intrinsic reactivity and biological activity.15 The regioselectivity of ring opening reaction has helped define the mechanism of DNA alkylation, where the preferred site of nucleophilic addition was found to depend on the stereoelectronic alignment of the cyclopropane (in this case cyclobutane) bonds with the cyclohexadienone π-system.
The solvolysis reactivity of N-Boc-CbBI (13) was followed spectrophotometrically by UV in pH 3, pH 2, and even pH 1 phosphate buffer, however no measurable solvolysis was observed. Treatment of 13 with 10% TFA or 10% F3CSO3H in MeOH led only to Boc cleavage over time (72 h) with no evidence of cyclobutane ring opening. The treatment of 13 with 4 N HCl in EtOAc (−78 °C to 25 °C) only afforded 17 resulting from Boc deprotection, with no observed chloride addition to the cyclobutane. In order to avoid the competitive acid-catalyzed Boc deprotection of 13 while assessing the relative reactivity of CbBI, the corresponding methyl carbamate 18 was prepared (eq 1). This was most easily accomplished by heating a solution of 12 at 160 °C in saturated aqueous NaHCO3/THF (1:1) for 30 min in a microwave reactor to provide 17 in 74% yield resulting from spirocyclization followed by in situ Boc deprotection. Deprotonation of 17 with sodium hydride (2.5 equiv, DMF, 30 min, 0 °C), followed by treatment with methyl chloroformate (5.0 equiv) added slowly afforded 18 in superb yield (96%). Analogous to observations made with 13, no measurable solvolysis of 18 was observed even in pH 1 phosphate buffer. Whereas treatment of 18 with 4 N HCl in EtOAc at −78 °C provided only recovered starting material, warming the solution to room temperature for 2 h provided a single chloride addition product 19 in quantitative yield (eq 2). Exclusive chloride addition to the less substituted cyclobutane carbon was observed, and no 7-membered ring addition product, resulting from attack on the more hindered carbon, was observed. Thus, the CbBI alkylation subunit exhibits a remarkable stability being unreactive to solvolysis even at pH 1. This contrasts N-Boc-CBI, which is stable at pH 7, but exhibits readily measurable solvolysis reactivity at pH 3 (k = 1.45 × 10−6, t1/2 = 133 h) and pH 2 (t1/2 = 12.5 h).16 A lower limit on the relative reactivity of CbBI versus CBI indicates that it is at least 100-times more stable than CBI toward acid-catalyzed solvolysis.
 |
(1) |
 |
(2) |
The remarkable stablity of CbBI is due in a large part to the stabilizing vinylogous amide conjugation. Most diagnostic of this conjugation is the shortened N2-C2a bond length (bond c, 1.382 Å) that shows a progressive shortening as one moves across the series of modified alkylation subunits illustrated in Figure 4.17–19 That is, the shorter c bond length corresponds to increased vinylogous amide conjugation that in turn correlates with remarkable progressive increases in compound stability. Similarly observed is the smaller χ1 dihedral angle and the longer amide bond length (bond d, 1.379 Å; indicative of less amide vs vinylogous amide conjugation) across the series. Further contributing to the stability of CbBI relative to CBI is the modestly diminished ring strain intrinsic in a cyclobutane versus cyclopropane (1.8 kcal/mol)7 that makes its cleavage less facile. The net effect is that CbBI exhibits a remarkable stability in which its intrinsic reactivity is masked in part by the strong stabilizing vinylogous amide conjugation. Additionally, the X-ray crystal structure indicates that the geometric alignment of the cyclobutane deviates slightly with respect to the plane bisecting the cyclohexadienone (Figure 5A). This results from the fusion of the 5-membered ring that precludes an ideal alignment and overlap between the cyclobutane orbitals and the cyclohexadienone.18
Figure 4.

X-ray crystal structure comparison of CBI analogues. Data taken from refs 17–19.
Figure 5.
