

Abstract
Persistently hyper-phosphorylated Akt contributes to human oncogenesis and resistance to therapy. TCN-P, the active metabolite of the Akt phosphorylation inhibitor triciribine (TCN), is in clinical trials, but the mechanism by which TCN-P inhibits Akt phosphorylation is unknown. Here we show that in vitro, TCN-P inhibits neither Akt activity nor the phosphorylation of Akt S473 and T308 by mTOR or PDK1, respectively. However, in intact cells, TCN inhibits EGF-stimulated Akt recruitment to the plasma membrane and phosphorylation of Akt. Surface plasmon resonance (SPR) demonstrates that TCN-P, but not TCN, binds Akt-derived pleckstrin homology (PH) domain (KD: 690 nM). Furthermore, nuclear magnetic resonance (NMR) spectroscopy shows that TCN-P, but not TCN, binds to the PH domain in the vicinity of the PIP3 binding pocket. Finally, constitutively active Akt mutants, Akt1-T308D/S473D and myr-Akt1, but not the transforming mutant Akt1-E17K, are resistant to TCN-P and rescue from TCN inhibition of proliferation and induction of apoptosis. Thus, our studies indicate that TCN-P binds to the PH domain of Akt and blocks its recruitment to the membrane and that the subsequent inhibition of Akt phosphorylation contributes to TCN-P anti-proliferative and pro-apoptotic activity, suggesting that this drug may be beneficial to patients whose tumors express persistently phosphorylated Akt.
The Ser/Thr-specific protein kinase Akt (also known as protein kinase B) was discovered independently in 1991 by three different groups 1-3 and has since emerged as an important promoter of tumor cell survival, proliferation as well as migration and invasion 4,5. In mammals, Akt is represented by three isoforms, Akt1, Akt2, and Akt3, which share 74 % sequence identity. Despite this high degree of homology, Akt isoforms may play distinct roles in development and normal physiology. For example, targeted gene deletions in mice have shown that loss of Akt1 function results in smaller body size and significant growth defects 6,7. Mice lacking Akt2 are unable to maintain glucose homeostasis and are diabetic 8, while Akt3 knockout mice have smaller brains but are otherwise normal 9.
For activity, Akt requires recruitment to the plasma membrane and subsequent phosphorylation at two critical sites, T308 and S473. These events are initiated by growth factors binding to their receptors, which activate phosphatidylinositol-3 kinase (PI3K) to convert phosphatidylinositol (4,5)-bisphosphate (PIP2) to phosphatidylinositol (3,4,5)-trisphosphate (PIP3). PIP3 recruits pleckstrin homology (PH) domain-containing proteins such as Akt to the plasma membrane, where it is phosphorylated at T308 by phosphoinositide-dependent kinase 1 (PDK1) 10. To be fully active, Akt needs to be phosphorylated at S473 as well. This site can be phosphorylated by several kinases including DNA-dependent protein kinase (DNA-PK) 11,12 and mTORC2 13. While mTORC2-mediated phosphorylation of S473 occurs at the membrane in response to growth factors 13, DNA-PK phosphorylates S473 in the nucleus in response to DNA damage 11,12. Whether phosphorylation at T308 and S473 in vivo occurs in a specific order is still controversial 5, but in vitro, T308 and S473 can be phosphorylated independently 14. While the lipid phosphatase and tumor suppressor PTEN antagonizes Akt signaling by dephosphorylating phosphoinositides, thereby preventing the activation of PDK1 and Akt, the mechanisms that directly terminate Akt signaling are not well characterized. pT308 is probably dephosphorylated by PP2A 15 and pS473 dephosphorylation may involve PP1 16 and/or novel PP2C-like phosphatases termed PHLPP1 and 2 17.
The PI3K/PTEN/Akt pathway is very frequently deregulated in human cancer, leading to persistent hyperphosphorylation-hyperactivation of Akt. This can be due to persistent activation of receptor tyrosine kinases, activating mutations in PI3K, inactivating mutations or deletion of PTEN 18,19, but also overexpression of Akt itself 20,21, and a recently discovered Akt mutation (E17K) that confers transforming and tumorigenic activity to Akt (however, this mutation is rare and was reported to occur in ~ 2, 6 or 8 % of ovarian, colon, or breast tumors, respectively) 22. Nonetheless, the specific roles of Akt isoforms in tumorigenesis are poorly understood. For instance, Akt1 is persistently activated in many cancers, and loss of Akt1 expression by antisense oligonucleotides results in inhibition of anchorage-independent growth and induction of apoptosis 23. Overexpression of Akt2, but not Akt1 or Akt3, results in an increase of PI3K-dependent invasion and metastasis of breast and ovarian cancer cells 24. Increased Akt3 expression and loss of PTEN result in the development of melanoma, and Akt3 siRNA stimulates apoptosis and inhibits melanoma development 25.
Akt contributes to malignant transformation and/or tumor progression by acting on a multitude of substrates (for recent reviews see refs. 4, 26-28) including IKKα, Bad, caspase-9, and forkhead transcription factors. Intriguingly, Akt phosphorylates Mdm2, thereby stimulating the subsequent degradation of p53. Furthermore, Akt has a direct role in promoting cell cycle progression by phosphorylating p21Cip1 and p27Kip1. Also, when growth factors are present, Akt can inhibit TSC1/2, eventually causing stimulation of mTOR, an important kinase that stimulates cell growth through promoting protein synthesis via S6K.
