

Abstract
Complement activation is an important aspect of systemic lupus erythematosus. In this study we investigated the role of C3a/C3a receptor (R) signaling in brains of the lupus model, MRL/lpr mice, by treating the mice with C3aR antagonist (a) from 13 to 19 weeks of age. C3aR mRNA (0.2 ± 0.027 versus 0.56 ± 0.19) and protein (0.16 ± 0.09 versus 0.63 ± 0.19) expression was increased in MRL/lpr brains compared with MRL+/+ controls. Apoptosis, a key feature in lupus brain, was significantly reduced by C3aRa treatment, as assessed by DNA laddering, TUNEL staining and caspase3 activity (48% of MRL/lpr mice). mRNA expression of proinflammatory molecules that cause apoptosis, TNFα (0.33 ± 0.07 versus 0.15 ± 0.1), MIP2 (3.8 ± 1.3 versus 1.7 ± 0.6), and INFγ (4.8 ± 1.0 versus 2.07 ± 1.28) are reduced in MRL/lpr brains with C3aRa treatment. In line with these results, Western blotting demonstrates the significant increase in phosphorylation of survival molecules Akt and Erk, decrease in PTEN and reduced iNOS expression. INFγ receptor (R) and AMPA-GluR1 co-localized, and concomitant with reduced INFγR expression, AMPA-GluR1 expression was also decreased by C3aR antagonist. All of these variables that modulate neuronal excitability and regulate synaptic plasticity are C3aR dependent in the MRL/lpr brains and suggest a potential therapeutic role for C3aR inhibition in CNS lupus.
Keywords: anaphylatoxins, brain, complement, inflammation, systemic lupus erythematosus
Introduction
Central nervous system (CNS) involvement occurs in 14–75% of systemic lupus erythematosus (SLE) patients and is associated with increased mortality.1 The exact underlying mechanism causing the CNS pathology remains unknown. The complement system is an innate immune mechanism that goes awry and contributes to increased or sustained inflammation and damage in SLE.2-6 Once activated, the complement system generates the anaphylatoxins, C3a and C5a.7-9 The systemic levels of anaphylatoxins correlate with CNS disease in SLE patients.10 Our previous studies demonstrate that complement inhibition by the upstream complement regulator, Crry, which inhibits the convertases, or by the absence of factor B, a ‘key’ protein of the alternative pathway significantly alleviates CNS disease in the experimental model, MRL/lpr mice.11,12 Given that inhibition of C3 convertases prevents generation of anaphylatoxins and other downstream products, it is conceivable that the therapeutic effects we observed were secondary to its effect to limit generation of C3a. Complement proteins can be neuroprotective or neuroinflammatory depending on the setting. C3a prevented neither serum deprivation-induced apoptotic neuronal death, nor AMPA/kainate-mediated excitotoxicity. However, in mixed cultures of neurons and astrocytes, C3a dose-dependently protected neurons against NMDA toxicity.13 Hence, in this study we assessed the role of C3a in CNS lupus using a small molecule inhibitor and the MRL/MpJ-Tnfrsf6lpr (MRL/lpr) strain, mice that are felt to closely reflect that which occurs in human SLE, including the neuropsychiatric manifestations.14
C3a binds to its G-coupled receptor, C3aR, located on different cell types15-17 and induces a wide range of inflammatory and immune effects.8,18,19 The anaphylatoxins can aggravate or exacerbate inflammation and apoptosis, key events in experimental lupus brain.20 When increased in circulation, it can lead to increased infiltration of proinflammatory cells, activation and proliferation of glial cells,21 generation of cytokines and chemokines and thereby apoptosis of nearby cells such as neurons. Apoptosis observed in MRL/lpr mice11,22 can be regulated by the PI3K/Akt pathway23,24 and its negative modulator, PTEN (phosphatase and tensin homolog deleted on chromosome ten)25,26 by acting on diverse downstream targets, including Bad and NF-κB.27,28 Downstream targets of NF-κB such as IL-1β, RANTES, MCP-1 and MIP-2 can mediate CNS functioning29 and play a role in neuropsychiatric (NP)-SLE.30 Caspase-3, the cysteine protease that executes apoptosis, was recently shown to directly cleave AMPA-GluR1 and modulate neuronal excitability, regulate synaptic plasticity and neuronal survival. AMPA-GluR1 forms unique calcium permeable complexes with INFγR on astrocytes. The presence of INFγ on one of the lupus susceptibility loci, its elevated mRNA expression and its ability to cause behavioral disturbances in both humans and animals suggest that the γINF pathway is important for the disease pathogenesis. The goal of this study was to determine the role of C3a/C3aR signaling on these variables.
Our results demonstrate for the first time that C3a produced on complement activation plays an important role in inducing the pathology seen in experimental CNS lupus. C3a acting through its receptor, C3aR, caused apoptosis and gliosis and altered neurotransmission in experimental lupus brain. Although further studies are needed to understand the signaling pathways involved, our results identify C3aR as a possible promising therapeutic target for lupus and other neurodegenerative diseases.
