

Abstract
Although the pathophysiology of diabetes is characterized by elevated levels of glucose and long-chain fatty acids (LCFA), nuclear mechanisms linking glucose and LCFA metabolism are poorly understood. As the liver fatty acid binding protein (L-FABP) shuttles LCFA to the nucleus, where L-FABP directly interacts with peroxisome proliferator-activated receptor-α (PPARα), the effect of glucose on these processes was examined. In vitro studies showed that L-FABP strongly bound glucose and glucose-1-phosphate (Kd = 103 ± 19 nM and Kd = 20 ± 3 nM, respectively), resulting in altered L-FABP conformation, increased affinity for lipid ligands, and enhanced interaction with PPARα. In living cells, glucose stimulated cellular uptake and nuclear localization of a nonmetabolizable fluorescent fatty acid analog (BODIPY C-16), particularly in the presence of L-FABP. These data suggest for the first time a direct role of glucose in facilitating L-FABP-mediated uptake and distribution of lipidic ligands to the nucleus for regulation of PPARα transcriptional activity.
Keywords: nuclear, transcription factor, cytoplasmic lipid binding protein
Elevated serum fatty acids and sugars are significant cardiovascular risk factors in diabetes, obesity, and metabolic syndrome (1–4). While these nutrients regulate transcription of multiple genes involved in their own metabolism, nuclear mechanistic links between sugar and fatty acid regulation remain elusive. Recent evidence suggests that peroxisome proliferator-activated receptors (PPARs), ligand-activated transcription factors belonging to the nuclear hormone receptor superfamily, play a central role in energy homeostasis by initiating transcription of multiple genes involved in fatty acid oxidation and glucose metabolism. In liver, PPARα induces transcription of genes involved in long-chain fatty acid (LCFA) uptake and transport (e.g., liver fatty acid binding protein [L-FABP]), fatty acid degradation by β-oxidation, and lipoprotein metabolism (4, 5). Thus, activation of PPARα induces transcription of a number of lipid metabolic proteins whose abnormal regulation may contribute to diabetes and obesity.
Although it is known that exogenous LCFAs activate PPARα and that certain PPARα-targeting drugs (fibrates) used in cardiovascular and diabetes therapy enhance glucose uptake, increase fatty acid metabolism, and improve insulin sensitivity (6, 7), the identity of endogenous, high-affinity PPARα ligands has proven more elusive. Only recently was it shown that PPARα exhibits high affinity for unsaturated (but not saturated) LCFA (8, 9) and all examined CoA thioesters of LCFA (LCFA-CoA) (9, 10). Upon binding these lipids, PPARα undergoes a conformational change and increased activation, consistent with LCFA and LCFA-CoA being endogenous ligands. The latter is especially likely as nuclear concentrations of LCFA and LCFA-CoA are in the range of PPARα affinity for these ligands (11, 12). New findings show that glucose is also an endogenous, high-affinity ligand of PPARα (13, 14)—thereby suggesting that PPARα may be responsible for maintaining energy homeostasis through concentration-dependent regulation by both lipids and sugars.
Parallel studies to these developments have begun to elucidate the mechanism whereby highly lipophilic, poorly water soluble ligands, such as LCFA and LCFA-CoA, are transported to the nucleus for interacting with and activating PPARα (reviewed in Ref. 15). Key in this regard is the finding that metabolically active tissues, such as liver, express high levels (2–3% of cytosolic protein) of L-FABP, which has high affinity for both LCFA and LCFA-CoA (16–18). L-FABP is thought to function as a shuttle that delivers these metabolites to the nucleus for interaction with nuclear receptors, such as PPARα (15, 19, 20). Since glucose potentiates PPARα activation by LCFA (13, 14), a potential effect of glucose on L-FABP and/or the L-FABP-PPARα interaction could play an important role in the maintenance of energy homeostasis. To begin to resolve the role L-FABP may play in such regulation, the effect of glucose directly on L-FABP and the PPARα-L-FABP complex was examined.
EXPERIMENTAL PROCEDURES
Protein expression and purification
Rat recombinant L-FABP protein was produced, purified, and delipidated as described (21). Mouse recombinant PPARαΔAB protein (plasmid generously provided by Dr. Noa Noy, Case Western Reserve University) was produced and purified as described (8, 13).
Glucose binding assay
The direct binding of glucose and glucose metabolites (glucose-1-phosphate [G-1-P] and glucose-6-phosphate [G-6-P]) or maltose to L-FABP (500 nM) was determined by quenching of intrinsic L-FABP aromatic amino acid fluorescence as previously described for PPARα (9, 13). Emission spectra from 300–400 nm were obtained at 24°C upon excitation at 280 nm with a PC1 photon counting spectrofluorometer (ISS, Champaign, IL). Data were corrected for background (buffer, ligands, and solvent effects), and maximal intensities were used to calculate the dissociation constant (Kd) with the ligand binding function of SigmaPlot (SPSS, Chicago, IL) utilizing the one-site saturation feature (22).
To determine if binding may also occur without resulting in tyrosine quenching, binding was also determined as quenching of fluorescently labeled L-FABP in the presence of glucose or glucose metabolites. Recombinant L-FABP protein was chemically labeled with Cy5 dye using a Fluorolink-antibody Cy5 labeling kit (Amersham Biosciences, Pittsburgh, PA) according to the manufacturer's recommendations. Absorbance measurements estimated the labeling efficiency to be approximately one dye per protein molecule. Cy5-L-FABP (25 nM) was titrated with increasing concentrations of glucose, glucose-1-phosphate, glucose-6-phosphate, or maltose at 24°C, and emission spectra from 655-700 nm were obtained upon excitation of the Cy5 dye at 645 nm in a PC1 photon counting spectrofluorometer (ISS, Champaign, IL). Data were corrected for background (buffer, ligands, and solvent effects) and maximal intensities were used to calculate the dissociation constant (Kd) with the ligand binding function of SigmaPlot (SPSS, Chicago, IL) utilizing the one-site saturation feature (22).
Fluorescent fatty acid and acyl-CoA binding
The fluorescent C16 fatty acid analog (BODIPY C-16), the naturally occurring fluorescent C18 cis-parinaric acid, and fluorescent C18 trans-parinaric acid were purchased from Molecular Probes (Eugene, OR). The cis-parinaroyl-CoA and BODIPY C16-CoA were synthesized from the respective parent fatty acids and purified by HPLC as previously described (9). To determine the effect of glucose and glucose metabolites on fluorescent LCFA or fluorescent LCFA-CoA binding to L-FABP, 100 nM L-FABP protein was mixed with 50 nM BODIPY C-16, BODIPY C16-CoA, trans-parinaric acid, or cis-parinaroyl-CoA, and the fluorescence intensity of the fluorophore was measured in the absence and presence of glucose. Emission spectra were obtained at 24°C with a PC1 photon counting spectrofluorometer (ISS Inc.). The BODIPY fluorophore was excited at 460 nm, and emission was measured from 490 nm to 540 nm. The parinaric lipids were excited at 317 nm, and emission measured from 380 nm to 450 nm. Data were corrected for background (buffer, fluorescent ligands, and solvent effects), and maximal intensities were used to calculate the percent change in LCFA or LCFA-CoA binding as previously described (13).
Circular dichroism
To determine glucose effects on protein secondary structure, circular dichroic (CD) spectra of 0.8 μM PPARαΔAB, 2.4 μM L-FABP, and equal amino acid molar concentrations of the two proteins [0.4 μM PPARαΔAB + 1.2 μM L-FABP] were taken in the absence and presence of glucose, maltose, and glucose metabolites with a J-815 spectropolarimeter (JASCO, Easton, MD) as previously described (13, 20). Ten scans per replicate were averaged for percent composition of secondary structures by three different methods (SELCON3, CDSSTR, and CONTIN/LL) with the software package CDPro (23) as previously described (13, 20). The CD spectrum of the mixed proteins was compared with a theoretical spectrum of combined but noninteracting proteins in the absence and presence of sugar. This spectrum was the average of each protein analyzed separately at a concentration equal to that in the mixture (24).
Co-immunoprecipitation assays
Briefly, 20 μg of each recombinant protein was mixed and allowed to incubate for 10 min in the absence or presence of glucose. Next, protein mixtures were incubated with antibody-linked resin (rabbit polyclonal anti-PPARα [Affinity BioReagents, Golden, CO] or goat polyclonal anti-L-FABP [Santa Cruz Biotechnology, Santa Cruz, CA]) for 1 1/2 h at room temperature according to the manufacturers' instructions (ProFoundTM mammalian co-immunoprecipitation kit, Pierce Biotechnology, Rockford, IL). Eluted proteins were examined by SDS-PAGE analysis as previously described (13). Values were normalized to the amount of protein detected from the control (e.g., the protein used for co-immunoprecipitation [CoIP]), and samples in absence of glucose were arbitrarily set to 100 percent.
Fluorescence resonance energy transfer
To further confirm effects of glucose on direct binding of PPARαΔAB and L-FABP, these recombinant proteins were fluorescently labeled with Cy3 and Cy5 dyes, respectively, using a Fluorolink-antibody Cy3 and Cy5 labeling kit (Amersham Biosciences, Pittsburgh, PA) according to the manufacturer's recommendations. Absorbance measurements were made of labeled and nonlabeled proteins to determine protein concentrations and dye to protein ratios. Cy3-PPARαΔAB protein (30 nM) was titrated with increasing concentrations of Cy5-L-FABP, and the emission spectra from 560 nm to 700 nm were measured upon excitation at 550 nm in a PC1 photon counting spectrofluorometer (ISS) as previously described (20, 25). This experiment was repeated in the presence of varying glucose concentrations. All spectra were corrected for background fluorescence (buffer and Cy5-L-FABP bleed-through), and maximal fluorescence intensities were used to calculate the affinity of the two proteins as well as the intermolecular distance between them as previously described (20, 26).