Because Akt is intimately involved in mediating many of the hallmarks of cancer, Akt has become a major anticancer drug target 18,19,29,30. Recently, we have discovered an Akt phosphorylation inhibitor, TCN and its active metabolite TCN-P, through screening the NCI Diversity Set 31. This compound does not inhibit Akt kinase activity per se, but in whole cells prevents phosphorylation of Akt1, Akt2 or Akt3 31. Furthermore, TCN does not inhibit PI3K, PDK1 and other protein kinases, but inhibits proliferation, induces apoptosis and inhibits tumor growth in animals much more potently in tumors that contain persistently hyper-phosphorylated Akt, suggesting that TCN is a selective Akt activation inhibitor 31. It is important to point out that TCN is a tricyclic nucleoside 32, which, once inside cells, is phosphorylated to its mono-phosphate derivative, TCN-P, by adenosine kinase. TCN-P is the active metabolite of TCN, as demonstrated by the finding that TCN is 5 000-fold less active in cells lacking adenosine kinase 33. TCN-P is presently undergoing human clinical trials in patients whose tumors contain high levels of phosphorylated Akt 34. However, the mechanism by which TCN-P inhibits the phosphorylation of Akt is not known. In this study, using a variety of approaches including biochemical and cell biological studies, surface plasmon resonance (SPR) and nuclear magnetic resonance (NMR) spectroscopy, we show that TCN-P, but not TCN, binds to the PH domain of Akt and prevents its recruitment to the plasma membrane and subsequent phosphorylation at T308 and S473. We also show that constitutively active Akt mutants, Akt1-T308D/S473D and myr-Akt, but not Akt1-E17K, rescue cells from TCN-P effects suggesting that the ability of TCN-P to inhibit tumor cell growth is due to its ability to prevent Akt phosphorylation.
Results
TCN-P inhibits in vitro neither Akt kinase activity nor the phosphorylation of purified Akt by PDK1 and mTORC2
TCN-P inhibits the phosphorylation of Akt in whole cells, suppresses tumor growth and induces apoptosis selectively in tumors that contain persistently hyper-phosphorylated Akt over those tumors that do not 31. This prompted a clinical trial in a population of patients whose tumors contain persistently hyper-phosphorylated Akt 34. Critical to the clinical development of TCN-P as a targeted agent is to understand the biochemical mechanism by which it inhibits the phosphorylation of Akt. Therefore, we have undertaken several biochemical, biophysical and cell biological studies to address this important question. First we determined whether in vitro, TCN-P inhibits Akt kinase activity or the ability of PDK1 and mTOR to phosphorylate Akt at T308 and S473, respectively, the two phosphorylation sites required for full activation of Akt. As shown in Figure 1a, TCN-P did not inhibit the ability of Akt1 to phosphorylate its substrate GSK3-β in vitro. Furthermore, purified PDK-1 and immuno-precipitated mTOR readily phosphorylated purified Akt1 at T308 and S473 in vitro, respectively, regardless of whether or not TCN-P was present in the reaction mixture (Figures 1b-c). These in vitro results demonstrate that TCN-P does not inhibit Akt kinase activity and that it does not directly interfere with the phosphorylation of Akt by either PDK-1 or mTOR.
Figure 1.
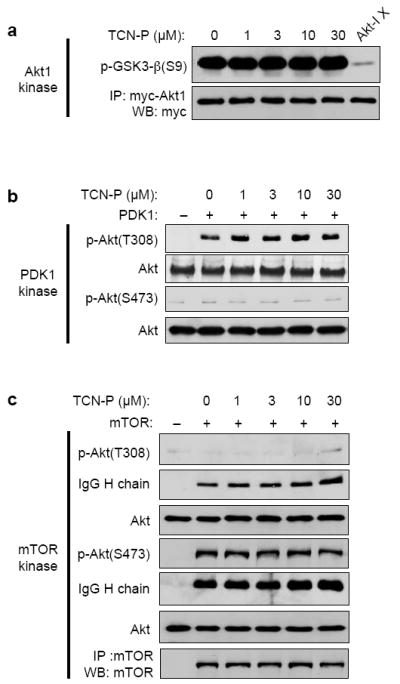
TCN-P does not inhibit the activity and phosphorylation of purified Akt in vitro. Kinase assays were performed with purified or immunoprecipitated proteins in the presence of 200 μM ATP in the absence or presence of increasing TCN-P concentrations, followed by Western blotting with the indicated antibodies as described in Materials and Methods. All experiments were performed independently at least three times. (a) Up to 30 μM TCN-P does not inhibit in vitro the kinase activity (as measured by phosphorylation of the substrate GSK3-β of Akt1 immunoprecipitated from HEK293T cells transfected with Myc-tagged Akt. Exposure to 10 μM Akt inhibitor X (lane 6) demonstrates that Akt activity could be inhibited efficiently. (b) Up to 30 μM TCN-P does not interfere with the phosphorylation of Akt1 at T308 by PDK-1. (c) Up to 30 μM TCN-P does not interfere with the phosphorylation of Akt1 at S473 by immunoprecipitated mTOR.
TCN prevents EGF-mediated Akt recruitment to the plasma membrane
Since TCN-P inhibits the phosphorylation of Akt in intact cells but not in vitro, we reasoned that a possible mechanism of action of TCN-P could be to prevent the translocation of Akt from the cytosol to the plasma membrane. To evaluate this possibility we used two approaches, immunofluorescence and subcellular fractionation/Western blotting. First, we transfected MDA-MB-468 cells with Myc-tagged Akt1, starved the cells, treated them with EGF, TCN or both and processed the cells for immunofluorescence as described under Materials and Methods. Figures 2a and b show that a major proportion (69.4 %) of vehicle-treated starved MDA-MB-468 cells contained Akt in the cytosol. However, these starved MDA-MB-468 cells contained 30.6 ± 5.7 % of Akt in the plasma membrane (Figure 2b). Treatment of these starved cells with TCN alone resulted in a decrease of cells with Akt in the plasma membrane from 30.6 ± 5.7 to 14.3 ± 3.0 %. In contrast, stimulation with EGF in the absence of TCN resulted in 100 % of the cells with Akt in the plasma membrane. Pre-treatment of the cells with TCN prior to EGF stimulation, dramatically reduced the percentage of cells with Akt in the plasma membrane from 100 to 20.0 ± 5.0 % (Figures 2a-b). Further, we have used subcellular fractionation to confirm these results. Figure 2c shows that, consistent with the immunofluorescence data of Figures 2a-b, TCN inhibited the ability of EGF to induce Akt recruitment to the plasma membrane in MDA-MB-468 cells.
Figure 2.