Materials and methods
Reagent and antagonist
All chemicals were procured from Sigma (St Louis, MO, USA) unless otherwise stated. Antibodies used were anti-C3aR, rabbit anti-GFAP (Dako, Carpinteria, CA, USA), rabbit anti-AMPA GLuR1 (Santa Cruz, USA), goat anti-INFγR (Pharmingen), and anti-PCNA. Rabbit anti-Akt, anti-PTEN, and anti-Erk (Cell Signaling) at 1:1000 dilution were used for Western blotting. FITC labeled donkey anti-rabbit (1:300; Jackson ImmunoResearch, USA), FITC anti-goat, and FITC anti-chicken (1:250; Sigma) were used as secondary antibodies.
C3aRa (N2-[(2,2-diphenylethoxy)acetyl]L-arginine) was obtained from Calbiochem and has an IC50 = 200 nM for the mouse C3aR. C3aRa has a short half-life (1.5 h) and therefore was administered continuously using subcutaneously placed osmotic pumps (Alzet model 2001; Durect). The dose of 60 mg/kg/day C3aRa was shown to be effective based on increased survival in these mice.31 In the same studies, C3aR blockade did not alter the autoimmune features in MRL/lpr mice such as circulating anti-dsDNA antibodies and immune complexes.
Treatment
To determine the role of C3aR in CNS lupus, MRL/lpr mice obtained from Jackson Laboratories were treated with the selective non-peptide antagonist of C3aR, C3aRa dissolved in 50% DMSO from the time of clinical onset of disease at 13 weeks of age to the time of LD50 at 19 weeks, when they were killed.31 Control mice were treated with non-specific garbled peptide in 50% DMSO from 13–19 weeks of age. The number of mice used in each group was large due to the pathological variation that may occur between them. Twenty-seven male MRL/lpr mice (The Jackson Laboratory) were randomly divided into two groups to receive C3aRa (n = 13) or vehicle alone (n = 14). Osmotic pumps were replaced weekly in order to maintain the circulating concentration of C3aRa constant. These studies were approved by the University of Chicago Animal Care and Use Committee.
Immunofluorescence microscopy
Cryosections (7 μm) obtained from these mice were fixed in 4% formaldehyde followed by 1:1 ether–ethanol and 95% ethanol. The sections were then exposed to antibodies to visualize the proteins of interest. Antibodies were used at a dilution of 1:100. Briefly, sections were incubated overnight in rabbit anti-C3aR, rabbit anti-MAP2, rabbit anti-AMPA GluR1, goat anti-INFγR, chicken anti-vimentin, or rabbit anti-GFAP. This was followed by incubation (2 h) in FITC donkey anti-rabbit (1:300) second antibody. Alexa-547 coupled PCNA and DAPI for nuclei were used for direct labeling. All assays included negative controls where the primary antibody was omitted. Upon completion of staining, all slides were coded so that the examiner was blind to the treatment group.
Ligase-mediated polymerase chain reaction
Brains were harvested and DNA laddering was amplified and detected by LM-PCR as described previously.20,32 DNA isolated from each animal was ligated with the supplied primer targets for 18 h at 16°C. The ligated DNA was then used as the substrate for PCR, using supplied primers and Advantage DNA polymerase (Clontech Laboratories, Palo Alto, CA) for 23 cycles of 94°C for 1 min and at 72°C for 3 min. The reaction product was electrophoresed through a 1.2% agarose gel, and ethidium bromide-stained bands were detected with UV light illumination.
Assay of caspase-3 activity
Brains were homogenized in cell lysis buffer (25 mM HEPES pH 7.4 buffer containing 2 mM DTT, 5 mM EDTA, and 10 mM digitonin).26 Lysates were incubated on ice for 15 min and protein concentrations were determined in the supernatant using the BCA assay. The supernatants were stored at −80°C. Proteins (50 μg) were incubated at 37°C with assay buffer (50 mM HEPES pH 7.4, 100 mM NaCl, 0.1% CHAPS, 10 mM dithiothreitol, 1 mM EDTA, 10% glycerol) and 200 mM Ac-DEVD-pNA (Biomol, Plymouth Meeting, PA). Hydrolysis of the DEVD-AFC substrate was followed for 15 min by fluorometry of the released AFC (excitation 400 nm, emission 505 nm) and activity was calculated from the slope. The addition of the caspase-3 inhibitor Ac-DEVD-CHO (0.1 mM; Biomol) to the reaction mixture was used to confirm the specificity of the assay.
Quantitative reverse-transcriptase polymerase chain reaction
RNA was isolated from brains using TRIzol reagent (Life Technologies, Grand Island, NY) and qRT-PCR performed as described previously.33 Real-time qRT-PCR for TNF-α, CXCL2/MIP-2, IFN-γ, and ICAM-1 were performed on RNA isolated from brain. All traces of genomic DNA were eliminated with RQ1 DNase (Promega, Madison, WI) at 37°C for 30 min. cDNA was generated from RNA using random hexamers with the SuperScript first-strand synthesis kit (Life Technologies), and qPCR was performed using a Smart Cycler (Cepheid, Sunnyvale, CA) and the SybrGreen intercalating dye method with Hot-Star DNA polymerase (PE Applied Biosystems) according to the manufacturer’s instructions. Each reaction (25 μl) was conducted with 1 μl TaqMan Master Mix (PE Applied Biosystems, Foster City, CA), 3 μl sample or standard cDNA and primers at 200 nM each. PCR was conducted with a hot start at 95°C (5 min), followed by 45 cycles of 95°C for 15s and 60°C for 30s. For each sample, the number of cycles required to generate a given threshold signal (Ct) was recorded. Using a standard curve generated from serial dilutions of splenic cDNA, the ratio of gene expression relative to GAPDH expression was calculated for each experimental and control animal. Primers were synthesized by Integrated DNA Technologies (Coralville, IA) and probes by Synthegen (Houston, TX). The sequences of primers/probes are given in Table 1.