Cell culture
Murine L-cells (L arpt-tk-), mock-transfected control L-cells, and L-cells transfected with cDNA encoding for L-FABP (27) were seeded onto Lab-Tek chambered cover glass and cultured as previously described (11). Culture media was replaced with PBS containing 1.25 μM Syto59 DNA-binding dye (Molecular Probes) and incubated at 37°C for 25min. Cells were washed with PBS, followed by the addition of 100 nM BODIPY C-16 (Molecular Probes) in the presence or absence of glucose, maltose, or sodium chloride at 37°C for 15 min, and then images were acquired in PBS at room temperature.
Laser scanning confocal microscopy
The real-time distribution of LCFA to nuclei (localization of PPARα) in response to glucose was determined by laser scanning confocal microscopy (LSCM) of BODIPY C-16, a poorly metabolizable fluorescent LCFA (11). LSCM studies were performed with a 63× Plan-Fluor oil immersion objective, N.A.1.45, an Axiovert 135 microscope (Zeiss, Carl Zeiss, Thornwood, NY), and MRC-1024 fluorescence imaging system (Bio-Rad, Hercules, CA) as described (11). Syto59 and BODIPY C-16 probes were excited at 488/568 lines with a krypton-argon laser (Coherent, Sunnyvale, CA). Emission from BODIPY-labeled fatty acid was recorded by a photomultiplier after passing through a 522/D35 emission filter, and emission from Syto59 was collected with a 680/32 emission filter, both under manual gain and black level control. The objective was focused to acquire 0.3 μm confocal slice images through a median section of cells in the field. Cells were excited for 0.1 s intervals, regulated by a computer-controlled shutter and Laser Sharp software (Bio-Rad). Cells incubated with each component individually were used as background controls and to optimize instrument settings. Images were analyzed using MetaMorph software (Advanced Scientific Imaging, Meraux, LA). Syto59 fluorescence was used to outline the nuclear region, so that the amount of BODIPY C-16 fluorescence could be measured both in the entire cell and just in the nucleus. The cellular uptake of BODIPY C-16 was calculated by dividing the BODIPY C-16 fluorescence of the whole cell by the area of the whole cell for each cell; the amount of BODIPY C-16 in the nucleus was calculated by dividing the BODIPY C-16 fluorescence inside the nucleus by the area of the nucleus for each cell; the percentage of BODIPY C-16 in the nucleus was calculated by dividing the BODIPY C-16 fluorescence intensity inside the nucleus by the BODIPY C-16 fluorescence intensity of the whole cell multiplied by 100 percent for each cell. The final data were averages of 50–200 cells after each experiment was replicated three to four times in separate chambers, using several images from different areas within each chamber.
Statistical analysis
All results are expressed as mean ± standard error. Statistical significance between samples in the presence or absence of glucose, glucose metabolites, or maltose was determined by using the Student's t-test or ANOVA with P < 0.05.
RESULTS
L-FABP exhibited high affinity for glucose and glucose metabolites
Although L-FABP binds LCFA and LCFA-CoA with high affinity (16–18), it is not known if L-FABP also binds glucose with high affinity. Therefore, this possibility was examined by a direct fluorescence binding assay taking advantage of the change in L-FABP intrinsic aromatic amino acid fluorescence intensity in response to ligand binding. Plotting the change in L-FABP amino acid fluorescence as a function of increasing glucose concentration yielded a sharp saturation curve with the maximal change in fluorescence intensity noted at approximately 400 nM (Fig. 1A). Transformation of the binding data into a linear reciprocal plot (Fig. 1A, inset) indicated high affinity binding at a single binding site. Multiple replicates (n = 4) showed that L-FABP strongly bound glucose (Kd = 103.1 ± 18.6 nM), in the same range as previously shown for L-FABP binding to LCFA and LCFA-CoA (16). L-FABP binding to glucose-1-phosphate (G-1-P) was also saturable (Fig. 1B) at a single binding site (Fig. 1B, inset) with even higher affinity as shown by Kd = 20.2 ± 3.5 nM. In contrast, glucose-6-phosphate (G-6-P) was only weakly bound (Fig. 1C). To determine the specificity of this binding, the effect of maltose on L-FABP intrinsic fluorescence was examined, and no binding was noted (Fig. 1G).
Fig. 1.

L-FABP binds glucose and glucose-1-phosphate with high affinity. Binding curves of the change in L-FABP intrinsic amino acid fluorescence at 310 nm upon excitation at 280 nm as a function of glucose (A), glucose-1-phosphate (B), and glucose-6-phosphate (C) concentration. Binding curves of the change in Cy5-L-FABP fluorescence emission intensity at 659 nm upon excitation at 645 nm as a function of glucose (D), glucose-1-phosphate (E), and glucose-6-phosphate (F) concentration. As negative controls, the change in L-FABP intrinsic amino acid fluorescence at 310 nm upon excitation at 280 nm (G) and the change in Cy5-L-FABP fluorescence emission intensity at 659 nm upon excitation at 645 nm (H) as a function of maltose concentration were examined. Values are presented as the mean value (n = 4) ± the SEM. Insets are double reciprocal plots of the mean binding curve data presented in each panel. L-FABP, liver fatty acid binding protein.
As it is possible for binding to occur without resulting in quenching of intrinsic amino acid fluorescence, the binding of glucose and glucose metabolites to Cy5-labeled L-FABP was examined. The change in Cy5 fluorescence intensity as a function of glucose (Fig. 1D) or G-1-P (Fig. 1E) concentration resulted in sharp saturation curves, whereas G-6-P binding resulted in a much shallower curve (Fig. 1F). Transformation of these data into linear reciprocal plots (Fig. 1D-F, insets) established high-affinity binding of glucose and G-1-P at a single binding site in L-FABP. Multiple replicates (n = 4) yielded binding affinities for glucose and G-1-P that were similar, although slightly weaker, to those obtained through the direct L-FABP aromatic amino acid fluorescence binding assay (Kd = 197.6 ± 53.4 nM for glucose and Kd = 26.5 ± 4.4 nM for G-1-P, respectively). While the Cy5-L-FABP binding assay was able to detect G-6-P binding to L-FABP, the affinity was three orders of magnitude weaker (Kd = 26.7 ± 7.9 μM), and no binding was noted for maltose (Fig. 1H). Taken together with the fact that these binding affinities of L-FABP for glucose, G-1-P, and G-6-P were in the range of physiological intracellular concentrations of these molecules (13), these data were consistent with glucose and especially its G-1-P metabolite being potential endogenous ligands of L-FABP.
L-FABP binding to glucose resulted in a glucose concentration-dependent alteration in L-FABP secondary structure
To determine whether glucose binding altered L-FABP conformation, CD was used to examine glucose effects on L-FABP secondary structure. The CD spectrum of L-FABP in the absence of glucose exhibited a large peak in molar ellipticity at 190 nm and a negative peak at 222 nm (Fig. 2A-E, •). CD spectra obtained over a range of glucose:L-FABP (Fig. 2A, C) and maltose:L-FABP (Fig. 2B, D) molar ratios demonstrated that glucose significantly altered the relative intensities of these peaks, while maltose had only minor changes on the L-FABP CD spectra. Analysis of these CD spectra for percent composition of individual secondary structural components demonstrated that low glucose (≤60 μM) concentrations elicited strong decreases in the proportion of α-helices and reciprocally strong increases in the proportion of β-sheets, while no significant changes were noted for low concentrations of maltose (Table 1). While the addition of glucose at near-physiological serum levels (6 mM) resulted in only minor alterations to L-FABP (Fig. 2C; 6 mM, ○), high glucose (20–30 mM) levels resulted in markedly decreased maxima at 190 nm (Fig. 2C; 20 mM, ▾), decreased proportion of distorted α-helices, increased distorted β-sheets, decreased regular β-sheets, and increased unordered structures compared with L-FABP in the absence of glucose (Table 1). Although maltose concentrations of 6 mM or lower resulted in only minor spectra changes and no significant changes to the secondary structural composition, maltose concentrations of 20 mM strongly altered the secondary structure (Fig. 2D) and percent composition (Table 1). This finding suggests that the structural changes resulting from low glucose levels are due to specific, high-affinity binding, while the differing effects seen at high sugar to protein ratios may be due to binding at other sites or changes in osmolarity. The glucose metabolites (Fig. 2E; G-1-P, ○; G-6-P, ▾) elicited weaker conformational changes than glucose itself, and only at higher concentrations. Upon analysis of spectra for percent composition, the addition of physiological concentrations of G-1-P (11 μM) or G-6-P (0.2 mM) resulted changes similar to μM glucose concentrations, with strongly decreased α-helical content and increased β-sheets (Table 1). These data suggested that glucose- and glucose metabolite-induced L-FABP structural changes might alter L-FABP's interaction with other ligands or other proteins, a possibility examined in a later section.
Fig. 2.

Binding of glucose and glucose metabolites alters L-FABP secondary structure. Far UV (UV) circular dichroic (CD) spectra of 2.4 μM L-FABP in the absence (A-E, •) and presence of 6 μM glucose (A, ○); 60 μM glucose (A, ▾); 6 μM maltose (B, ○); 60 μM maltose (B, ▾); 6 mM glucose (C, ○); 20 mM glucose (C, ▾); 6 mM maltose (D, ○); 20 mM maltose (D, ▾); 11 μM G-1-P (E, ○); and 0.2 mM G-6-P (E, ▾). Each spectrum represents an average of 10 scans for a given representative spectrum, n = 3–6. G-1-P, glucose-1-phosphate; G-6-P, glucose-6-phosphate; L-FABP, liver fatty acid binding protein.