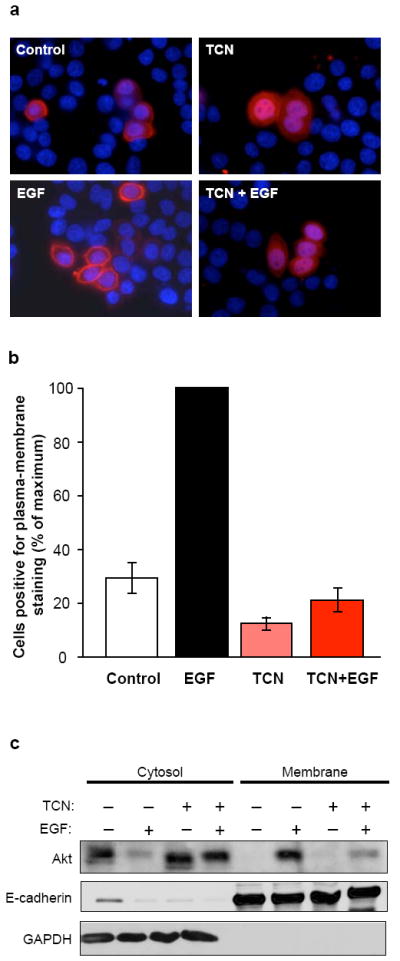
TCN inhibits EGF-induced recruitment of AKT1 to the plasma membrane. (a-c) Serum-starved MDA-MB-468 cells exogenously expressing Myc-tagged wild-type Akt1 were treated with vehicle, EGF, TCN or TCN + EGF, and further analyzed by immunofluorescence (a-b) and subcellular fractionation (c) as described in Materials and Methods. (a) Fluorescent images representative of three independent experiments show that in control cells, Akt (red) was mainly found in the cytosol, while in EGF-stimulated cells, Akt clearly relocated to the membrane. Cells treated with TCN showed cytosolic staining with or without stimulation by EGF. (b) Quantification of membrane localization of Akt1 was carried out as follows. Approximately 100 cells were scored in each of three randomly selected fields. Results are presented as percentage of Myc-Akt1-transfected cells that are positive for plasma membrane-located Akt1. Each column represents the mean of three independent experiments ± standard deviation. (c) Membrane and cytosolic fractions were prepared as described in Materials and Methods and analyzed by Western blotting with the indicated antibodies (representative of two independent
TCN-P, but not TCN, binds to the PH domain of Akt as demonstrated by SPR
The results described in Figures 1-2 suggest that a possible mechanism by which TCN inhibits Akt phosphorylation in intact cells is that TCN-P binds to the PH domain of Akt, thereby preventing its recruitment to the plasma membrane. To evaluate this possibility, we carried out binding experiments using SPR and NMR with Akt-derived PH domain as described in Materials and Methods. First we determined by SPR whether TCN-P and/or TCN bind the PH domain of Akt. Figure 3a shows a typical sensorgram obtained with 6 different TCN-P concentrations (46 nM to 11.1 μM), indicating that TCN-P bound to the PH domain of Akt in a dose-dependent manner. TCN-P bound to the Akt PH domain with a ka of 366 (M-1 s-1), a kd of 2.52×10-4 (s-1) and a KD of 690 nM (Figure 3a). In contrast, TCN did not bind the Akt PH domain (Figure 3b). Figure 3c shows the responses after a contact time of 300 sec for TCN-P and TCN as a function of their concentration, further demonstrating that TCN-P, but not TCN, binds to the Akt PH domain. To validate these results, we carried out several control experiments. Table 1 summarizes the kinetic data obtained with several compounds and the Akt-PH domain. As a positive control, we used Akt inhibitor VI, a peptide derived from the TCL1 oncoprotein that was reported to bind the PH-domain of Akt 35, and found in our experiments that it binds the Akt-derived PH domain with a KD of 1.4 μM (Table 1). Furthermore, PIP3, the plasma membrane lipid that recruits Akt to the plasma membrane, bound the Akt-PH domain with a ka of 4 251 (M-1 s-1), a kd of 2.05 ×10-4 (s-1) and a KD of 48 nM (Table 1).
Figure 3.
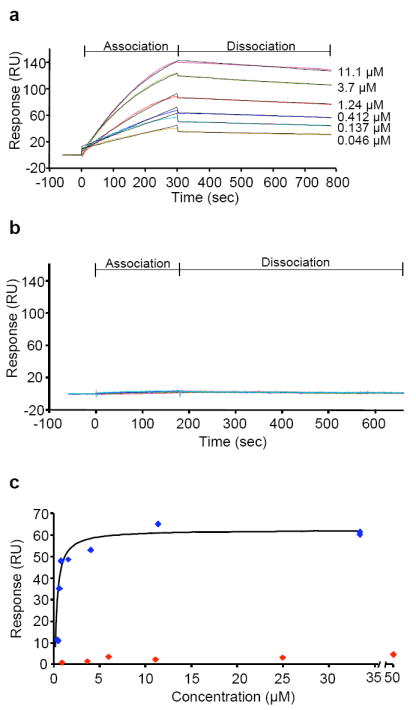
TCN-P, but not TCN, binds to the PH domain of Akt. The interaction between TCN and TCN-P, respectively, and Akt PH domain was investigated with the Biacore T100 system. The graphs shown are representative of at least two independent experiments. (a-b) Akt1-derived PH domain was immobilized to the surface via ligand thiol coupling as described in Materials and Methods. (a) Representative example of sensorgrams for the interaction between TCN-P and Akt1-PH domain, showing an increased response with increasing concentrations. (b) Representative example of sensorgrams for the interaction between TCN and Akt1-PH domain, indicating that there is no significant interaction. (c) Dose-dependent interaction at 300 sec with NTA-captured His-tagged Akt1 PH domain was observed between TCN-P (blue), but not TCN (red).
Table 1.
Interactions between TCNP and Akt1-derived PH domain, immobilized via thiol coupling
| Compound | Rexp/Rmax | ka (M-1s-1) | kd (s-1) | KD |
|---|---|---|---|---|
| TCN-P | 80/91 | 366 | 0.000252 | 690 nM |
| PIP3 | 31/186 | 4,251 | 0.000205 | 48 nM |
| Akti-VI | 48/422 | 80 | 0.000116 | 1.4 μM |
Ligand binding studies by NMR chemical shift mapping reveal that TCN-P, but not TCN, binds Akt-PH domain
To further characterize the binding properties of TCN-P and TCN to the Akt PH domain, we conducted ligand binding studies using nuclear magnetic resonance (NMR). Chemical shift mapping data suggest that Akt-PH domain residues in the PIP3 binding pocket 36, 37 were perturbed upon complexation with TCN-P, but perturbation also extended outside this region (Figures 4a-b), possibly due to some small conformational changes upon binding. For example, G16, I19, R23, R25 and I36 all show chemical shift perturbation. On the contrary, no appreciable chemical shift perturbation could be observed with the compound TCN (Figures 4a-b). This is consistent with the SPR results and suggests that the presence of the phosphate group is essential for binding (see more below).