Table 1.
Primers used in quantitative reverse transcriptase polymerase chain reaction
| Genes | Primer 1 (Forward) | Primer 2 (Reverse) |
|---|---|---|
| C3aR | 5′-taaccagatgagcaccacca-3′ | 5′-tgtgaatgttgtgtgcatgg-3′ |
| MIP-2 | 5′-caccaaccaccaggctac-3′ | 5′-gcccttgagagtggctatga-3′ |
| ICAM-1 | 5′-cgcaagtccaattcacactga-3′ | 5′-cagagcggcagagcaaaag-3′ |
| TNF-α | 5′-ccgatgggttgtaccttgtc-3′ | 5′-gtgggtgaggagcacgtagt-3′ |
| IFN-γ | 5′-actggcaaaaggatggtgac-3′ | 5′-tgagctcattgaatgcttgg-3′ |
| GAPDH | 5′-gcaaattcaacggcacagt-3′ | 5′-agatggtgatgggcttccc-3′ |
Western blotting
Tissue samples were homogenized in cold RIPA buffer (with a protease inhibitor cocktail from Sigma). Protein content was measured using the BCA reagent and bovine serum albumin as standard. We electrophoresed 30 μg of protein by SDS–PAGE and electroblotted to PVDF membranes for detection. Membranes were blocked with Tris-buffered saline (TBS) containing 5% dry milk, rinsed with TBS plus Tween (T, 0.05%), then incubated with primary antibodies: anti-Akt, anti-PTEN, and anti-Erk in TBST and bovine serum albumin (0.2%) overnight at 4°C. The primary antibody was removed, membranes washed and peroxidase-labeled anti-rabbit secondary antibody added for 2 h. Following further washes with TBST, bands were visualized with an ECL Western Blotting Analysis System (Amersham, Pierce, Rockford, IL, USA). The Western blots were quantitated using Image J software.
Statistical analyses
Data are expressed as mean ± SEM and were analyzed using Minitab (version 12; Minitab) software. For the comparison between two groups, t testing was used for parametric data, and Mann–Whitney testing was used for non-parametric data. Significance was determined as p < 0.05.
Results
C3aR expression is up-regulated in brains of MRL/lpr mice
C3aR mRNA expression, assessed by real-time PCR, was markedly up-regulated in brains of MRL/lpr mice relative to the age-matched, 19-week-old, MRL/+ controls (p < 0.05). The observed increase in C3aR mRNA was translated into protein as shown by Western blotting (p < 0.05, Figure 1A). Representative cortical sections were stained for C3aR. C3aR was observed to be membrane-bound and localized on the surface of the cells (Figure 1B, A). Higher expression of C3aR was present in MRL/lpr mice (Figure 1B, B) compared with controls Figure 1B, A. C3aR is present on neurons in brains of MRL/lpr mice as indicated by co-staining with anti-MAP2 antibody (Figure 1C).
Figure 1.

C3aR expression is up-regulated in brains of MRL/lpr mice. (A) A significant increase in C3aR mRNA (p < 0.04) and protein expression (p < 0.03) occurred in brains of MRL/lpr mice compared with the congenic MRL/+ controls. mRNA expression of C3aR as determined by RT-PCR was normalized to GAPDH (arrow) and protein expression assessed by Western blotting was normalized to actin (arrow). (B) Representative brain sections stained with Alexa-488 labeled anti-C3aR indicate increased C3aR expression in brains of MRL/lpr mice (B) compared with MRL/+ controls (A). Inset is a cell at higher magnification (40× under oil) showing C3aR receptor expression mainly on the cell-surface. (C) Representative brain sections co-stained with anti-C3aR (A, green) and anti-MAP2 (B, red) and merge (C) indicate C3aR expression in brains of MRL/lpr mice is localized on neurons. Inset are cells (arrow head) at higher magnification (40× under oil).
C3aR inhibition reduces neuronal cell death in MRL/lpr mice
Our earlier studies demonstrate complement-dependent neuronal cell death in brains of MRL/lpr mice compared with their MRL+/+ counterparts.11,20 DNA fragmentation (LM-PCR) techniques show reduced laddering in brains of MRL/lpr mice when treated with C3aRa (Figure 2A, a). Dual staining with specific cell-marker NeuN demonstrates that the main TUNEL-positive cells were neurons (Figure 2b, c and d). The TUNEL positive cells were observed in all tissue sections assessed and the figures shown are representative for the mice in each group. Caspase-3, a critical mediator of apoptosis, was increased in MRL/lpr mouse brains, as assessed by hydrolysis of the DEVD-AFC substrate. Treatment with C3aRa significantly reduced (48% of MRL/lpr mice, p < 0.05) caspase-3 activity in lupus brain (Figure 2B). These findings further support the concept of complement-dependent neuronal apoptosis and demonstrate the important role of C3a/C3aR signaling in causing neurodegeneration in lupus.