TABLE 1.
Percent composition of L-FABP secondary structures in the presence of glucose and glucose metabolites
| Regular α-helices | Distorted α-helices | Regular β-sheets | Distorted β-sheets | Turns | Unordered | |
|---|---|---|---|---|---|---|
| % | % | % | % | % | % | |
| L-FABP | 10.2 ± 0.3 | 9.8 ± 0.2 | 19.4 ± 0.6 | 10.5 ± 0.2 | 21.8 ± 0.3 | 27.3 ± 0.3 |
| 0.0006 mM Glucose | 1.6 ± 0.6a | 5.5 ± 0.2a | 28.9 ± 0.6a | 12.4 ± 0.1a | 20.7 ± 0.5c | 29.9 ± 0.2a |
| 0.006 mM Glucose | 3.3 ± 1.0a | 6.7 ± 0.6a | 25.4 ± 1.6a | 12.3 ± 0.2a | 22.5 ± 0.9 | 29.3 ± 0.5c |
| 0.06 mM Glucose | 0.7 ± 0.1a | 4.9 ± 0.2a | 29.8 ± 0.5a | 12.1 ± 0.2a | 20.7 ± 0.5c | 29.7 ± 0.4b |
| 0.6 mM Glucose | 11.6 ± 0.7 | 10.3 ± 0.4 | 17.7 ± 1.0 | 9.7 ± 0.4 | 22.1 ± 0.4 | 28.2 ± 0.2c |
| 6 mM Glucose | 10.2 ± 0.2 | 9.6 ± 0.2 | 19.0 ± 0.6 | 11.3 ± 0.2b | 23.5 ± 0.2a | 26.5 ± 0.6 |
| 20 mM Glucose | 7.8 ± 1.3 | 5.7 ± 1.4b | 14.6 ± 3.0 | 13.4 ± 0.4a | 25.1 ± 1.1c | 33.2 ± 3.7 |
| 30 mM Glucose | 11.0 ± 5.3 | 4.6 ± 1.4b | 6.3 ± 1.9a | 13.9 ± 1.0b | 21.1 ± 2.4 | 43.2 ± 5.7c |
| 0.0006 mM Maltose | 9.8 ± 0.7 | 9.5 ± 0.6 | 21.9 ± 1.5 | 11.0 ± 0.2 | 20.6 ± 0.7 | 26.7 ± 0.7 |
| 0.006 mM Maltose | 8.9 ± 0.5 | 9.2 ± 0.3 | 21.7 ± 0.9 | 11.5 ± 0.7 | 22.0 ± 0.6 | 26.0 ± 0.3 |
| 0.06 mM Maltose | 10.2 ± 0.2 | 9.7 ± 0.2 | 20.2 ± 0.6 | 11.0 ± 0.4 | 22.2 ± 0.3 | 26.4 ± 0.7 |
| 0.6 mM Maltose | 8.8 ± 0.8 | 9.0 ± 0.3 | 19.6 ± 0.6 | 11.3 ± 0.3c | 23.0 ± 0.8 | 27.8 ± 0.2 |
| 6 mM Maltose | 8.5 ± 1.8 | 10.7 ± 1.6 | 21.2 ± 4.4 | 11.2 ± 2.6 | 22.3 ± 3.1 | 28.7 ± 1.4 |
| 20 mM Maltose | 2.2 ± 0.8a | 5.4 ± 0.7a | 26.0 ± 0.9a | 13.0 ± 0.4a | 23.4 ± 0.9 | 29.6 ± 0.4c |
| 30 mM Maltose | 9.6 ± 1.6 | 8.1 ± 3.1 | 20.9 ± 2.1 | 11.5 ± 1.9 | 21.9 ± 3.4 | 28.1 ± 2.1 |
| Glucose-1-Phosphate | 3.8 ± 0.8a | 6.2 ± 0.5a | 26.2 ± 1.2a | 11.5 ± 0.1a | 20.7 ± 0.4b | 27.8 ± 0.6 |
| Glucose-6-Phosphate | 4.6 ± 0.8a | 6.5 ± 0.5a | 25.4 ± 1.2a | 11.4 ± 0.2a | 21.2 ± 0.3c | 27.8 ± 0.6 |
Significant differences between L-FABP structural composition in the presence of sugar compared with L-FABP structural composition in the absence of sugar (L-FABP). L-FABP, liver fatty acid binding protein.
P < 0.001, n = 3–6.
P < 0.01, n = 3–6.
P < 0.05, n = 3–6.
Glucose increased L-FABP binding capacity for LCFA and LCFA-CoA
To determine if the high-affinity binding of glucose altered lipidic ligand binding to L-FABP, the ability of glucose and glucose metabolites to displace an L-FABP-bound fluorescent LCFA and/or fluorescent LCFA-CoA from the L-FABP binding pocket was examined. This was accomplished by use of BODIPY C-16 (fluorescent LCFA analog) and BODIPY C16-CoA (fluorescent LCFA-CoA analog), both of which were previously shown to be bound by L-FABP (12). Glucose significantly increased BODIPY C-16 (Fig. 3A, ▪) and BODIPY C16-CoA (Fig. 3A, □) binding to L-FABP up to 2-fold. In contrast, glucose metabolites (G-1-P and G-6-P) or phosphate (negative control) did not significantly alter BODIPY-C16 binding; only G-1-P slightly decreased BODIPY C16-CoA binding to L-FABP (Fig. 3A, □).
Fig. 3.

Glucose increases L-FABP binding capacity for LCFA and LCFA-CoA. (A) L-FABP (100 nM) was mixed with 50 nM BODIPY C-16 (▪) or BODIPY C16-CoA (□), and fluorescence intensities were measured at 490–540 nm upon excitation at 460 nm. (B) L-FABP (100 nM) was mixed with 50 nM trans-parinaric acid (▪) or cis-parinaroyl-CoA (□), and the fluorescence intensities were measured at 380–450 nm upon excitation at 317 nm. After the initial fluorescence measurements (0 mM glucose) were obtained, glucose or glucose metabolites were added, and fluorescence intensities measured to determine the effect of glucose on lipid binding. All values are presented as the mean percent change in maximal fluorescence intensity (n = 4) ± SEM. Asterisks (*) indicate significant differences from the 0 mM glucose controls; P < 0.05. G-1-P, glucose-1-phosphate; G-6-P, glucose-6-phosphate; L-FABP, liver fatty acid binding protein; LCFA, long-chain fatty acid; LCFA-CoA, long-chain fatty acyl-CoA.
To determine whether the effect of glucose in enhancing binding of BODIPY-labeled LCFA and LCFA-CoA to L-FABP was due to the presence of BODIPY in these synthetic fluorescent LCFA and LCFA-CoA derivatives, the effect of glucose on displacement of two naturally occurring fluorescent ligands [LCFA (trans-parinaric acid) and LCFA-CoA (cis-parinaroyl-CoA)] bound with high affinity by L-FABP (16), was examined. Addition of glucose strongly increased trans-parinaric acid binding (Fig. 3B, ▪) and less so cis-parinaroyl-CoA binding (Fig. 3B, □). These findings suggested that the effect of glucose on enhancing lipidic ligand binding to L-FABP was not just an artifact of the fluorescent BODIPY moiety but in fact represented the effect one might observe on the binding of naturally occurring LCFA or LCFA-CoA.
Glucose enhanced L-FABP interaction with PPARα by co-immunoprecipitation
To determine whether the glucose-induced alterations in secondary structures observed by CD would increase not only lipidic ligand binding but also L-FABP interaction with the nuclear receptor (PPARα) regulated by these ligands, the effect of glucose on co-immunoprecipitation of L-FABP with PPARα was examined. Although low levels of glucose had only minor effects, high glucose concentrations (20–30 mM) increased co-immunoprecipitation of L-FABP with PPARα (Fig. 4). Although the CD results at these higher sugar concentrations suggested that high sugar-to-protein ratios might result in additional effects, the amount of protein in these assays was more than 10-fold greater than in the CD experiments, suggesting that these effects were both relevant and specific. These data suggest that although low glucose levels have minor effects on L-FABP interaction with PPARα, higher glucose concentrations, such as those seen in diabetes, increased L-FABP interaction with PPARα.
Fig. 4.