Figure 4.

TCN-P, but not TCN, causes chemical shift perturbation of amino acids in the Akt PH domain. (a) Superposition of the HSQC spectra of the Akt PH domain in presence of TCN (blue) and of TCN-P (green) onto the spectrum of apo-protein (red). (b) Summary of the chemical shift perturbation values in the presence of TCN-P (gray) and TCN (green). Three dashed lines indicate Δppm positions of 0.015, 0.03, and 0.06 respectively. (c) Structure of TCN-P (top) and model of Akt PH domain (ribbons) in complex with TCN-P (bottom). The pose of TCN-P was obtained from the docking software GOLD. The overall ribbon structures of Akt PH domain in complex with inositol (1,3,4,5)-tetrakisphosphate (IP4) (PDB code: 1H10) 36 were colored in gray except for residues showing significant chemical shift perturbation. These were colored based on Δppm values: red, Δppm > 0.06; magenta, 0.06 > Δppm > 0.03; yellow, 0.03 > Δppm >0.015. (d) Ribbon representation of the structure of Akt PH domain in complex with IP4 (PDB code: 1H10) 36. The overall orientation is the same as that in (c). (e) Surface representation of the docking complex Akt/TCN-P and (f) of the complex Akt/IP4. In both panels, the overall surface of Akt was colored according to lipophilicity (blue, less lipophilic; brown, more lipophilic). The hydrogen bonds predicted to be formed between Akt and the compounds are displayed as yellow dashed lines. The position of residues involved in the formation of hydrogen bonds is shown in white and yellow. In (e), the position of residues showing chemical shifting perturbation in (c) is also shown using the color code as in (c).
Hence, based on the chemical shift mapping studies, we performed molecular docking studies to provide further possible binding poses of TCN-P in the PIP3 binding site of Akt PH domain. A possible binding mode for the compound was obtained using the program GOLD version 2.1 and the structure of Akt PH domain (PDB code: 1H10) 36. The TCN-P binding site was defined within a 15-Å radius around the residue K14 36, located at the PIP3 binding site. A total 100 runs were performed and the conformation of TCN-P with the best GOLD score is shown in Figure 4c, along with the binding mode of IP4, the well-accepted head group of PIP3 that binds to the Akt PH domain 36,37 (Figure 4d). For comparison, Figures 4e-f show space-filling models and the interacting amino acid residues for both TCN-P and PIP3, respectively. While ultimately X-ray diffraction studies will be needed to further delineate the intermolecular contacts at the atomic level, the docking pose obtained further suggests that TCN-P partially mimics the binding interactions provided by PIP3. Indeed, in the docking models, the phosphate of TCN-P is positioned at the location of the D3 phosphate of PIP3 and is surrounded by the residues K14, R23, R25 and N53 (Figures 4e-f). These are the same residues that have been shown to interact with the D3 phosphate of PIP3 in the crystal structure of Akt PH domain and PIP3 36. Furthermore, the five-member sugar ring of TCN-P (Figures 4c and 4e) can almost be superimposed to the six-member ring of PIP3 (Figures 4d and 4f).
Constitutively active Akt1-T308D/S473D and myr-Akt1 but not Akt1-E17K rescue from TCN-mediated inhibition of Akt activation, loss of cell viability and induction of apoptosis
The results from Figures 1-4 demonstrated that TCN-P binds to the PH domain of Akt and inhibits its recruitment to the plasma membrane, and suggest this as a mechanism by which TCN treatment of cells results in inhibition of Akt phosphorylation and subsequent activation. If this is the case, we reasoned that an Akt mutant that no longer requires recruitment to the plasma membrane and activation by phosphorylation would be resistant to TCN-P, and rescues cells from TCN-P-mediated inhibition of proliferation and induction of apoptosis. To evaluate this possibility, we stably expressed in COS-7 cells either Myc-tagged wild-type Akt1 or Myc-tagged Akt1-T308D/S473D (Akt1-DD), a constitutively active Akt mutant that does not require plasma membrane binding or phosphorylation at T308 and S473 for activation. Figure 5a (left panel) shows that both wild-type Akt1- and Akt1-DD-, but not the vector-transfected cells expressed the Myc-tagged Akt proteins. First, we determined whether the Akt1-DD kinase activity in intact cells is resistant to TCN. To this end, we treated wild-type Akt1 and Akt1-DD stably expressing cells with TCN, immunoprecipitated Akt with Myc antibody and assayed in vitro the Akt1 kinase activity using GSK3-β fusion protein as a substrate. Figure 5a (right panel) shows that treatment of the stably transfected cells with TCN resulted in dose-dependent inhibition of wild-type Akt1 but not Akt1-DD kinase activity. In addition to the Akt1-DD mutant, we also used two other constitutively active mutants of Akt. The first is myristoylated Akt1 (myr-Akt1), which does not require its PH domain for recruitment to the plasma membrane. The other mutant of Akt is the naturally occurring, transforming mutant Akt1-E17K, which requires the PH domain for plasma membrane recruitment. As expected, in the absence of the growth factor EGF, cells expressing wild-type Akt1 had much lower levels of phospho-Akt than those expressing either Akt1-E17K or myr-Akt1, whereas cells expressing Akt1-DD had no detectable phospho-Akt (Figure 5b). Upon stimulation with EGF, wild-type phospho-Akt levels increased and TCN treatment decreased these wild-type phospho-Akt levels. TCN also inhibited the phospho-Akt levels in cells expressing Akt1-E17K, but to a lesser degree. In contrast, TCN had no effect on phospho-Akt levels in cells expressing myr-Akt1. Furthermore, TCN inhibited endogenous phospho-GSK3-β in cells expressing wild-type Akt1, but not Akt1-DD or myr-Akt1. TCN inhibited phospho-GSK3-β in Akt1-E17K expressing cells, but to a lesser degree (Figure 5b). We next determined the effect of TCN treatment for 24 h on the proliferation of these same cells by MTT assay. As shown in Figure 5c, cells transfected with vector alone were not sensitive to TCN. Since these cells have virtually undetectable levels of phospho-Akt (Figure 5d), this is in agreement with our previous report showing that human cancer cells low in phospho-Akt levels are less sensitive to TCN than those with high phospho-Akt levels 31. Stable expression of wild-type Akt1 rendered these cells sensitive to TCN. However, both of the constitutively active mutants, Akt1-DD and myr-Akt1, rescued these cells from TCN-mediated inhibition of viability, whereas cells ectopically expressing Akt1-E17K were sensitive towards TCN, but not as sensitive as those expressing wild-type Akt1. Thus, Akt1-DD and myr-Akt1 are resistant to TCN and rescue from TCN effects. Consistent with these results, TCN treatment of COS-7 cells expressing vector, Akt1-DD or myr-Akt1 failed to induce any noticeable caspase-3 activity, whereas cells expressing wild-type Akt1 showed a 2- to 11-fold activation of caspase-3 in response to increasing concentrations of TCN. Furthermore, in cells expressing Akt1-E17K, TCN activated caspase-3 by up to 4-fold (Figure 5e). Taken together these results suggest that TCN-induced apoptosis depends on its ability to prevent Akt phosphorylation.