Figure 2.

C3aR inhibition reduces apoptosis in experimental lupus brain. (a) Representative gel demonstrates increased apoptosis by LM-PCR in MRL/lpr brains (lanes 1–3). Inhibition of C3aR reduced apoptosis in lupus brains (lanes 4–6). Each lane represents an animal. Shown are representative samples from MRL/lpr mice treated with C3aRa and MRL/lpr mice treated with vehicle alone. Sections from brains of MRL/lpr mice were stained for neurons with anti-neurofilament Ab (c) and for TUNEL-stained apoptotic nuclei (b). Merge of images (b) and (c) show that the cells undergoing apoptosis are predominantly neurons (d). (B) Caspase-3 activity in brains of MRL/lpr mice is reduced by C3aR inhibition. The activity of caspase-3, as assessed by hydrolysis of DEVD-AFC substrate was significantly reduced to 50% in brains of MRL/lpr treated with C3aRa (n = 13) compared with control MRL/lpr mice (n = 14). Data are expressed as mean ± SD. *p < 0.05 compared with matched MRL/lpr mice.
C3aRa treatment alters survival signaling pathways
Since Akt and MAPK/Erk pathways are essential for growth and survival of neurons, we evaluated the role of C3a in these pathways. Western blot analysis revealed that phospho (p)-Akt (phosphorylated at serine 473) was increased in mice treated with C3aRa (p < 0.03) compared with the untreated MRL/lpr mice. In contrast, PTEN, the negative modulator of p-Akt, was substantially reduced in C3aRa treated MRL/lpr mice (p < 0.02) (Figure 3). C3aRa treated mice had increased phosphorylation of Erk (p < 0.02) similar to that observed for Akt indicating that C3aR related (but Fas-independent) signals lead to apoptosis, which may involve PKB/Akt and Erk signaling pathways.
Figure 3.
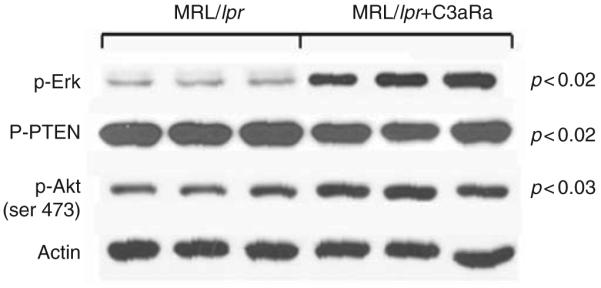
Anaphylatoxin-dependent Akt signaling pathway plays a key role in central nervous system (CNS) apoptosis in lupus. Phosphorylated Akt was increased (p < 0.03) and phospho-PTEN decreased (p < 0.02) in MRL/lpr mice treated from 13 to 19 weeks with C3aRa. Equivalent quantities of CNS protein from individual mice were subjected to SDS-PAGE and immunoblotted with specific Abs for p-Akt (serine 473), p-PTEN (serine 380), and phospho-Erk and normalized to actin. Blockade of the anaphylatoxin receptors led to increased p-Akt and -Erk and reduced p-PTEN. Shown are representative immunoblots from three separate experiments.
Inhibition of C3aR reduces the expression of inflammatory mediators in lupus brain
Increased infiltrating cells in MRL/lpr brains can induce an inflammatory response. In line with this, earlier studies showed reduction of neutrophil infiltration into brains of MRL/lpr mice reduced the severity of disease. However, our results show that C3aR inhibition caused a reduction in neutrophil number which did not reach statistical significance (data not shown). C3a binds to its G-coupled receptor, present on glial and infiltrating cells and initiates inflammatory functions. Since one of the predominant effects is the induction of cytokine signaling, we studied the mRNA expression of inflammatory mediators in the brains of these mice. IFNg-, TNFa-, ICAM-1, and CXCL2/MIP-2 were measured to provide insight into the potential mechanism(s) that result from complement activation and thereby increased C3a expression. As shown in Figure 4, the expression of TNFa- (0.33 ± 0.07 versus 0.15 ± 0.1, p < 0.05), MIP-2 (3.8 ± 1.3 versus 1.7 ± 0.6, p < 0.02), and IFNg- (4.83 ± 1.0 versus 2.07 ± 1.28, p < 0.02) in control and C3aRa-treated groups, respectively, were significantly changed, while ICAM-1 (5.0 ± 1.6 versus 5.0 ± 2.3) remained unaltered in C3aR-inhibited lupus brain, indicating that the expression of these cytokines appears to be mediated, at least in part, through signals delivered through C3aR.
Figure 4.

C3a alters the expression of inflammatory mediators in MRL/lpr brains. mRNA expression of the inflammatory mediators TNFα, MIP2, γ-INF, and ICAM-1 in MRL/lpr mice treated from 13 to 19 weeks of age with C3aRa (n = 13) or vehicle (n = 14) were assessed by qRT-PCR. The expression of the mediators was significantly decreased in the brains of antagonist treated mice, *p < 0.05, compared with vehicle-treated control MRL/lpr mice except ICAM-1 expression which remained the same in both groups. Data presented as units are expression relative to 18S RNA measured in the same sample.