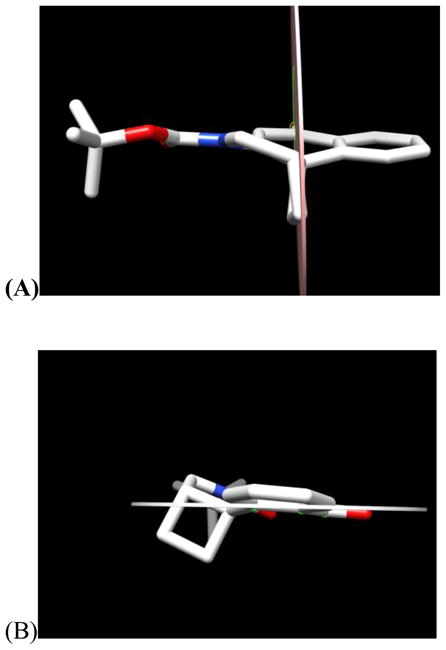
(A) Back view of 13 and (B) Side view of 13
The reaction regioselectivity originates in the stereoelectronic alignment of the two available cyclobutane bonds with the cyclohexadienone π-system. As seen in Figure 5B, the orbital of the cyclobutane bond extending to the least substituted carbon overlaps best with the developing π-system of the phenol reaction product. In contrast, the bond extending to the more substituted carbon nearly lies in the plane of this developing π-system and is not stereoelectronically aligned to undergo cleavage. Notably, this stereoelectronic control overrides any intrinsic preference to place a developing positive charge on a preferred secondary versus primary center (ring expansion). Further and diagnostic of the regioselectivity of nucleophilic addition, the cleaved bond is longer (1.590 Å vs 1.551 Å) and weaker than the bond extending to the more substituted carbon, reflecting its conjugation with the π-system. Steric factors resulting from preferential SN2 attack on the least hindered site may also play a role.
Cytotoxic Activity
Compounds 15 and 16 were assayed for cytotoxic activity against the L1210 tumor cell line (mouse leukemia cell line). These compounds were found to display IC50’s of 1.2 μM and 6.3 μM, respectively. This represents a 106 drop in potency when compared to the natural products themselves (2, IC50 = 6–10 pM) and a 105–106 fold loss in activity when compared to CBI-TMI (IC50 = 30 pM),20 a testament to the remarkable stability of these CbBI derivatives. Although relatively inactive, this level of cytotoxic activity is in line with expectations based on the exceptional stability of compounds that preclude their effective alkylation of duplex DNA and follows trends depicted in the parabolic relationship between reactivity and cytotoxic activity.15
Conclusions
The subtle change in the structure (CbBI vs CBI) that results in such a dramatic alteration of the chemical and biological properties of the compound highlight the remarkable constellation of properties that are incorporated into the compact natural product structures (1 and 2) and are used to mask or tame an otherwise reactive electrophile.21 Even though CbBI could be expected to be more stable than CBI, the remarkable stability exhibited by N-acyl-CbBI (stable at pH 1) is not intuitively obvious from a cursory inspection of its structure. Contributing to this unusual stability is the dominate vinylogous amide conjugation,22 the non-ideal cyclobutane alignment with the activating cyclohexadienone imposed by the fused 5-membered ring,18 and the modestly reduced strain energy (1.8 kcal/mol)7 intrinsic in a 4-membered cyclobutane versus 3-membered cyclopropane ring.
Supplementary Material
Acknowledgments
We gratefully acknowledge the financial support of the National Institutes of Health (CA 41986) and the Skaggs Institute for Chemical Biology and we thank William M. Robertson for conducting the cytotoxic activity assays. JPL is a Skaggs Fellow.
Footnotes
Supporting Information Available. Full experimental details and compound characterizations are provided.