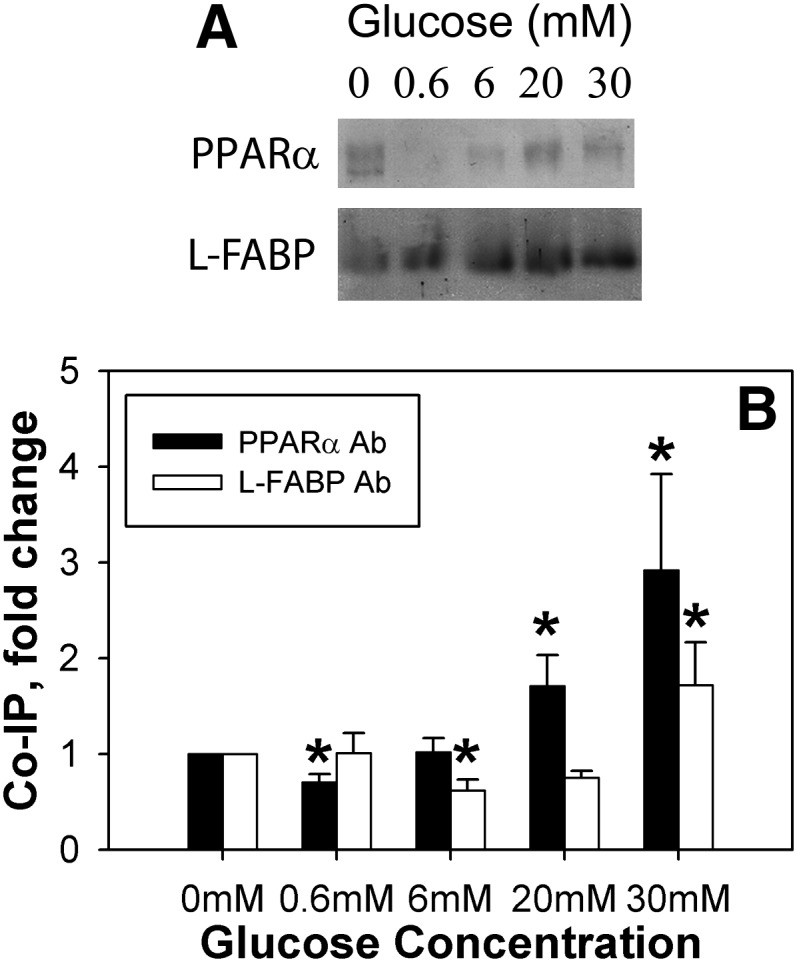
High glucose concentrations increase PPARα and L-FABP co-immunoprecipitation. Equal concentrations of PPARα and L-FABP proteins were mixed in the absence and presence of glucose, and the resultant mixtures were co-immunoprecipitated with the PPARα antibody and assayed for the amount of L-FABP protein (A, L-FABP; B, ▪) or co-immunoprecipitated with the L-FABP antibody and assayed for the amount of PPARα protein (A, PPARα; B, □). A: Representative image of co-immunoprecipitation results for PPARα or L-FABP protein. B: Values are presented as the mean fold change ± SEM compared with the 0 mM glucose controls, n = 4. Asterisks (*) indicate significant differences from the 0 mM controls; P < 0.05. Co-IP, co-immunoprecipitation; L-FABP, liver fatty acid binding protein; PPAR, peroxisome proliferator-activated receptor.
Glucose altered the secondary structures of the L-FABP complex with PPARα
To further confirm that glucose increased interaction of L-FABP with PPARα, the effect of glucose on secondary structure was examined by CD. The actual spectra of both proteins (PPARα + L-FABP) in the presence of 6 μM (Fig. 5A, ○) and 60 μM (Fig. 5B, ○) glucose differed from the spectra of (PPARα + L-FABP) in the absence of glucose (•) but were almost superimposable over the theoretical spectra of (PPARα + L-FABP) in the presence of glucose if no protein interaction occurs (▾). When examined for percent composition, the addition of 0.6 μM or 6 μM glucose resulted in values that were not significantly different from those calculated for the theoretical values if no protein-protein interaction occurred (Table 2), suggesting that low glucose concentrations prevented the conformational change induced by PPARα binding with L-FABP. To examine the specificity of glucose on this interaction, the effect of low concentrations of maltose was examined. No significant differences in spectra (Fig. 5C) or percent composition (Table 2) were noted for maltose at 6 μM or 60 μM, suggesting that low maltose concentrations had no effect on the PPARα-L-FABP interaction.
Fig. 5.

Glucose, but not maltose, alters PPARα interaction L-FABP. Far UV (UV) circular dichroic (CD) spectra of PPARα and L-FABP in the absence (A-H, •) and presence (○) of glucose compared with the theoretically obtained spectrum for PPARα and L-FABP in the presence of glucose if no interaction occurred between the proteins (▾) (A) 6 μM glucose, (B) 60 μM glucose, (C) 6 μM maltose (○) and 60 μM maltose (▾), (D) 6 mM glucose, (E) 20 mM glucose, (G) 6 mM maltose (○) and 20 mM maltose (▾), (G) 11 μM G-1-P, and (H) 0.2 mM G-6-P. Each spectrum represents an average of ten scans for a given representative spectrum, n = 3–4. G-1-P, glucose-1-phosphate; G-6-P, glucose-6-phosphate; L-FABP, liver fatty acid binding protein; PPARα, peroxisome proliferator-activated receptor alpha.
TABLE 2.
Secondary structures of PPARα and L-FABP proteins in the presence of glucose metabolites
| Regular α-helices | Distorted α-helices | Regular β-sheets | Distorted β-sheets | Turns | Unordered | |
|---|---|---|---|---|---|---|
| % | % | % | % | % | % | |
| PPARα and L-FABP | 21.1 ± 0.6 | 14.7 ± 0.3 | 10.5 ± 0.4 | 8.2 ± 0.2 | 20.9 ± 0.4 | 25.0 ± 0.4 |
| 0.0006 mM Glucose | 9.7 ± 0.1a | 9.9 ± 0.2a | 19.7 ± 0.4a | 9.0 ± 0.6c | 21.6 ± 0.2 | 28.7 ± 0.2a |
| Theoretical | 9.4 ± 0.2 | 9.4 ± 0.2 | 20.1 ± 0.4 | 9.9 ± 0.2 | 21.8 ± 0.2 | 27.7 ± 0.5 |
| 0.006 mM Glucose | 10.2 ± 0.4a | 10.3 ± 0.3a | 19.3 ± 0.4a | 10.2 ± 0.2a | 21.4 ± 0.3 | 27.5 ± 0.6b |
| Theoretical | 9.68 ± 0.39 | 10.4 ± 0.3 | 19.7 ± 0.3 | 10.1 ± 0.2 | 22.0 ± 0.5 | 27.2 ± 0.6 |
| 0.06 mM Glucose | 11.8 ± 0.1a | 10.7 ± 0.1a | 16.3 ± 0.7a | 9.3 ± 0.1a | 22.9 ± 0.3c | 28.8 ± 0.2a |
| Theoretical | 9.8 ± 0.3d | 9.4 ± 0.2d | 19.7 ± 0.6e | 10.1 ± 0.1d | 21.7 ± 0.3 | 27.7 ± 0.3f |
| 0.6 mM Glucose | 12.2 ± 0.1a | 10.4 ± 0.1a | 16.5 ± 1.0a | 9.5 ± 0.2a | 22.0 ± 0.7 | 28.8 ± 0.6a |
| Theoretical | 15.6 ± 0.5d | 11.9 ± 0.2d | 13.50 ± 0.63f | 8.7 ± 0.2e | 22.2 ± 0.6 | 27.9 ± 0.5 |
| 6 mM Glucose | 10.5 ± 0.2a | 9.7 ± 0.2a | 18.2 ± 0.1a | 10.7 ± 0.1a | 22.3 ± 0.1b | 28.0 ± 0.1a |
| Theoretical | 16.0 ± 0.6d | 12.5 ± 0.2d | 13.4 ± 0.3d | 9.0 ± 0.1d | 22.4 ± 0.5 | 27.3 ± 0.3 |
| 20 mM Glucose | 11.1 ± 0.2a | 10.3 ± 0.2a | 16.7 ± 0.3a | 10.9 ± 0.2a | 22.9 ± 0.1a | 28.0 ± 0.2a |
| Theoretical | 8.9 ± 0.3d | 9.2 ± 0.4f | 19.3 ± 0.7e | 12.4 ± 0.3d | 22.7 ± 0.5 | 27.6 ± 0.4 |
| 30 mM Glucose | 12.1 ± 5.3c | 15.8 ± 10.3 | 10.0 ± 2.0 | 13.1 ± 0.8a | 20.3 ± 2.1 | 38.8 ± 5.3b |
| Theoretical | 11.0 ± 4.8 | 6.1 ± 1.6 | 12.0 ± 2.1 | 12.8 ± 0.6 | 20.1 ± 2.2 | 36.9 ± 4.9 |
| 0.006 mM Maltose | 18.8 ± 0.9 | 13.3 ± 0.5 | 10.9 ± 0.7 | 9.2 ± 0.4 | 23.2 ± 0.5 | 25.4 ± 0.9 |
| 0.06 mM Maltose | 18.9 ± 0.5 | 12.9 ± 0.6 | 11.4 ± 0.6 | 8.9 ± 0.4 | 22.5 ± 0.5 | 25.8 ± 0.5 |
| 0.6 mM Maltose | 22.2 ± 1.6 | 16.6 ± 1.9 | 8.1 ± 1.2 | 8.7 ± 0.6 | 22.0 ± 1.4 | 22.7 ± 2.1 |
| 6 mM Maltose | 18.7 ± 5.2 | 13.9 ± 2.9 | 10.1 ± 3.0 | 9.2 ± 2.1 | 23.5 ± 1.8 | 24.8 ± 1.5 |
| 20 mM Maltose | 6.2 ± 1.2b | 7.4 ± 1.2b | 22.6 ± 1.6 | 13.8 ± 1.6b | 23.4 ± 1.3 | 24.7 ± 1.8 |
| G-1-P | 11.0 ± 0.2a | 10.2 ± 0.2a | 17.9 ± 0.7a | 9.68 ± 0.13a | 22.1 ± 0.3c | 28.1 ± 0.2a |
| Theoretical | 9.6 ± 0.3e | 9.7 ± 0.4 | 20.3 ± 0.8f | 9.7 ± 0.3 | 21.8 ± 0.3 | 27.7 ± 0.2 |
| G-6-P | 12.8 ± 0.2a | 10.9 ± 0.2a | 15.1 ± 0.7a | 9.1 ± 0.2a | 22.5 ± 0.4b | 28.8 ± 0.4a |
| Theoretical | 10.9 ± 0.2d | 10.2 ± 0.2f | 17.7 ± 0.7f | 9.5 ± 0.2 | 22.3 ± 0.3 | 28.4 ± 0.3 |
Significant difference between experimental values and values for protein interaction without sugar or metabolite (PPARα and L-FABP); aP < 0.001, bP < 0.01, cP < 0.05. Significant difference between predicted values for protein mixtures with sugar or metabolite assuming no protein interaction (theoretical) and experimental values; dP < 0.001, eP < 0.01, fP < 0.05, n = 4–8. G-1-P, glucose-1-phosphate; G-6-P, glucose-6-phosphate; L-FABP, liver fatty acid binding protein; PPAR, peroxisome proliferator-activated receptor.