C3aRa treatment alters AMPA-GluR1 and INFγ signaling
Our earlier studies showed that AMPA-GluR1 is altered in CNS lupus while the studies of several others demonstrated that the INF-pathway is important for the disease pathogenesis. Immunofluorescence shows that expression of both the AMPA-GluR1 (red, Figure 5A and B) and INFγR (green, Figure 5C and D) co-localize (merge, Figure 5E and F) and they are significantly reduced in brains of MRL/lpr mice treated with C3aRa (Figure 5B, D and F) compared with their untreated counterparts (Figure 5A, C and E).
Figure 5.
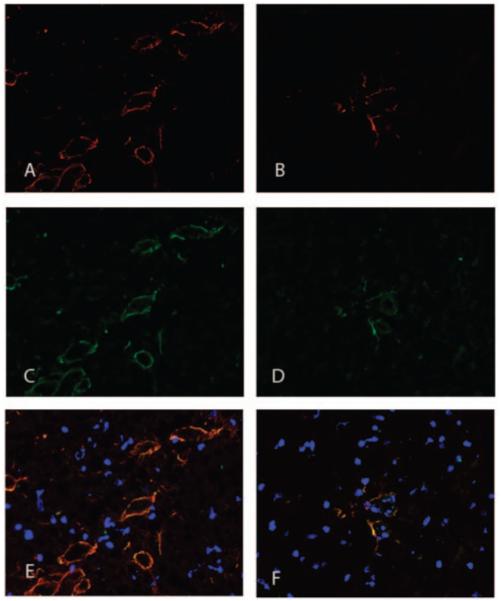
Expression of INFγR and AMPA-GluR1 are C3aR-dependent. Representative 7 μm cortical cryosections stained for INFγR (red) and AMPA-GluR1 (green) show that they co-localize in brain. Sections were counterstained with DAPI (blue). Both INFγR and AMPA-GluR1 were highly expressed in MRL/lpr brain (A, C and E) compared with brains from C3aR-inhibited mice (B, D and F).
C3aRa treatment reduces iNOS mRNA expression in lupus mice
IFN-γ is a potent inducer of NO, and can induce iNOS expression. In this study using qRT-PCR we examined and found that inhibition of C3aR significantly reduced iNOS mRNA expression in brains of lupus mice (Figure 6, p < 0.05).
Figure 6.

Expression of iNOS is decreased in brains of MRL/lpr mice treated with C3aRa. As shown by qRT-PCR, iNOS mRNA expression was decreased in brains from MRL/lpr mice treated with C3aRa compared with MRL/lpr mice treated with vehicle alone. Data are shown as mean values from C3aRa (n = 13) or vehicle (n = 14) treated mice. *p < 0.05.
C3aR inhibition reduces gliosis in MRL/lpr mice
Since INFγR and AMPA-GluR1 were observed on astrocytes, we studied the involvement of these cells in lupus by immunostaining for glial fibrillary acidic protein (GFAP) and vimentin in brain sections. The astrocytes in MRL/lpr mice show marked reduction of GFAP and vimentin immunoreactivity, on C3aR inhibition (Figure 7B and D). Both GFAP and vimentin co-localized on the astrocytes. However, vimentin containing astrocytes exhibited fewer long and straight processes and was predominant around the microvasculature (Figure 7C, arrow). This was accompanied by decreased proliferation as indicated by PCNA staining (red, Figure 8) with DAPI in blue.
Figure 7.

C3aR antagonist treatment reduces astrogliosis, in the brains of lupus mice. Representative sections immunostained for GFAP and vimentin to determine the degree of astrocyte involvement. Sections from MRL/lpr mice treated with vehicle (A and C) or with C3aRa (B and D) are given here. Vehicle-administered mice had numerous GFAP (A) and vimentin (C)-labeled astrocytes compared with those treated with C3aRa (B and D). Photomicrographs were taken at 10X magnification with a Zeiss camera. All images are typical and representative of each group. (B) Panel A shows vimentin (green) strongly expressed around the micro-vasculature compared with GFAP (red) that was present around the periphery. However, panel B shows that both GFAP and vimentin co-localized in the astrocytes. Inset shows that GFAP is predominant in the astrocytic processes. Nuclei are stained in blue (DAPI).
Figure 8.
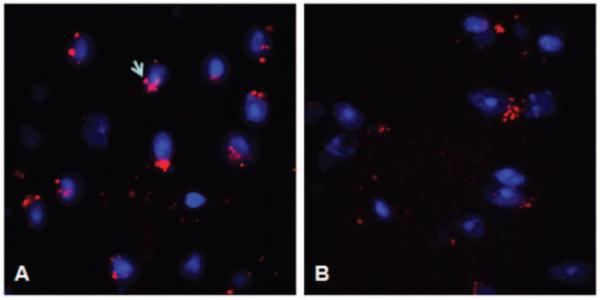
Proliferation is reduced in C3aR-inhibited mice. Proliferation (PCNA) was measured in MRL/lpr mice treated with vehicle (A) or C3aRa (B). Representative cortical cryosections stained with DAPI (blue) and PCNA (red) indicate that proliferation is reduced in MRL/lpr mice treated with C3aRa compared with those given vehicle alone.