Figure 5.

Constitutively active forms of Akt1 rescue cells from TCN-induced loss of viability and apoptosis. COS-7 cells expressing vector, Myc-Akt1, Myc-Akt1-DD, Myc-Akt1-E17K or myr-Akt1 were exposed to TCN. (a) The level of expression of Akt1 constructs is shown in the left panel. In the right panel, exogenously expressed Akt1 was immunoprecipitated from cells with Myc antibody, and in vitro Akt1 kinase activity was assayed by its ability to phosphorylate S9 in GSK3-β. (b) Serum-starved cells were treated with different doses of TCN for 1 h, with or without subsequent stimulation by 30 ng/ml EGF for 20 min. Levels of endogenous phospho-Akt (S473) and phospho-GSK3-β(S9) were examined by Western blotting, with vinculin serving as loading control. The Myc blots demonstrate that the recombinant proteins were being expressed at comparable levels. (c) COS-7 cells expressing Akt1 constructs were exposed to TCN for 24 h and then subjected to MTT assay 41 to determine their proliferation/viability. The graph is representative of three independent experiments. (d) COS-7 cells have very low basal levels of phospho-Akt. To assess basal levels of S473 phosphorylation in Akt of COS-7 cells, we compared the phospho-Akt levels in COS-7 cells stably transfected with vector or wild-type Akt1, following exposure to vehicle (0.1 % DMSO, lanes 1 and 6) or increasing concentrations of TCN for 24 h. Cells were lysed as described in Materials and Methods, and analyzed by Western blotting with the indicated antibodies. (e) COS-7 cells expressing vector, wt-Akt1, Akt1-DD, Akt1-E17K or myr-Akt1 were exposed to TCN for 24 h and then lysed. Triplicate aliquots of these lysates were analyzed by caspase-3 assay as described in Materials and Methods. This was calculated, separately for each of the three cell types, by dividing the average fluorescence intensity at a given TCN concentration by the average fluorescence intensity in the absence of TCN. The graph is representative of two independent experiments.
Discussion
The PI3K/PTEN/Akt pathway is one of the most frequently deregulated pathways in human cancer. This justifies the need for potent and specific inhibitors capable of downregulating Akt. Targeting Akt directly would also be expected to neutralize malfunctions due to overexpression or activating mutations of PI3K and non-functional PTEN, which are often responsible for Akt hyperactivation. We have recently identified a compound, TCN, that has antitumor activity selectively in tumor cells that contain persistently hyper-phosphorylated Akt over those tumors that do not 31. Since TCN does not inhibit Akt kinase activity in vitro, but prevents the phosphorylation of Akt in intact cells, TCN is thought to function as an Akt activation inhibitor rather than an Akt inhibitor, but its precise mechanism of action remains to be determined. Since Akt regulation is complex (see Introduction), in theory several strategies exist to inhibit Akt, namely i) direct inhibition of Akt enzyme activity, ii) inhibition of Akt phosphorylation, iii) activation of Akt dephosphorylation, or iv) prevention of Akt translocation to the plasma membrane, which is a prerequisite for the critical phosphorylations at T308 and S473. In this paper, we provide evidence that TCN functions primarily through the latter mechanism.
Using SPR and NMR (see Figures 3-4), we have shown that TCN-P, which is known to be the active metabolite of TCN inside cells 33, can directly bind to the PH domain of Akt1 or Akt2. In contrast, TCN did not bind, demonstrating that the phosphate group is important for binding. The fact that TCN-P, but not TCN, binds to the PH domain of Akt supports our proposed mechanism of TCN-P competing with PIP3 and preventing Akt membrane binding. This is consistent with previous reports demonstrating that cells unable to convert TCN to TCN-P are highly resistant to TCN 33. Furthermore, our NMR studies also demonstrated that TCN-P, but not TCN, interacted with amino acids that are involved in PIP3 binding, which is in agreement with molecular docking studies suggesting that the Akt-PH domain PIP3 binding site can accommodate TCN-P. For example, TCN-P, but not TCN, caused chemical shifts perturbation in Akt-PH domain amino acids known to be involved in PIP3 binding. Furthermore, molecular docking shows that the phosphate group of TCN-P occupies the binding pocket of the D3 phosphate in PIP3 involving residues K14, R23, R25 and N53 of the Akt-PH domain. Finally, the sugar ring of TCN-P occupies the binding site of the six-member ring of PIP3. Collectively, the results of SPR and NMR experiments, combined with computational docking studies, explain why TCN-P did not prevent the phosphorylation of Akt by either PDK1 or mTOR in vitro (Figure 1), but TCN prevented the translocation of Akt to the plasma membrane following EGF stimulation in intact starved cells (Figure 2). This is most likely due to TCN-P preventing PIP3 from recruiting Akt to the plasma membrane, possibly either by competing with PIP3 for binding to the Akt PH domain or by binding to an adjacent pocket that induces conformational changes preventing PIP3 binding.