In contrast, higher glucose concentrations resulted in spectra and structural component contributions indicative of significant additional conformational alterations in the PPARα-L-FABP complex. The spectra of (PPARα + L-FABP) in the presence of 0.6 mM (spectrum not shown), 6 mM (Fig. 5D, ○) and 20 mM (Fig. 5E, ○) glucose all exhibited decreased maxima at 190 nm and minima at 222 nm compared with the spectrum of (PPARα + L-FABP) in the absence of glucose (•). Although the spectra at each of these concentrations also varied from the theoretical spectra for no PPARα-L-FABP interaction (▾), the largest spectral changes were noted for (PPARα + L-FABP) in the presence of 6 mM glucose. When analyzed for percent composition, decreased α-helices and increased β-sheets were noted compared with the no-glucose controls, and (PPARα + L-FABP) in the presence of 0.6 mM or 6 mM glucose resulted in even lower α-helical content and increased β-sheets than the theoretical (Table 2). However, while 6 mM maltose resulted in spectra that were superimposable over the spectrum of (PPARα + L-FABP) in the absence of sugar (Fig. 5F) and had no significant alterations to the percent composition, 20 mM maltose strongly altered the CD spectra and resulted in decreased α-helical content and increased β-sheets (Table 2). Taken together, these data suggested that glucose concentrations between 6 μM and 6 mM elicited more marked conformational changes in the PPARα complex with L-FABP and that the ratio of sugar to protein may be an important regulatory mechanism in affecting these protein-protein interactions.
To resolve whether glucose metabolites might also affect the PPARα-L-FABP interaction detected by CD, spectra of (PPARα+L-FABP) were recorded in the presence of 11 μM G-1-P (Fig. 5G) and 0.2 mM G-6-P (Fig. 5H). These spectra (○) differed strongly from the spectrum of (PPARα + L-FABP) in the absence of glucose (•), with decreased minima and maxima similar to those observed for 6 μM glucose (Fig. 5A, ○), and only slight variation was noted between the actual and theoretical spectrum for no protein interaction (▾). Both G-1-P and G-6-P decreased the α-helical content and increased the β-sheet content of the PPARα-L-FABP complex similar to changes obtained with 60 μM glucose (Table 2). These data suggested that not only glucose but also both glucose metabolites can elicit conformational changes in the PPARα complex with L-FABP.
Glucose-induced conformational changes in the PPARα complex with L-FABP as shown by fluorescence resonance energy transfer
To determine if the glucose-mediated increases in secondary structure of the PPARα complex with L-FABP might also extend to changes in tertiary structure, a fluorescence resonance energy transfer (FRET) study was undertaken using Cy3 and Cy5 dyes, which are small fluorescent dyes that form an efficient FRET donor/acceptor pair (24, 28). As FRET efficiency varies inversely as the 6th root of intermolecular distance, energy transfer will only occur if Cy3-PPARα and Cy5-L-FABP are in close proximity (i.e., 0–100Å). The recombinant proteins were chemically labeled with Cy3 (PPARα) or Cy5 (L-FABP) dyes, and the labeling ratios of fluorescent dye to protein were maintained low to ensure an average of one dye per protein molecule. FRET between these fluorescently labeled proteins was then used to determine the intermolecular distance between the fluorescent tags in the PPARα-L-FABP complex. In the absence (Fig. 6A) and presence of glucose (6 mM, Fig. 6C; 20 mM, Fig. 6E; 30 mM, Fig. 6G) or glucose metabolites (G-1-P, Fig. 7A; G-6-P, Fig. 7C), FRET was observed to quench donor Cy3-PPARα fluorescence and to increase the appearance of sensitized emission of acceptor Cy5-L-FABP. In the absence of glucose, the intermolecular distance between Cy3-PPARα and Cy5-L-FABP was determined to be about 49 Å (Table 3), which is consistent with close molecular interaction. High glucose (20–30 mM), as well as physiological levels of glucose metabolites (G-1-P and G-6-P) all increased the intermolecular distance between Cy3-PPARα and Cy5-L-FABP by as much as 11Å (Table 3). Taken together, these FRET data suggested that glucose binding altered not only the secondary structure (determined by CD) but also the tertiary conformation of the PPARα-L-FABP complex.
Fig. 6.

High glucose increases PPARα affinity for L-FABP. FRET from donor Cy3-labeled PPARα to acceptor Cy5-labeled L-FABP was detected as quenching of Cy3 fluorescence emission (near 570 nm) and the concomitant appearance of sensitized emission from Cy5 (near 680 nm). Emission spectra of Cy3-PPARα and Cy5-L-FABP upon excitation of Cy3 at 550 nm in the presence of 0 mM (A), 6 mM (C), 20 mM (E), and 30 mM (G) glucose titrated with increasing Cy5-L-FABP concentrations (solid line, 0 nM; larger dashes to dots, 10 nM, 50 nM, 100 nM, and 200 nM Cy5-L-FABP, respectively). Plot of the average change in maximal fluorescence intensity at 570 nm (F0-F) of Cy3-PPARα upon excitation at 550 nm as a function of Cy5-L-FABP concentration (with the inset referring to a linear plot of the binding curve) in the presence of 0 mM (B), 6 mM (D), 20 mM (F), and 30 mM (H) glucose. Values represent the mean ± SE, n = 4. FRET, fluorescence resonance energy transfer; L-FABP, liver fatty acid binding protein; PPAR, peroxisome proliferator-activated receptor.
Fig. 7.

Glucose metabolites oppositely affect fluorescence resonance energy transfer. FRET from donor Cy3-labeled PPARα to acceptor Cy5-labeled L-FABP was detected as quenching of Cy3 fluorescence emission (near 570 nm) and the concomitant appearance of sensitized emission from Cy5 (near 680 nm). Emission spectra of Cy3-PPARα and Cy5-L-FABP upon excitation of Cy3 at 550 nm in the presence of 11 μM G-1-P (A) and 0.2 mM G-6-P (C) titrated with increasing Cy5-L-FABP concentrations (solid line, 0 nM; larger dashes to dots, 10 nM, 50 nM, 100 nM, and 200 nM Cy5-L-FABP, respectively). Plot of the average change in maximal fluorescence intensity at 570 nm (F0-F) of Cy3-PPARα upon excitation at 550 nm as a function of Cy5-L-FABP concentration (with the inset referring to a linear plot of the binding curve) in the presence of 11 μM G-1-P (B) and 0.2 mM G-6-P (D). Values represent the mean ± SE, n = 4. FRET, fluorescence resonance energy transfer; G-1-P, glucose-1-phosphate; G-6-P, glucose-6-phosphate; L-FABP, liver fatty acid binding protein; PPAR, peroxisome proliferator-activated receptor.
TABLE 3.
Binding affinity, energy transfer efficiency, and molecular distance between Cy3-labeled PPARα and Cy-5-labeled L-FABP proteins in the presence of glucose metabolites determined by FRET as a function of donor quenching
| Metabolite | Kd (nM) | E (%) | r (Å) |
|---|---|---|---|
| None | 31.3 ± 5.7 | 53.2 ± 4.5 | 49.0 ± 1.5 |
| 6 mM Glucose | 26.6 ± 4.4 | 59.6 ± 7.8 | 46.7 ± 2.7 |
| 20 mM Glucose | 12.5 ± 1.7 | 26.0 ± 1.2 | 59.6 ± 0.7 |
| 30 mM Glucose | 8.6 ± 1.2 | 27.7 ± 2.1 | 59.3 ± 1.1 |
| G-1-P | 47.9 ± 6.4 | 29.5 ± 3.0 | 58.0 ± 1.5 |
| G-6-P | 11.3 ± 1.9 | 25.0 ± 3.7 | 60.4 ± 1.9 |
FRET, fluorescence resonance energy transfer; G-1-P, glucose-1-phosphate; G-6-P, glucose-6-phosphate; L-FABP, liver fatty acid binding protein; PPAR, peroxisome proliferator-activated receptor.
Glucose increased PPARα affinity for L-FABP as shown by FRET
To determine if the glucose-induced alterations (determined by CD and FRET intermolecular distance) in the PPARα-L-FABP complex were associated with altered strength of binding between these proteins, FRET between Cy3-PPARα and Cy5-L-FABP was used to determine their binding affinity in the absence and presence of glucose. By plotting the change in fluorescence intensity of the donor (excitation 550 nm, emission 570 nm) as a function of increasing acceptor concentration, the affinity of PPARα for L-FABP was calculated (Table 3). In the absence of glucose (Fig. 6B) or in the presence of glucose (6 mM, Fig. 6D; 20 mM, Fig. 6F; 30 mM, Fig. 6H) or glucose metabolites (G-1-P, Fig. 7B; G-6-P, Fig. 7D), a saturable binding curve was obtained, suggesting strong affinity binding. Multiple replicates (n = 4) revealed that PPARα bound L-FABP with high affinity in the absence of glucose, Kd = 31.3 ± 5.7 nM (Table 3). Increasing concentrations of glucose or G-6-P increased the binding affinity 3–4-fold (Table 3). In contrast, G-1-P reduced this affinity by 45% as shown by 1.5-fold weaker Kd (Table 3). This suggests that even though glucose, G-1-P, and G-6-P all altered the overall protein conformation, the conformational changes elicited by G-1-P binding were different and resulted in an opposite effect on the PPARα-L-FABP interaction compared with the effects of glucose and G-6-P.