Discussion
The present study extends our previous observations and provides new insights on the role of complement in lupus brain. Our earlier studies showed that complement inhibition with Crry or factor B deletion exerted a positive effect and reduced both the pathological changes in the brain of lupus mice and the resulting behavioral alterations.11,20 The present study, designed to identify effective downstream therapeutic targets, demonstrates the potential of C3a as a strong therapeutic candidate. C3aR expression is increased in the brains of MRL/lpr mice, suggesting a role for this receptor and its ligand in lupus brain.
Gliosis can be beneficial or harmful and its manipulation could provide an appropriate environment for neuronal regeneration. Significant activation of astrocytes characterized by hyperplasia, hypertrophy, and increased GFAP content and vimentin expression occurs in MRL/lpr brain.33 Astrocyte activation or astrogliosis was observed mainly in the hippocampus and cortical regions of the brain. In line with gliosis our results show C3a-dependent increase in proinflammatory cytokines. The expression of ICAM-1 which regulates the transport of cells across membranes is increased in MRL/lpr brains and was not affected by C3aR inhibition. Concomitantly, there was no significant change in the infiltration of neutrophils into brain. The increase of cytokines γ-INF, TNFα, and MIP2 observed in lupus brain could aggravate inflammation and cause apoptosis. Neuronal apoptosis is a key event in experimental lupus brain.11 In a complex, systemic disease such as lupus, several factors could cause neuronal death including cytokines, oxygen free radicals, and autoantibodies. C3aR inhibition maintained the survival signaling molecules, including phosphorylated Erk, Akt (serine 473), and its negative modulator PTEN at normal levels in lupus brains. Since Fas protein is absent in MRL/lpr mice, it is conceivable that C3a regulates apoptosis through Akt and Erk signaling pathways independent of Fas, in the setting of lupus.
IFN and IFN-inducible gene signatures with elevated levels of mRNA are present in both lupus patients and mouse models, suggesting that they may play a role in the disease pathogenesis.34,35 IFN-γRs are expressed by both neurons and glia36,37 and form a unique, neuron-specific, calcium-permeable receptor complex with AMPA receptor subunit GluR1.38 In addition, IFN-γ is a potent inducer of NO,39 and can induce both calcium-dependent NO synthase in neurons (nNOS) or the inducible NOS (iNOS). NO can amplify the release of glutamate from presynaptic sites40 and inhibit glutamate uptake by astrocytes concomitant with the increased glutamate levels observed in these mice.41 NO produced along with the phosphorylated AMPA-GluR1 can alter Ca(2+) influx, decrease ATP production, and cause neuronal toxicity. Increase in the transcription factor c-Fos is regulated by glutamate and indicates neuronal activation, astrocytic alteration, and stress in the brain. Future studies are required to decipher the crosstalk between apoptosis and gliosis following insult in these mice.
In this study, we have shown for the first time that C3a plays an important role in CNS lupus, signaling through its receptor C3aR. Inhibition of C3aR alleviated two key events, neuronal apoptosis and gliosis, in this complex setting. In addition, our studies suggest that the neuronal toxicity could be through the INFγR–AMPA–GluR1 complex. Since this study has addressed the changes occurring globally in brain, further studies are required to understand the cellular localization of the signaling crosstalk in the lupus brain.
Acknowledgements
We thank Ms Miglena Petkova for excellent technical assistance. This work was supported by National Institutes of Health Grant R01DK055357 (to RJQ).
Abbreviations
- Akt/PKB
protein kinase B
- C3aRa
(N2-[(2, 2-diphenylethoxy)acetyl]L-arginine)
- CNS
central nervous system
- GFAP
glial fibrillary acidic protein
- LM-PCR
ligase-mediated polymerase chain reaction
- MRL/lpr
MRL/MpJ-Tnfrsf6lpr
- NP-SLE
neuropsychiatric systemic lupus erythematosus
- PTEN
phosphatase and tensin homolog deleted on chromosome ten
- qRT-PCR
quantitative reverse-transcriptase polymerase chain reaction
- SDS
sodium dodecyl sulfate
- SLE
systemic lupus erythematosus
References
- 1.Huizinga TW, Diamond B. Lupus and the central nervous system. Lupus. 2008;17:376–379. doi: 10.1177/0961203308090112. [DOI] [PubMed] [Google Scholar]