The behavior of the two mutants Akt1-DD and myr-Akt1 supports this interpretation. Figure 5 demonstrates that these forms of Akt1 do not depend on growth factor-dependent phosphorylation for activation and suggests neither Akt1-DD nor myr-Akt1 depend on growth factor-dependent recruitment to the plasma membrane or on membrane-associated phosphorylation. Therefore, both Akt1-DD and myr-Akt1 are constitutively active. Interestingly, ectopic expression of wild-type Akt1 conferred TCN sensitivity to COS-7 cells (Figures 5c and 5e). This is consistent with previous data that showed that parental NIH 3T3 cells are resistant to TCN whereas Akt2 overexpressing NIH 3T3 cells are sensitive to TCN 31. This is also consistent with the observation that human cancer cells with low phospho-Akt levels (OVCAR5, DU-145, T47D, Colo357, WM852) are much less sensitive than human cancer cells with high levels of phospho-Akt (OVCAR3, OVCAR8, PANC1, LNCaP, PC-3, MDA-MB-468, WM35) 31. In contrast, Akt1-DD was no longer phosphorylated at T308 or S473 and its ability to phosphorylate the Akt substrate GSK3-β was resistant to TCN. Similarly, in cells expressing myr-Akt1, TCN was unable to inhibit Akt1 phosphorylation at S473 or to inhibit phosphorylation of GSK3-β. In agreement with the failure of TCN to affect Akt phosphorylation and kinase activity, cells that express these constitutively active Akt mutants were resistant to TCN-induced loss of viability and apoptosis. These data support the notion that the mechanism by which TCN inhibits tumor survival and growth is by preventing the phosphorylation of Akt. Furthermore, cells expressing the transforming mutant Akt1-E17K were less sensitive to TCN-induced inhibition of Akt and also less sensitive to TCN inhibition of proliferation and induction of apoptosis. A possible explanation for the reduced sensitivity of this mutant is that replacement of a negative with a positive charge associated with the E-to-K mutation in position 17 is predicted to result in tighter binding of Akt1-E17K to the phospholipids of the plasma membrane 22, and therefore slightly reduced, but did not abolish the ability of TCN to interfere with the function Akt1-E17K.
In summary, our study has delineated a mechanism of action for the anti-cancer drug and chemical probe TCN-P. This small molecule appears to function primarily by binding to the PH domain of Akt and preventing the recruitment of Akt to the cell membrane, where this protein is normally activated by phosphorylation. Our results also support a mechanism of anti-tumor activity of TCN-P that involves inhibition of Akt phosphorylation, and suggest that in future TCN-P clinical trials, it would be important to evaluate the levels of phospho-Akt as a biomarker for TCN-P activity in patients whose tumors harbor high levels of phospho-Akt.
Materials and Methods
Materials
Human recombinant Akt and PDK1 enzymes were purchased from the following companies: Inactive Akt1 and active Akt1 (Invitrogen, Carlsbad, CA), PDK1 (R&D Systems, Minneapolis, MN). The active version of Akt1 was phosphorylated in T308 and S473, respectively. TCN and TCN-P were obtained from the National Cancer Institute. Antibodies were from the following sources: Akt, pAkt(T308), pAkt(S473) and pGSK3-β(S9) (Cell Signaling, Danvers, MA), mTOR (Millipore, Temecula, CA), E-cadherin (BD Biosciences, San José, CA), GAPDH (Covance, Princeton, NJ), Myc monoclonal antibody 9E10 (Santa Cruz Biotechnology, Santa Cruz, CA), AlexaFluor594 goat anti-mouse IgG (Invitrogen), β-actin and vinculin (Sigma, St. Louis, MO). EGF was purchased from Invitrogen. The caspase-3 substrate Ac-DEVD-AMC, Akt inhibitor X and Akt inhibitor VI, a 15 residue peptide derived from the oncogene TCL1 35, were purchased from Calbiochem, San Diego, CA. PIP3 was from Cayman Chemicals, Ann Arbor, MI.
Cells and cell culture
MDA-MB-468 breast cancer cells, HEK-293T cells and COS-7 cells were grown in Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), and cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2.
Generation of recombinant Akt constructs
The DNA sequence of the human Akt1 PH domain (coding for residues S2-S128, accession number: NP_005154.2) was PCR-amplified using the full-length Akt1 sequence (codon-optimized for the expression in E.coli and purchased from GeneArt, Regensburg, Germany) as a template. In the PCR reaction, the DNA oligomer ATCGCATATGGGATCCTCTGATGTTGCCATTGTTAAAGA was used as the 5’ end primer with an NdeI and BamHI restriction site and the DNA oligomer GCTAGCGGCCGCTTAGCTCGGGCTACCGCTACG as the 3’ end primer with a NotI restriction site. The PCR product was subcloned into the pET28a vector (Novagen, San Diego, CA) via the BamHI and NotI site coding for an N-terminally His6-tagged Akt1 PH domain S2-S128 construct with a 24 residue spacer region (bold letters) between the His6 tag and the PH domain: MGSSHHHHHHSSGLVPRGSHMASMTGGQQMGRGS-S2-PH-S128.
E. coli BL21(DE3) cells were transformed with the pET28a-His6-spacer-PH(S2-S128) plasmid and grown at 37°C in 4 l of Luria Bertani medium supplemented with 30 μg/ml kanamycin until the cell culture showed an OD600 of 0.5-0.7. The expression of the His6-tagged PH-Akt1 construct was induced by adding 250 μM isopropyl-d-thiogalactopyranoside (IPTG). Cells were further incubated at 22°C - 25°C for 16 h.