References
- 1.For CC-1065, see: Martin DG, Biles C, Gerpheide SA, Hanka LJ, Krueger WC, McGovren JP, Mizsak SA, Neil GL, Stewart JC, Visser J. J Antibiot. 1981;34:1119. doi: 10.7164/antibiotics.34.1119.For duocarmycin SA, see: Ichimura M, Ogawa T, Takahashi K, Kobayashi E, Kawamoto I, Yasuzawa T, Takahashi I, Nakano H. J Antibiot. 1990;43:1037. doi: 10.7164/antibiotics.43.1037.For duocarmycin A, see: Takahashi I, Takahashi K, Ichimura M, Morimoto M, Asano K, Kawamoto I, Tomita F, Nakano H. J Antibiot. 1988;41:1915. doi: 10.7164/antibiotics.41.1915.For yatakemycin, see: Igarashi Y, Futamata K, Fujita T, Sekine A, Senda H, Naoki H, Furumai T. J Antibiot. 2003;56:107. doi: 10.7164/antibiotics.56.107.
- 2.For duocarmycin SA, see: Boger DL, Johnson DS, Yun W. J Am Chem Soc. 1994;116:1635.For yatakemycin, see: Parrish JP, Kastrinsky DB, Wolkenberg SE, Igarashi Y, Boger DL. J Am Chem Soc. 2003;125:10971. doi: 10.1021/ja035984h.Trzupek JD, Gottesfeld JM, Boger DL. Nat Chem Biol. 2006;2:79. doi: 10.1038/nchembio761.Tichenor MS, MacMillan KS, Trzupek JD, Rayl TJ, Hwang I, Boger DL. J Am Chem Soc. 2007;129:10858. doi: 10.1021/ja072777z.For CC-1065, see: Hurley LH, Lee CS, McGovren JP, Warpehoski MA, Mitchell MA, Kelly RC, Aristoff PA. Biochemistry. 1988;27:3886. doi: 10.1021/bi00410a054.Hurley LH, Warpehoski MA, Lee CS, McGovren JP, Scahill TA, Kelly RC, Mitchell MA, Wicnienski NA, Gebhard I, Johnson PD, Bradford VS. J Am Chem Soc. 1990;112:4633.Boger DL, Johnson DS, Yun W, Tarby CM. Bioorg Med Chem. 1994;2:115. doi: 10.1016/s0968-0896(00)82007-6.Boger DL, Coleman RS, Invergo BJ, Sakya SM, Ishizaki T, Munk SA, Zarrinmayeh H, Kitos PA, Thompson SC. J Am Chem Soc. 1990;112:4623.For duocarmycin A, see: Boger DL, Ishizaki T, Zarrinmayeh H, Munk SA, Kitos PA, Suntornwat O. J Am Chem Soc. 1990;112:8961.Boger DL, Ishizaki T, Zarrinmayeh H. J Am Chem Soc. 1991;113:6645.Boger DL, Yun W, Terashima S, Fukuda Y, Nakatani K, Kitos PA, Jin Q. Bioorg Med Chem Lett. 1992;2:759.Boger DL, Yun W. J Am Chem Soc. 1993;115:9872.Boger DL, Munk SA, Zarrinmayeh H, Ishizaki T, Haught J, Bina M. Tetrahedron. 1991;47:2661.
- 3.Reviews: Boger DL, Johnson DS. Angew Chem Int Ed Engl. 1996;35:1438.Boger DL. Acc Chem Res. 1995;28:20.Boger DL, Johnson DS. Proc Natl Acad Sci USA. 1995;92:3642. doi: 10.1073/pnas.92.9.3642.Boger DL, Garbaccio RM. Acc Chem Res. 1999;32:1043.Tichenor MS, Boger DL. Natural Prod Rep. 2008;25:220. doi: 10.1039/b705665f.MacMillan KS, Boger DL. J Med Chem. 2009;52:5771. doi: 10.1021/jm9006214.Searcey M. Curr Pharm Des. 2002;8:1375. doi: 10.2174/1381612023394539.