- 2.Wyss-Coray T, Yan F, Lin AH, et al. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer’s mice. Proc Natl Acad Sci USA. 2002;99:10837–10842. doi: 10.1073/pnas.162350199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tenner AJ, Fonseca MI. The double-edged flower: Roles of complement protein C1q in neurodegenerative diseases. Adv Exp Med Biol. 2006;586:153–176. doi: 10.1007/0-387-34134-X_11. [DOI] [PubMed] [Google Scholar]
- 4.Tenner AJ. Complement in Alzheimer’s disease: Opportunities for modulating protective and pathogenic events. Neurobiol Aging. 2001;22:849–861. doi: 10.1016/s0197-4580(01)00301-3. [DOI] [PubMed] [Google Scholar]
- 5.Davoust N, Nataf S, Reiman R, Holers V, Campbell IL, Barnum SR. Central nervous system-targeted expression of the complement inhibitor sCrry prevents experimental allergic encephalomyelitis. J Immunol. 1999;163:6551–6556. [PubMed] [Google Scholar]
- 6.Alexander JJ, Quigg RJ. Systemic lupus erythematosus and the brain: What mice are telling us. Neurochem Int. 2007;50:5–11. doi: 10.1016/j.neuint.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 7.van Beek J, Bernaudin M, Petit E, et al. Expression of receptors for complement anaphylatoxins C3a and C5a following permanent focal cerebral ischemia in the mouse. Exp Neurol. 2000;161:373–382. doi: 10.1006/exnr.1999.7273. [DOI] [PubMed] [Google Scholar]
- 8.Barnum SR, Ames RS, Maycox PR, et al. Expression of the complement C3a and C5a receptors after permanent focal ischemia: An alternative interpretation. Glia. 2002;38:169–173. doi: 10.1002/glia.10069. [DOI] [PubMed] [Google Scholar]
- 9.Chenoweth DE, Cooper SW, Hugli TE, Stewart RW, Blackstone EH, Kirklin JW. Complement activation during cardiopulmonary bypass: Evidence for generation of C3a and C5a anaphylatoxins. N Engl J Med. 1981;304:497–503. doi: 10.1056/NEJM198102263040901. [DOI] [PubMed] [Google Scholar]
- 10.Belmont HM, Hopkins P, Edelson HS, et al. Complement activation during systemic lupus erythematosus. C3a and C5a anaphylatoxins circulate during exacerbations of disease. Arthritis Rheum. 1986;29:1085–1089. doi: 10.1002/art.1780290905. [DOI] [PubMed] [Google Scholar]
- 11.Alexander JJ, Jacob A, Bao L, MacDonald RL, Quigg RJ. Complement-dependent apoptosis and inflammatory gene changes in murine lupus cerebritis. J Immunol. 2005;175:8312–8319. doi: 10.4049/jimmunol.175.12.8312. [DOI] [PubMed] [Google Scholar]
- 12.Alexander JJ, Bao L, Jacob A, Kraus DM, Holers VM, Quigg RJ. Administration of the soluble complement inhibitor, Crry-Ig, reduces inflammation and aquaporin 4 expression in lupus cerebritis. Biochim Biophys Acta. 2003;1639:169–176. doi: 10.1016/j.bbadis.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 13.van Beek J, Nicole O, Ali C, et al. Complement anaphylatoxin C3a is selectively protective against NMDA-induced neuronal cell death. Neuroreport. 2001;12(2):289–293. doi: 10.1097/00001756-200102120-00022. [DOI] [PubMed] [Google Scholar]; Proc Natl Acad Sci USA. 1993;90:1756–1760. doi: 10.1073/pnas.90.5.1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356:314–317. doi: 10.1038/356314a0. [DOI] [PubMed] [Google Scholar]
- 15.Davoust N, Jones J, Stahel PF, Ames RS, Barnum SR. Receptor for the C3a anaphylatoxin is expressed by neurons and glial cells. Glia. 1999;26:201–211. doi: 10.1002/(sici)1098-1136(199905)26:3<201::aid-glia2>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 16.Gasque P, Singhrao SK, Neal JW, et al. The receptor for complement anaphylatoxin C3a is expressed by myeloid cells and non-myeloid cells in inflamed human central nervous system: Analysis in multiple sclerosis and bacterial meningitis. J Immunol. 1998;160:3543–3554. [PubMed] [Google Scholar]
- 17.Ischenko A, Sayah S, Patte C, et al. Expression of a functional anaphylatoxin C3a receptor by astrocytes. J Neurochem. 1998;71:2487–2496. doi: 10.1046/j.1471-4159.1998.71062487.x. [DOI] [PubMed] [Google Scholar]
- 18.Nataf S, Stahel PF, Davoust N, Barnum SR. Complement anaphylatoxin receptors on neurons: new tricks for old receptors? Trends Neurosci. 1999;22:397–402. doi: 10.1016/s0166-2236(98)01390-3. [DOI] [PubMed] [Google Scholar]
- 19.van Beek J, Bernaudin M, Petit E, et al. Expression of receptors for complement anaphylatoxins C3a and C5a following permanent focal cerebral ischemia in the mouse. Exp Neurol. 2000;161:373–382. doi: 10.1006/exnr.1999.7273. [DOI] [PubMed] [Google Scholar]
- 20.Alexander JJ, Jacob A, Vezina P, Sekine H, Gilkeson GS, Quigg RJ. Absence of functional alternative complement pathway alleviates lupus cerebritis. Eur J Immunol. 2007;37:1691–1701. doi: 10.1002/eji.200636638. [DOI] [PubMed] [Google Scholar]
- 21.Eng LF, Ghirnikar RS, Lee YL. Glial fibrillary acidic protein: GFAP-thirty-one years (1969-2000) Neurochem Res. 2000;25:1439–1451. doi: 10.1023/a:1007677003387. [DOI] [PubMed] [Google Scholar]