Cells were harvested by centrifugation and lysed by resuspension in 0.1 M Na/K-phosphate buffer pH 7.0, 300 mM NaCl, 10 mM imidazole, 0.5 mg/ml lysozyme, 0.01 % Triton X-100. After 1 h incubation at 4°C, cells were disrupted by sonication. Soluble protein was separated from cell debris and insoluble protein by centrifugation for 1 h at 53 000 g. The supernatant was loaded onto a Ni-NTA column (Qiagen), and the His6-tagged PH-Akt1 was eluted using 10 volumes of a 10-250 mM imidazole gradient. The peak fractions eluting at imidazole concentrations between 170 and 220 mM consisted of almost pure His6-tagged PH-Akt1 (≥ 98%). The fractions were pooled, and loaded onto a Superdex 200 gel filtration column (GE Healthcare) to remove miss-folded aggregated protein. The His6-tagged PH-Akt1 was concentrated to ca. 20 mg/ml using appropriate Amicon centrifugal filter units (Millipore). The concentrated protein was aliquoted, shock frozen in liquid nitrogen and stored at -80° C.
The phospho-mimicking constitutively active Myc-tagged Akt1-DD and the transforming mutant Akt1-E17K were generated using the pCMV-Tag3B vector and the QuikChange Site-directed Mutagenesis kit from Stratagene, La Jolla, CA. As a template, we used human Myc-tagged wild-type Akt1 (accession number: NM_001014431.1). Myr-Akt1 was prepared as described previously 38. The DNA sequences of the wild-type and mutant cDNAs encoding the recombinant constructs were validated by our Molecular Biology Core Facility and was found to be correct.
In vitro phosphorylation assays
The effect of TCN on the phosphorylation of Akt or the kinase activity of Akt was essentially determined in two ways: by using either purified recombinant proteins or lysates or proteins immunoprecipitated from cells transfected with recombinant Akt.
Non-radioactive in vitro Akt kinase assay was performed in the presence of 200 μM ATP with GSK-3β as a substrate according to the manufacturer’s instructions (Cell Signaling), except that we used Myc antibody instead of immobilized Akt antibody to immunoprecipitate Akt.
To assess phosphorylation of Akt by PDK1, human recombinant inactive Akt1 (30 ng) was incubated with PDK1 (100 ng) in the presence of 200 μM ATP for the indicated time at 30°C in a buffer consisting of 5 mM MOPS pH 7.2, 2.5 mM β-glycerophosphate, 1 mM EGTA, 0.4 mM EDTA, 5 mM MgCl2 and 0.05 mM DTT. The reaction was terminated by adding 8 μl of 4X SDS sample buffer to 30 μl of the reaction mixture and heating at 95°C for 5 min. Samples were subjected to 10% SDS-PAGE. Phosphorylation at T308 and S473 was monitored by subsequent Western blotting with the phosphorylation site-specific antibodies and developed using Chemiluminescence reagents (PerkinElmer, Boston, MA).
For in vitro kinase assays following exposure of cells to TCN, cells were transfected with vector, myc-wt-Akt1, myc-Akt1-DD or myc-Akt1-E17K using LipofectAMINE-2000. COS-7 cells stably transfected with these Akt1 constructs were selected by G418 (Sigma). Myr-Akt1 was transiently transfected into COS-7 cells.
The phosphorylation of Akt at S473 was assayed with immunoprecipitated mTOR as described by Sarbassov et al.39 with slight modifications. Briefly, 293T cells transfected with myc-tagged mTOR were lysed in lysis buffer containing 40 mM HEPES pH 7.5, 120 mM NaCl, 0.3% CHAPS, 1 mM EDTA, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, and protease inhibitor cocktail. Lysate (1 mg) was pre-cleared by incubating with 20 μl protein G-Sepharose conjugated to pre-immune IgG. The lysates were then incubated with 25 μl of Protein G and mTOR antibody. Immune complexes were incubated for 4 h at 4°C on a vibrating platform. Immunoprecipitates were washed four times with the above lysis buffer, followed by two washes with the mTOR kinase buffer (25 mM HEPES pH 7.5, 100 mM potassium acetate, 1 mM MgCl2). Immunoprecipitates were incubated in 30 μl of the mTOR kinase buffer containing 300 ng inactive Akt1 and 500 μM ATP at 37°C. The reaction was stopped by the addition of 8 μl of 4X SDS sample buffer and analysis of Akt phosphorylation by Western blotting was performed as described above.
Immunofluorescence analyses of Akt1 membrane localization and phosphorylation
MDA-MB-468 cells growing on cover slips in 6-well plates were transiently transfected with pCMV-Myc-wt-Akt1 by LipofectAMINE-2000 (Invitrogen) according to the manufacturer’s instructions. 48 h post-transfection cells were serum-starved overnight and treated with 20 μM TCN for 1 h and/or 100 ng/ml EGF for 15 min. Cells were washed three times with cold phosphate-buffered saline and fixed in 4 % formaldehyde for 30 min. Fixed cells were mounted onto glass slides with Vectashield Mounting Medium (Vector Labs, Burlingame, CA) containing DAPI (1.5 μg/ml) and stained with Myc antibody (1 μg/ml), followed by AlexaFluor594-conjugated secondary antibody (1 μg/ml). Fluorescence was observed using a Zeiss Axiovert Upright Fluorescent microscope with a 60X oil-immersion lens. For each experiment, around 100 Akt1-expressing cells were scored to quantify membrane localization of Akt1. We scored as positive for Myc-Akt1 membrane localization those cells whose perimeter showed a brightly red stain that was distinctly set apart from the nuclear blue DAPI stain. Results are presented as percentage of membrane-positive cells per total Akt1-expressing cells. Each bar represents the mean of three experiments ± standard deviation. To assess Akt phosphorylation at S473, we used Akt antibody and phospho-Akt(S473) (193H12) Alexa Fluor 488 conjugated secondary antibody, using similar procedures as described above.
Cell fractionation
Cytosol and membrane fractions were prepared as described by Fukuda et al.40. Briefly, cells were suspended in hypotonic lysis buffer (25 mM Tris/HCl, pH 7.4, 5 mM EDTA, 1 mM dithiothreitol, 1 mM Na3VO4, 2.5 mM Na4P2O7, and protease inhibitors) and disrupted by five passages through a 27-gauge needle. Large cell debris was pelleted by centrifugation at 2 000 g for 5 min at 4°C. The supernatants were centrifuged at 100 000 g for 20 min at 4°C. The resulting supernatant (cytosol) was collected, and the pellet was washed once with hypotonic lysis buffer, then resuspended in hypotonic lysis buffer containing 1% Triton X-100. The lysate was again centrifuged at 100 000 g for 20 min at 4°C, and the supernatant (membrane) was collected. The distribution of proteins in the cytosol and membrane fractions was analyzed by Western blotting.