- 4.(a) Boger DL, Coleman RS. J Am Chem Soc. 1988;110:1321. [Google Scholar]; (b) Boger DL, Coleman RS. J Am Chem Soc. 1988;110:4796. [Google Scholar]; (c) Boger DL, Machiya K. J Am Chem Soc. 1992;114:10056. [Google Scholar]; (d) Boger DL, Machiya K, Hertzog DL, Kitos PA, Holmes D. J Am Chem Soc. 1993;115:9025. [Google Scholar]; (e) Boger DL, McKie JA, Nishi T, Ogiku T. J Am Chem Soc. 1996;118:2301. [Google Scholar]; (f) Boger DL, McKie JA, Nishi T, Ogiku T. J Am Chem Soc. 1997;119:311. [Google Scholar]; (g) Tichenor MS, Kastrinsky DB, Boger DL. J Am Chem Soc. 2004;126:8346. doi: 10.1021/ja0472735. [DOI] [PubMed] [Google Scholar]; (h) Tichenor MS, Trzupek JD, Kastrinsky DB, Shiga F, Hwang I, Boger DL. J Am Chem Soc. 2006;128:15683. doi: 10.1021/ja064228j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Boger DL, Ishizaki T, Kitos PA, Suntornwat O. J Org Chem. 1990;55:5823. [Google Scholar]; (b) Boger DL, Ishizaki T, Wysocki RJ, Jr, Munk SA, Kitos PA, Suntornwat O. J Am Chem Soc. 1989;111:6461. [Google Scholar]; (c) Boger DL, Ishizaki T. Tetrahedron Lett. 1990;31:793. [Google Scholar]; (d) Boger DL, Ishizaki T, Zarrinmayeh H, Kitos PA, Suntornwat O. Bioorg Med Chem Lett. 1991;1:55. [Google Scholar]; (e) Boger DL, Ishizaki T, Sakya SM, Munk SA, Kitos PA, Jin Q, Besterman JM. Bioorg Med Chem Lett. 1991;1:115. [Google Scholar]; (f) Boger DL, Munk SA, Ishizaki T. J Am Chem Soc. 1991;113:2779. [Google Scholar]; (g) Boger DL, Munk SA. J Am Chem Soc. 1992;114:5487. [Google Scholar]; (h) Boger DL, Yun W. J Am Chem Soc. 1994;116:5523. [Google Scholar]; (i) Boger DL, Yun W, Han N, Johnson DS. Bioorg Med Chem. 1995;3:611. doi: 10.1016/0968-0896(95)00048-l. [DOI] [PubMed] [Google Scholar]; (j) Boger DL, Yun W, Cai H, Han N. Bioorg Med Chem. 1995;3:761. doi: 10.1016/0968-0896(95)00066-p. [DOI] [PubMed] [Google Scholar]; (k) Boger DL, Yun W, Han N. Bioorg Med Chem. 1995;3:1429. doi: 10.1016/0968-0896(95)00130-9. [DOI] [PubMed] [Google Scholar]; (l) Drost KJ, Cava MP. J Org Chem. 1991;56:2240. [Google Scholar]; (m) Aristoff PA, Johnson PD. J Org Chem. 1992;57:6234. [Google Scholar]
- 6.(a) Boger DL, Jenkins TJ. J Am Chem Soc. 1996;118:8860. [Google Scholar]; (b) Boger DL, Palanki MSS. J Am Chem Soc. 1992;114:9318. [Google Scholar]; (c) Boger DL, Johnson DS, Palanki MS, Kitos PA, Chang J, Dowell P. Bioorg Med Chem. 1993;1:27. doi: 10.1016/s0968-0896(00)82100-8. [DOI] [PubMed] [Google Scholar]; (d) Tietze LF, Herzig T, Fecher A, Haunert F, Schuberth I. ChemBioChem. 2001;2:758. doi: 10.1002/1439-7633(20011001)2:10<758::AID-CBIC758>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 7.Dudev T, Lim C. J Am Chem Soc. 1998;120:4450. [Google Scholar]