Glucose and L-FABP increase LCFA uptake and nuclear localization in cultured cells
L-FABP is known to enhance transport of bound LCFA to the nucleus (11, 12, 29). As the above data showed that glucose increased L-FABP's affinity for lipidic ligands (LCFA and LCFA-CoA) and for PPARα, the possibility that glucose might thereby alter the distribution of LCFA to the nucleus was examined using BODIPY C-16, a fluorescent fatty acid analog that is bound with high affinity by both PPARα (13) and L-FABP (12). Uptake and nuclear distribution were examined in cells devoid of L-FABP (mock-transfected control L-cells; “control”) and transfected cells expressing L-FABP (transfected L-cells overexpressing L-FABP; “L-FABP”) (11, 12). These cells were stained with the nuclear SYTO59 stain (Fig. 8, red pixels) as a colocalization marker for BODIPY C-16 distribution to nuclei. In control cells not expressing L-FABP, total BODIPY C-16 uptake (Fig. 8A1, green pixels) and the amount of BODIPY C-16 localizing to the nucleus (observed as colocalized pixels with the SYTO59 nuclear stain, Fig. 8A2, yellow pixels) was relatively low in the absence of glucose. However, addition of glucose significantly increased BODIPY C-16 uptake (Fig. 8C1, green pixels; 8E1, green pixels; 8G1, green pixels; 8I1, green pixels; 9A, ▪) and nuclear localization (Fig. 8C2, yellow pixels; 8E2, yellow pixels; 8G2, yellow pixels; 8I2, yellow pixels; 9B, ▪), with 20 mM glucose eliciting the maximal increase.
Fig. 8.

L-FABP expression and addition of glucose both increase nuclear BODIPY C-16 localization. Mock-transfected control (A, C, E, G, and I) and L-FABP overexpressing (B, D, F, H, and J) L-cells were incubated with the nonmetabolizable fluorescent fatty acid BODIPY C-16 (green pixels) and the nuclear dye SYTO59 (red pixels) in the presence of 0 mM (A and B), 0.6 mM (C and D), 6 mM (E and F), 20 mM (G and H), and 30 mM (I and J) glucose and imaged by LSCM. Panels numbered 1 show the pixels of BODIPY C-16 (green) and SYTO59 (red), and panels numbered 2 show BODIPY C-16 pixels colocalizing with the STYO59 stain for each condition in yellow. L-FABP, liver fatty acid binding protein.
Overexpression of L-FABP, in the absence of glucose, increased both total uptake (Fig. 8B1, green pixels) and nuclear localization (Fig. 8B2, yellow pixels) of BODIPY C-16. These increases corresponded to a 1.5-fold increase in the amount of uptake (Fig. 9A, □) and an almost 3-fold increase in nuclear localization (Fig. 9B, □) compared with mock-transfected control cells with no L-FABP expression (filled bars). High concentrations of glucose exacerbated the effect of L-FABP overexpression on cellular BODIPY C-16 uptake (Fig. 8F1, green pixels; 8H1, green pixels; 8J1, green pixels; 9A, □). Furthermore, both control and L-FABP overexpressing cells showed similar maximal fatty acid uptake at 20 mM, suggesting that at high glucose levels the L-FABP effect on uptake may be diminished. In addition, glucose increased nuclear BODIPY C-16 levels in the L-FABP overexpressing cells (Fig. 8D2, yellow pixels; 8F2, yellow pixels; 8H2, yellow pixels; 8J2, yellow pixels; 9B, □) as well as the control cells. These data suggested that both glucose and L-FABP expression functioned to increase LCFA uptake and increase LCFA nuclear localization, with the effect being greatest when both glucose and L-FABP were high.
Fig. 9.

L-FABP expression and addition of glucose synergistically increase BODIPY C-16 uptake and nuclear concentrations of BODIPY C-16 in a concentration-dependent manner. (A) Cellular BODIPY C-16 fluorescence from control (□) and L-FABP-expressing (▪) L-cells was calculated per cell by taking the BODIPY C-16 fluorescence intensity of the whole cell divided by the area of the whole cell (to compensate for variation due to cell size); these values were then averaged over n = 50–100 cells. (B) Using the Syto59 fluorescence, the nuclear perimeter was marked, allowing for analysis of the BODIPY C-16 levels in the nucleus. Nuclear BODIPY C-16 fluorescence from control (□) and L-FABP-expressing (▪) L-cells was calculated per nucleus by taking the BODIPY C-16 fluorescence intensity of each nucleus divided by the area of the nucleus (to compensate for variation due to cell size); these values were then averaged over n = 50–200 cells. (C) The percentage of BODIPY C-16 localizing to the nucleus, calculated by dividing the total BODIPY C-16 fluorescence in the nucleus by the total fluorescence in the whole cell and multiplying by 100%. Values are presented as the mean value ± SEM, from 50–100 cells from two to three separate replicates. Asterisks (*) indicate significant differences from the 0 mM controls; P < 0.05. Pound symbols (#) indicate significant differences due to the presence of L-FABP compared with wild-type cells under the same conditions; P < 0.05. L-FABP, liver fatty acid binding protein.
To determine whether these effects might be due to changes in osmolarity, rather than glucose binding, these experiments were repeated in the presence of 6 mM maltose or 3 mM sodium chloride (Fig. 9). Although the effects of 6 mM maltose and 3 mM sodium chloride on cellular BODIPY C-16 uptake were similar to each other, with L-FABP-expressing cells having decreased uptake compared with control cells, these results varied significantly from those obtained with glucose (Fig. 9A). Further, in the presence of L-FABP, the addition of 6 mM maltose or 3 mM sodium chloride had no effect on BODIPY C-16 uptake or distribution to nuclei compared with cells in PBS. This suggested that effects from glucose are not just due to changes in osmolarity.
L-FABP altered the percentage of LCFA localizing to the nucleus
To determine if increased nuclear BODIPY C-16 amounts (concentration) corresponded with increased preferential nuclear BODIPY C-16 distribution (i.e., increased percentage in the nucleus), rather than simply being a consequence of increased overall uptake (total concentration), the proportion of BODIPY C-16 in the nucleus (versus the rest of the cell) was examined and expressed as a percentage of that taken up. In all cases, L-FABP increased the percentage of BODIPY C-16 localizing to the nucleus (Fig. 9C). Glucose only seemed to affect the percent distribution at high concentrations and in the presence of L-FABP, while maltose in the absence of L-FABP actually decreased the percent distribution, and sodium chloride had no effect (Fig. 9C).
Thus, glucose and L-FABP modified the percentage of BODIPY C-16 in the nucleus in a complex, concentration-dependent manner. For example, although the addition of 20 mM glucose to the control cells resulted in relatively the same amount of nuclear BODIPY C-16 as L-FABP-expressing cells in the absence of glucose (Fig. 9B), the distribution of fatty acid within the nucleus was different. The control cells showed a higher percentage of the BODIPY C-16 located at the nuclear envelope membranes (Fig. 8G2), while the L-FABP-expressing cells (L-FABP) showed a more even dispersion of BODIPY C-16 within the nucleoplasm (Fig. 8B2). Addition of glucose to the L-FABP-expressing cells (L-FABP) resulted in a higher level of BODIPY C-16 throughout the nucleoplasm, as well as around the nuclear membrane (Fig. 8; 0.6 mM, D2; 6 mM, F2; 20 mM, H2), while the addition of 30 mM glucose resulted in less BODIPY C-16 at the nuclear membrane (Fig. 8J2). Based on earlier studies showing that L-FABP cotransports bound LCFA into the nucleus (29), this variation in BODIPY C-16 distribution could be due to BODIPY C-16 entering the nucleus through diffusion versus L-FABP facilitated transport (e.g., free fatty acid versus carrier protein bound).
DISCUSSION
It has been proposed that cytoplasmic lipid binding proteins, such as L-FABP, might function in long-term regulation of hepatic LCFA β-oxidation by transporting bound LCFA or LCFA-CoA into the nucleus, directly interacting with nuclear receptors, such as PPARα, and transferring these ligands to PPARα for activating transcription of multiple genes involved in LCFA β-oxidation (rev. in Ref. 15). However, recent data show that although lipidic ligands, such as LCFA, activate PPARγ (another PPAR isoform), ligand binding is not required for PPARγ functionality (30). Taken together these findings suggest that cytoplasmic lipid binding proteins, such as L-FABP, may activate PPARα, either by delivering bound LCFA to the nuclear receptor and/or by directly interacting with the nuclear receptor to change its conformation and coactivator association. Consistent with this possibility, in the absence of lipidic ligands, L-FABP directly interacts with PPARα to alter conformation and coactivator recruitment (20). However, in addition to the above mechanisms of lipid-dependent and lipid-independent PPARα activation, recent findings indicate that PPARα also exhibits high-affinity binding of and activation by glucose (13, 14). Thus, PPARα is unique among the ligand-activated nuclear receptors in that its activity is regulated by two very different classes of nutrients/endogenous ligands: hydrophobic lipids (LCFA, LCFA-CoA, and fibrates) (8–10) and hydrophilic sugars (glucose and glucose metabolites) (13, 14). As L-FABP is the predominant LCFA and LCFA-CoA binding protein in liver (31) and directly interacts with PPARα (20), it was important to examine the effect of glucose on these processes. The data obtained herein demonstrated for the first time that (i) L-FABP bound glucose with high affinity, resulting in increased LCFA/LCFA-CoA binding; (ii) Glucose increased PPARα's affinity for L-FABP; (iii) Glucose increased nuclear localization and relative distribution of LCFA in a concentration-dependent manner, both in the presence and absence of L-FABP; and (iv) A complex interaction exists between glucose, lipid, and L-FABP levels in the regulation of PPARα. These studies yielded several new insights regarding the regulation of PPARα by L-FABP.