- 22.Sakic B, Maric I, Koeberle PD, et al. Increased TUNEL staining in brains of autoimmune Fas-deficient mice. J Neuroimmunol. 2000;104:147–154. doi: 10.1016/s0165-5728(99)00277-5. [DOI] [PubMed] [Google Scholar]
- 23.Downward J. PI 3-kinase, Akt and cell survival. Semin Cell Dev Biol. 2004;15:177–182. doi: 10.1016/j.semcdb.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 24.Scheid MP, Woodgett JR. Unravelling the activation mechanisms of protein kinase B/Akt. FEBS Lett. 2003;546:108–112. doi: 10.1016/s0014-5793(03)00562-3. [DOI] [PubMed] [Google Scholar]
- 25.Sulis ML, Parsons R. PTEN: From pathology to biology. Trends Cell Biol. 2003;13:478–483. doi: 10.1016/s0962-8924(03)00175-2. [DOI] [PubMed] [Google Scholar]
- 26.Vazquez F, Ramaswamy S, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol. 2000;20:5010–5018. doi: 10.1128/mcb.20.14.5010-5018.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goswami R, Kilkus J, Dawson SA, Dawson G. Overexpression of Akt (protein kinase B) confers protection against apoptosis and prevents formation of ceramide in response to pro-apoptotic stimuli. J Neurosci Res. 1999;57:884–893. [PubMed] [Google Scholar]
- 28.Harada N, Hatano E, Koizumi N, et al. Akt activation protects rat liver from ischemia/reperfusion injury. J Surg Res. 2004;121:159–170. doi: 10.1016/j.jss.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 29.Shintani F, Kanba S, Nakaki T, et al. Interleukin-1 beta augments release of norepinephrine, dopamine, and serotonin in the rat anterior hypothalamus. J Neurosci. 1993;13:3574–3581. doi: 10.1523/JNEUROSCI.13-08-03574.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hirohata S, Miyamoto T. Elevated levels of interleukin-6 in cere-brospinal fluid from patients with systemic lupus erythematosus and central nervous system involvement. Arthritis Rheum. 1990;33:644–649. doi: 10.1002/art.1780330506. [DOI] [PubMed] [Google Scholar]
- 31.Bao L, Osawe I, Haas M, Quigg RJ. Signaling through up-regulated C3a receptor is key to the development of experimental lupus nephritis. J Immunol. 2005;175:1947–1955. doi: 10.4049/jimmunol.175.3.1947. [DOI] [PubMed] [Google Scholar]
- 32.Jacob A, Hensley LK, Safratowich BD, Quigg RJ, Alexander JJ. The role of the complement cascade in endotoxin-induced septic encephalopathy. Lab Invest. 2007;87:1186–1194. doi: 10.1038/labinvest.3700686. [DOI] [PubMed] [Google Scholar]
- 33.Eng LF, Yu AC, Lee YL. Astrocytic response to injury. Prog Brain Res. 1992;94:353–365. doi: 10.1016/s0079-6123(08)61764-1. [DOI] [PubMed] [Google Scholar]
- 34.Baechler EC, Batliwalla FM, Karypis G, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA. 2003;100:2610–2615. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bennett L, Palucka AK, Arce E, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197:711–723. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vikman K, Robertson B, Grant G, Liljeborg A, Kristensson K. Interferon-gamma receptors are expressed at synapses in the rat superficial dorsal horn and lateral spinal nucleus. J Neurocytol. 1998;27:749–759. doi: 10.1023/a:1006903002044. [DOI] [PubMed] [Google Scholar]
- 37.Wang Y, Zhou CF. Involvement of interferon-gamma and its receptor in the activation of astrocytes in the mouse hippocampus following entorhinal deafferentation. Glia. 2005;50:56–65. doi: 10.1002/glia.20152. [DOI] [PubMed] [Google Scholar]
- 38.Mizuno T, Zhang G, Takeuchi H, et al. Interferon-gamma directly induces neurotoxicity through a neuron specific, calcium-permeable complex of IFN-gamma receptor and AMPA GluR1 receptor. FASEB J. 2008;22:1797–1806. doi: 10.1096/fj.07-099499. [DOI] [PubMed] [Google Scholar]
- 39.Vikman KS, Owe-Larsson B, Brask J, Kristensson KS, Hill RH. Interferon-gamma-induced changes in synaptic activity and AMPA receptor clustering in hippocampal cultures. Brain Res. 2001;896:18–29. doi: 10.1016/s0006-8993(00)03238-8. [DOI] [PubMed] [Google Scholar]
- 40.Neumann H, Schmidt H, Cavalie A, Jenne D, Wekerle H. Major histocompatibility complex (MHC) class I gene expression in single neurons of the central nervous system: differential regulation by interferon (IFN)-gamma and tumor necrosis factor (TNF)-alpha. J Exp Med. 1997;185:305–316. doi: 10.1084/jem.185.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alexander JJ, Zwingmann C, Quigg R. MRL/lpr mice have alterations in brain metabolism as shown with [(1)H-(13)C] NMR spectroscopy. Neurochem Int. 2005;47:143–151. doi: 10.1016/j.neuint.2005.04.016. [DOI] [PubMed] [Google Scholar]