- 8.(a) Winstein S, Baird R. J Am Chem Soc. 1957;79:756. [Google Scholar]; (b) Baird R, Winstein S. J Am Chem Soc. 1957;79:4238. [Google Scholar]; (c) Baird R, Winstein S. J Am Chem Soc. 1962;84:788. [Google Scholar]; (d) Baird R, Winstein S. J Am Chem Soc. 1963;85:567. [Google Scholar]
- 9.(a) Boger DL, Yun W, Teegarden BR. J Org Chem. 1992;57:2873. [Google Scholar]; (b) Boger DL, McKie JA. J Org Chem. 1995;60:1271. [Google Scholar]; (c) Boger DL, McKie JA, Boyce CW. Synlett. 1997:515. [Google Scholar]; (d) Kastrinsky DB, Boger DL. J Org Chem. 2004;69:2284. doi: 10.1021/jo035465x. [DOI] [PubMed] [Google Scholar]
- 10.Boger DL, Boyce CW, Garbaccio RM, Goldberg JA. Chem Rev. 1997;97:787. doi: 10.1021/cr960095g. [DOI] [PubMed] [Google Scholar]
- 11.Boger DL, Boyce CW, Garbaccio RM, Searcey M. Tetrahedron Lett. 1998;39:2227. [Google Scholar]
- 12.The X-ray structure of 13 has been deposited with the Cambridge Crystallographic Data Centre (CCDC787358).
- 13.Boger DL, Ishizaki T, Zarrinmayeh H, Kitos PA, Suntornwat O. J Org Chem. 1990;55:4499. [Google Scholar]
- 14.(a) Boger DL, Hertzog DL, Bollinger B, Johnson DS, Cai H, Goldberg J, Turnbull P. J Am Chem Soc. 1997;119:4977. [Google Scholar]; (b) Boger DL, Bollinger B, Hertzog DL, Johnson DS, Cai H, Mesini P, Garbaccio RM, Jin Q, Kitos PA. J Am Chem Soc. 1997;119:4487. [Google Scholar]; (c) Boger DL, Garbaccio RM. Bioorg Med Chem. 1997;5:263. doi: 10.1016/s0968-0896(96)00238-6. [DOI] [PubMed] [Google Scholar]
- 15.Parrish JP, Hughes TV, Hwang I, Boger DL. J Am Chem Soc. 2004;126:80. doi: 10.1021/ja038162t. [DOI] [PubMed] [Google Scholar]
- 16.Boger DL, Garbaccio RM. J Org Chem. 1999;64:5666. doi: 10.1021/jo990762g. [DOI] [PubMed] [Google Scholar]
- 17.For CBI, see: Boger DL, Ishizaki T, Kitos PA, Suntornwat O. J Org Chem. 1990;55:5823.
- 18.For CBQ, see: Boger DL, Mesini P, Tarby CM. J Am Chem Soc. 1994;116:6461.Boger DL, Mesini P. J Am Chem Soc. 1994;116:11335.
- 19.For CNA, see: Boger DL, Turnbull P. J Org Chem. 1997;62:5849.
- 20.Boger DL, Yun W. J Am Chem Soc. 1994;116:7996. [Google Scholar]
- 21.(a) Wolkenberg SE, Boger DL. Chem Rev. 2002;102:2477. doi: 10.1021/cr010046q. [DOI] [PubMed] [Google Scholar]; (b) Tse WC, Boger DL. Chem Biol. 2004;11:1607. doi: 10.1016/j.chembiol.2003.08.012. [DOI] [PubMed] [Google Scholar]; (c) Tse WC, Boger DL. Acc Chem Res. 2004;37:61. doi: 10.1021/ar030113y. [DOI] [PubMed] [Google Scholar]
- 22.Boger DL, Turnbull P. J Org Chem. 1998;63:8004. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.