First, L-FABP may function in transporting not only bound lipidic ligands (LCFA and LCFA-CoA) but also bound glucose to PPARα in the nucleus. Consistent with this possibility as shown herein, L-FABP bound glucose with high affinity (Kd = 103.1 ± 18.6 nM), approximately 50-fold weaker than previously reported for PPARα binding of glucose (13). Furthermore, glucose enhanced the interaction between PPARα and L-FABP. Since L-FABP is known to enter nuclei to directly interact with PPARα (12, 20), these findings suggest L-FABP-bound glucose may be transported to and influence transcriptional activity of PPARα. Based on the glucose binding sites of other glucose binding proteins (32), it appears that a combination of asparagine (N), aspartic acid (D), or glutamic acid (E) molecules are required for glucose binding to occur, and phenylalanine (F) residues may stabilize such interactions. The X-ray crystal structure of holo-L-FABP reveals not only the expected two LCFA binding sites but also an additional surface site occupied by a small molecule, butyric acid (33). This site is localized near the end of the first α-helix of L-FABP N-terminus. Furthermore, even with two LCFA molecules bound within the L-FABP binding pocket, there is still space within the binding cavity for several small molecules, and three small subcavities exist (33). Although the exact binding site for glucose is unknown, based upon the D, E, N, or F amino acid requirement, it is possible that glucose binding occurs through hydrogen bonding at one of two locations: between residues E13, F15, and F18 or near residues E62, F63, E72, and F95.
Second, glucose altered L-FABP conformation to enhance binding of lipidic ligands, as shown by CD and ligand binding assays in the presence of glucose. Furthermore, glucose-induced L-FABP structural changes were stronger than previously reported for glucose binding to PPARα (13). Unlike the glucose-induced PPARα conformational changes, which did not alter LCFA binding and decreased LCFA-CoA binding, the structural changes resulting from glucose binding by L-FABP increased binding of both LCFA and LCFA-CoA to L-FABP. These data suggest that glucose could function to increase cellular uptake and transport of lipids through its interaction with L-FABP and that L-FABP could simultaneously transport both glucose and LCFA into the nucleus for interaction with and activation of PPARα. Consistent with this possibility, glucose enhanced cellular uptake of exogenous LCFA uptake and facilitated the delivery of LCFA from the cytoplasm into the nucleus, a process potentiated by L-FABP. Studies in cultured cells have shown that cytoplasmic glucose levels are 2–8-fold higher than nucleoplasmic glucose concentrations, depending on the extracellular glucose concentration (13). The difference between nucleoplasmic and cytoplasmic glucose levels, together with the higher binding affinity of PPARα for glucose, may favor L-FABP offloading of bound LCFA in the nucleus for binding to and activating PPARα.
Third, L-FABP and PPARα differed markedly in their binding of glucose metabolites produced as a result of hexokinase action within cells. Although PPARα binds not only glucose but also G-1-P and G-6-P with high affinity, PPARα binds glucose with approximately 10-fold stronger affinity than G-1-P and approximately 30-fold stronger affinity than G-6-P (13). In contrast, L-FABP affinity for G-6-P was extremely weak, almost 250-fold less than for glucose. Further, L-FABP binding of G-1-P was approximately 5-fold higher than glucose binding by L-FABP and about the same as previously reported for PPARα binding of G-1-P (13). Additionally, the glucose metabolites differed in their ability to affect L-FABP binding to LCFA/LCFA-CoA and in their ability to affect the PPARα-L-FABP interaction. L-FABP binding of G-1-P resulted in decreased LCFA-CoA binding, similar to the effect of G-1-P on PPARα binding to LCFA-CoA (13), while G-6-P had no effect on L-FABP binding of LCFA or LCFA-CoA. While both G-1-P and G-6-P induced conformational changes to the PPARα-L-FABP complex that were different than the glucose-induced changes, G-1-P decreased PPARα's affinity for L-FABP while G-6-P increased this affinity, a result similar to that obtained with glucose. The concentration of these metabolites in serum is estimated to be several orders of magnitude lower than glucose concentrations (11 μM for G-1-P and 0.2 mM for G-6-P) (34), while G-6-P concentrations in muscle are even lower, approximately 0.1 mM (35). Thus, depending on cell type and availability of glucose transporters or hexokinases (glucokinase in the case of liver), the low-binding affinity of L-FABP for G-6-P suggests that these effects might not be physiologically relevant. However, the higher affinity of L-FABP for G-1-P suggests that this effect could be physiologically relevant in some cell types. These data suggest that in the presence of hexokinases, glucose metabolites (especially G-1-P) might compete with glucose for binding to L-FABP and/or PPARα to affect lipid binding and/or transport to the nucleus.
Taken together these findings suggest that glucose may enhance the ability of L-FABP to bind lipidic ligands, transport lipidic ligands to the nucleus, and form a complex with PPARα. Further, these data suggest a complex interaction between glucose, lipids, and L-FABP concentrations in the regulation of PPARα. Liver cell concentrations of L-FABP are high, in the range of 200–400 μM, while nuclear concentrations of L-FABP are estimated to be approximately 43 μM (11, 12), well within the range of nucleoplasmic glucose concentrations reported for some cell types, i.e., 6–22 μM (13), and nucleoplasmic glucose concentrations are likely even higher in liver due to the properties of the hepatocyte glucose transporter-2 and glucokinase (36). As L-FABP has high affinity for glucose (Kd = 103.1 ± 18.6 nM), these data suggest that a substantial amount of nuclear L-FABP may be liganded with glucose. Furthermore, as nuclear PPARα levels are estimated to be 100–300 μM (13), glucose and L-FABP-mediated effects on PPARα are not likely to be saturated but rather respond to normal physiological conditions. However, not only glucose but also L-FABP levels are known to dramatically increase (e.g., 2–5-fold) in insulin-dependent diabetes and gestational diabetes in humans, streptozotocin-induced diabetes or obesity in rats, type 1 diabetes in mice, and certain gene-targeted mice (37–41). Thus, the overall effect of glucose and L-FABP on PPARα is expected to depend on (i) which LCFA/LCFA-CoA molecular species are available, (ii) PPARα's affinity versus L-FABP's affinity for those lipids, (iii) the amount of glucose in the nucleus, and (iv) the relative abundance of PPARα versus L-FABP in the nucleus. Although it is beyond the scope of the present study, whether other lipid binding proteins are also affected by glucose; either through direct binding or through PPARα-mediated regulation, remains to be determined. In summary, the data presented herein suggest the existence of dynamic interrelationships between glucose, lipidic ligands (LCFA, LCFA-CoA, fibrates, etc.), and lipid binding proteins (PPARα, L-FABP, etc.) in the nucleus.
Acknowledgments
The authors thank Dr. Noa Noy (Case Western University) for the PPARα bacterial expression vector.
Footnotes
Abbreviations:
- AOX
- acyl-CoA oxidase
- CD
- circular dichroism
- co-IP
- co-immunoprecipitation
- CPT-I
- carnitine palmitoyltransferase I
- FRET
- fluorescence resonance energy transfer
- G-1-P
- glucose-1-phosphate
- G-6-P
- glucose-6-phosphate
- L-FABP
- liver fatty acid binding protein
- LCFA
- long-chain fatty acid
- LCFA-CoA
- long-chain fatty acyl-CoA
- LCSM
- laser scanning confocal microscopy
- PPAR
- peroxisome proliferator-activated receptor
This work was supported by National Institutes of Health Grants DK-77573 (H.A.H.) and DK-41402 (F.S. and A.B.K.). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health. Startup funds from Boonshoft School of Medicine, Wright State University, were used to purchase the Jasco J-815 spectropolarimeter (H.A.H.).
REFERENCES
- 1.Zhang Y., Lee F. Y., Barrera G., Lee H., Vales C., Gonzalez F. J., Willson T. M., Edwards P. A. 2006. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc. Natl. Acad. Sci. USA. 103: 1006–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rutledge A. C., Adeli K. 2007. Fructose and the metabolic syndrome: pathophysiology and molecular mechanisms. Nutr. Rev. 65: S13–S23. [DOI] [PubMed] [Google Scholar]
- 3.Wei Y., Wang D., Topczewski F., Pagliassotti M. J. 2007. Fructose-mediated stress signaling in the liver: implications for hepatic insulin resistance. J. Nutr. Biochem. 18: 1–9. [DOI] [PubMed] [Google Scholar]
- 4.Desvergne B., Michalik L., Wahli W. 2004. Be fit or be sick: peroxisome proliferator-activated receptors are down the road. Mol. Endocrinol. 18: 1321–1332. [DOI] [PubMed] [Google Scholar]
- 5.Mangelsdorf D. J., Evans R. M. 1995. The RXR heterodimers and orphan receptors. Cell. 83: 841–850. [DOI] [PubMed] [Google Scholar]
- 6.Frederiksen K. S., Wulf E. M., Wassermann K., Sauerberg P., Fleckner J. 2003. Identification of hepatic transcriptional changes in insulin-resistant rats treated with peroxisome proliferator activated receptor-alpha agonists. J. Mol. Endocrinol. 30: 317–329. [DOI] [PubMed] [Google Scholar]
- 7.Adida A., Spener F. 2002. Intracellular lipid binding proteins and nuclear receptors inolved in branched-chain fatty acid signaling. Prostaglandins Leukot. Essent. Fatty Acids. Fatty Acids. 67: 91–98. [DOI] [PubMed] [Google Scholar]
- 8.Lin Q., Ruuska S. E., Shaw N. S., Dong D., Noy N. 1999. Ligand selectivity of the peroxisome proliferator-activated receptor alpha. Biochem. 38: 185–190. [DOI] [PubMed] [Google Scholar]
- 9.Hostetler H. A., Petrescu A. D., Kier A. B., Schroeder F. 2005. Peroxisome proliferator activated receptor alpha (PPARalpha) interacts with high affinity and is conformationally responsive to endogenous ligands. J. Biol. Chem. 280: 18667–18682. [DOI] [PubMed] [Google Scholar]
- 10.Hostetler H. A., Kier A. B., Schroeder F. 2006. Very-long-chain and branched-chain fatty acyl CoAs are high affinity ligands for the peroxisome proliferator-activated receptor alpha (PPARalpha). Biochemistry. 45: 7669–7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang H., Starodub O., McIntosh A., Kier A. B., Schroeder F. 2002. Liver fatty acid binding protein targets fatty acids to the nucleus: real-time confocal and multiphoton fluorescence imaging in living cells. J. Biol. Chem. 277: 29139–29151. [DOI] [PubMed] [Google Scholar]
- 12.Huang H., Starodub O., McIntosh A., Atshaves B. P., Woldegiorgis G., Kier A. B., Schroeder F. 2004. Liver fatty acid binding protein colocalizes with peroxisome proliferator receptor alpha and enhances ligand distribution to nuclei of living cells. Biochemistry. 43: 2484–2500. [DOI] [PubMed] [Google Scholar]
- 13.Hostetler H. A., Huang H., Kier A. B., Schroeder F. 2008. Glucose directly links to lipid metabolism through high-affinity interactionwith peroxisome proliferator activated receptor-alpha. J. Biol. Chem. 283: 2246–2254. [DOI] [PubMed] [Google Scholar]
- 14.Hostetler H. A., Syler L. R., Hall L. N., Zhu G., Schroeder F., Kier A. B. 2008. A novel high-throughput screening assay for putative anti-diabetic agents through PPARa interactions. J. Biomol. Screen. 13: 855–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schroeder F., Petrescu A. D., Huang H., Atshaves B. P., McIntosh A. L., Martin G. G., Hostetler H. A., Vespa A., Landrock K., Landrock D., et al. 2008. Role of fatty acid binding proteins and long chain fatty acids in modulating nuclear receptors and gene transcription. Lipids. 43: 1–17. [DOI] [PubMed] [Google Scholar]
- 16.Frolov A., Cho T. H., Murphy E. J., Schroeder F. 1997. Isoforms of rat liver fatty acid binding protein differ in structure and affinity for fatty acids and fatty acyl CoAs. Biochemistry. 36: 6545–6555. [DOI] [PubMed] [Google Scholar]
- 17.Schroeder F., Jolly C. A., Cho T. H., Frolov A. A. 1998. Fatty acid binding protein isoforms: structure and function. Chem. Phys. Lipids. 92: 1–25. [DOI] [PubMed] [Google Scholar]
- 18.Richieri G. V., Ogata R. T., Kleinfeld A. M. 1994. Equilibrium constants for the binding of fatty acids with fatty acid binding proteins from adipocyte, intestine, heart, and liver measured with the flourscent probe ADIFAB. J. Biol. Chem. 269: 23918–23930. [PubMed] [Google Scholar]
- 19.Wolfrum C., Borrmann C. M., Borchers T., Spener F. 2001. Fatty acids and hypolipidemic drugs regulate PPARalpha and PPARgamma gene expresion via L-FABP: a signaling path to the nucleus. Proc. Natl. Acad. Sci. USA. 98: 2323–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hostetler H. A., McIntosh A. L., Atshaves B. P., Storey S. M., Payne H. R., Kier A. B., Schroeder F. 2009. Liver type fatty acid binding protein (L-FABP) interacts with peroxisome proliferator activated receptor-α in cultured primary hepatocytes. J. Lipid Res. 50: 1663–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murphy E. J., Edmondson R. D., Russell D. H., Schroeder F. 1999. Isolation and characterization of two distinct forms of liver fatty acid binding protein from the rat. Biochim. Biophys. Acta. 1436: 413–425. [DOI] [PubMed] [Google Scholar]
- 22.McIntosh A. L., Atshaves B. P., Hostetler H. A., Huang H., Davis J., Lyuksyutova O. I., Landrock D., Kier A. B., Schroeder F. 2009. Liver type fatty acid binding protein (L-FABP) gene ablation reduces nuclear ligand distribution and peroxisome proliferator activated receptor-alpha activity in cultured primary hepatocytes. Arch. Biochem. Biophys. 485: 160–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sreerama N., Woody R. 2000. Estimation of protein secondary structure from circular dichroism spectra; Comparison of CONTIN, SELCON, and DCSSTR methods with an expanded reference set. Anal. Biochem. 287: 252–260. [DOI] [PubMed] [Google Scholar]
- 24.Petrescu A. D., Payne H. R., Boedeker A. L., Chao H., Hertz R., Bar-Tana J., Schroeder F., Kier A. B. 2003. Physical and functional interaction of acyl CoA binding protein (ACBP) with hepatocyte nuclear factor-4alpha (HNF4alpha). J. Biol. Chem. 278: 51813–51824. [DOI] [PubMed] [Google Scholar]
- 25.Martin G. G., Hostetler H. A., McIntosh A. L., Tichy S. E., Williams B. J., Russell D. H., Berg J. M., Spencer T. A., Ball J. A., Kier A. B., et al. 2008. Structure and function of the sterol carrier protein-2 N-terminal presequence. Biochem. 47: 5915–5934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petrescu A. D., Huang H., Hostetler H. A., Schroeder F., Kier A. B. 2008. Structural and functional characterization of a new recombinant histidine-tagged acyl CoA binding protein (ACBP) from mouse. Protein Expr. Purif. 58: 184–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jefferson J. R., Slotte J. P., Nemecz G., Pastuszyn A., Scallen T. J., Schroeder F. 1991. Intracellular sterol distribution in transfected mouse L-cell fibroblasts expressing rat liver fatty acid binding protein. J. Biol. Chem. 266: 5486–5496. [PubMed] [Google Scholar]
- 28.Wouters F. S., Bastiaens P. I., Wirtz K. W., Jovin T. M. 1998. FRET microscopy demonstrates molecular association of non-specific lipid transfer protein (nsL-TP) with fatty acid oxidation enzymes. EMBO J. 17: 7179–7189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lawrence J. W., Kroll D. J., Eacho P. I. 2000. Ligand dependent interaction of hepatic fatty acid binding protein with the nucleus. J. Lipid Res. 41: 1390–1401. [PubMed] [Google Scholar]
- 30.Walkey C. J., Spiegelman B. M. 2008. A functional peroxisome proliferator activated receptor gamma ligand binding domina is not required for adipogenesis. J. Biol. Chem. 283: 24290–24294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McArthur M. J., Atshaves B. P., Frolov A., Foxworth W. D., Kier A. B., Schroeder F. 1999. Cellular uptake and intracellular trafficking of long chain fatty acids. J. Lipid Res. 40: 1371–1383. [PubMed] [Google Scholar]
- 32.Li T., Lee H. B., Park K. 1998. Comparative stereochemical analysis of glucose-binding proteins for rational design of glucose specific agents. J. Biomater. Sci. Polym. Ed. 9: 327–344. [DOI] [PubMed] [Google Scholar]
- 33.Thompson J., Winter N., Terwey D., Bratt J., Banaszak L. 1997. The crystal structure of the liver fatty acid-binding protein. J. Biol. Chem. 272: 7140–7150. [DOI] [PubMed] [Google Scholar]
- 34.Stewart J. M., Dewling V. F., Wright T. G. 1998. Fatty acid binding to rat liver fatty acid-binding protein is modulated by early glycolytic intermediates. Biochim. Biophys. Acta. 1391: 1–6. [DOI] [PubMed] [Google Scholar]
- 35.Dresner A., Laurent D., Marcucci M., Griffin M. E., Dufour S., Cline G. W., Slezak L. A., Andersen D. K., Hundal R. S., Rothman D. L., et al. 1999. Effects of free fatty acids on glucose transport and IRS-1 associated phosphatidylinositol-3-kinase activity. J. Clin. Invest. 103: 253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harris R. A. 2006. Carbohydrate metabolism I: major metabolic pathways and their control. Textbook of Biochemistry with Clinical Correlations. 6th ed Devlin T. M., editor John Wiley and Sons, Hoboken, NJ: 581–635. [Google Scholar]
- 37.Magnusson A. L., Waterman I. J., Jansson T., Powell T. L. 2004. Triglyceride hydrolase activities and expression of fatty acid binding proteins in the human placenta in pregnancies complicated by intrauterine growth restriction and diabetes. J. Clin. Endocrinol. Metab. 89: 4607–4614. [DOI] [PubMed] [Google Scholar]
- 38.Engels W., van Bilsen M., Wolffenbuttel B. H. R., Van der Vusse G. J., Glatz J. F. 1999. Cytochrome P450, peroxisome proliferation, and cytoplasmic fatty acid binding protein content in liver, heart, and kidney of the diabetic rat. Mol. Cell. Biochem. 192: 53–61. [PubMed] [Google Scholar]
- 39.Morrow F. D., Allen C. E., Martin R. J. 1979. Intracellular fatty acid-binding protein: hepatic levels in lean and obese rats. Fed. Proc. 38: 280. [Google Scholar]
- 40.Kamijo-Ikemori A., Sugaya T., Sekizuka A., Hirata K., Kimura K. 2009. Amelioration of diabetic tubulointerstitial damage in liver type fatty acid binding protein transgenic mice. Nephrol. Dial. Transplant. : 788–800. [DOI] [PubMed] [Google Scholar]
- 41.Seedorf U., Raabe M., Ellinghaus P., Kannenberg F., Fobker M., Engel T., Denis S., Wouters F., Wirtz K. W. A., Wanders R. J. A., et al. 1998. Defective peroxisomal catabolism of branched fatty acyl coenzyme A in mice lacking the sterol carrier protein-2/sterol carrier protein-x gene function. Genes Dev. 12: 1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]