Surface plasmon resonance (SPR)
The Biacore T100 instrument, the Series S sensor chips CM5 and NTA, the amine and thiol coupling kits, Biacore T100 Control and Evaluation Software were purchased from GE Healthcare (Princeton, NJ). The Akt inhibitor VI (reported KD for Akt1 PH domain = 18 μM 35) was used as a positive control.
Akt1-derived PH domain (100 μg/ml 100 mM sodium phosphate, pH 6.0) was immobilized on the active surface by ligand thiol coupling, which engages SH groups in the protein. In this case, a reference surface was prepared by “blank immobilization”. All kinetic/affinity experiments were carried out at 25°C with at least 5 different analyte concentrations as well as solvent alone (which served as a blank) with a flow rate of 30 μl/min. The contact time was typically ≥ 3 min and the dissociation time ≥ 6 min. All curves and diagrams shown are reference- and blank-subtracted. Compounds were prepared in running buffer, which was either PBS (for TCN-P, PIP3 and Akt inhibitor VI) or PBS containing 1 % DMSO (for TCN). For each immobilization, a theoretical maximal response (Rmax) was calculated for all compounds according to the formula: Rmax = (MWcompound/MWprotein) × RI, where MW = molecular weight and RI = immobilization level.
Nuclear magnetic resonance (NMR)
Human Akt2 protein fragment encompassing residues 1-118 was cloned into the expression vector pGEX2T and transfected into E. coli. Uniformly 15N-labeled protein was produced by growing the expression strain in the M9 medium with 15N NH4Cl as the sole nitrogen source. The protein was purified using GST affinity chromatography and then cleaved with thrombin. The resulting protein product has two extra amino acid residues (GS) at the N-terminus. The NMR samples contained 100 mM of protein in 100 mM phosphate buffer, pH 7.0 in 95% H2O / 5% D2O. All NMR experiments were performed at 300K on a 700 MHz Bruker Avance III spectrometer (Billerica, MA) equipped with a triple-resonance cryo-probe.
Molecular docking studies
The docking studies of Akt2 and TCN-P were performed using the program GOLD version 2.1 (CCDC Software Ltd., Cambridge, UK). The structure of Akt in complex with inositol (1,3,4,5)-tetrakisphosphate (IP4) (PDB code: 1H10) 36 was adopted to prepare the docking studies. The TCN-P binding site was defined within a 15-Å radius around the residue K14. A total 100 runs were performed and the conformation of TCN-P with the best GOLD score was selected for display.
Caspase-3 activation assay
300 000 COS-7 cells stably expressing vector, wt Akt1 or Akt1-DD were exposed to 0.6, 6 or 60 μM TCN for 24 h, lysed in a buffer consisting of 50 mM tris/Cl pH 7.5, 5 mM EDTA, 150 mM NaCl and 0.5 % NP-40. Lysate containing 20 μg protein was then incubated in triplicate for 1 h at 37°C with 20 μM of the fluorogenic caspase-3 substrate Ac-DEVD-AMC in the presence of 50 mM tris/Cl pH 7.5 in a total volume of 100 μl. Fluorescence was monitored with a Wallac Victor2 multiplate reader (ex: 355 nm, em: 460 nm).
Acknowledgments
This work was funded in part by NIH grant RO1-CA098473. We thank the Microscopy, High-Throughput Screening and Molecular Biology Core Facilities at the Moffitt Cancer Center for their expert technical assistance.
Footnotes
Conflict of interest statement The authors declare no conflict of interest.
References
- 1.Jones PF, Jakubowicz T, Pitossi FJ, Maurer F, Hemmings BA. Molecular cloning and identification of a serine/threonine protein kinase of the second-messenger subfamily. Proc Natl Acad Sci USA. 1991;88:4171–4175. doi: 10.1073/pnas.88.10.4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coffer PJ, Woodgett JR. Molecular cloning and characterisation of a novel putative protein-serine kinase related to the cAMP-dependent and protein kinase C families. Eur J Biochem. 1991;201:475–481. doi: 10.1111/j.1432-1033.1991.tb16305.x. [DOI] [PubMed] [Google Scholar]
- 3.Bellacosa A, Testa HR, Staal SP, Tsichlis PN. A retroviral oncogene, akt, encoding a serine-threonine kinase containing an SH2-like region. Science. 1991;254:274–277. doi: 10.1126/science.254.5029.274. [DOI] [PubMed] [Google Scholar]
- 4.Manning BD, Cantley LC. AKT/PKB signaling: Navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qiao M, Sheng S, Pardee AB. Metastasis and AKT activation. Cell Cycle. 2008;7:2991–2996. doi: 10.4161/cc.7.19.6784. [DOI] [PubMed] [Google Scholar]
- 6.Chen WS, Xu PZ, Gottlob K, Chen ML, Sokol K, Shiyanova T, et al. Growth retardation and increased apoptosis in mice with homozygous disruption of the akt1 gene. Genes Dev. 2001;15:2203–2208. doi: 10.1101/gad.913901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBα is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001;276:38349–38352. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
- 8.Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu QW, Crenshaw EB, Iii, et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKBβ) Science. 2001;292:1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- 9.Easton RM, Cho H, Roovers K, Shineman DW, Mizrahi M, Forman Ms, et al. Role for Akt3/protein kinase By in attainment of normal brain size. Mol Cell Biol. 2005;25:1869–1878. doi: 10.1128/MCB.25.5.1869-1878.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PRJ, Reese CB, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 11.Feng J, Park J, Cron P, Hess D, Hemmings BA. Identification of a PkB/Akt hydrophobic motif Ser-473 kinase as DNA-dependent protein kinase. J Biol Chem. 2004;279:41189–41196. doi: 10.1074/jbc.M406731200. [DOI] [PubMed] [Google Scholar]
- 12.Bozulic L, Surucu B, Hynx D, Hemmings BA. PKBα/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol Cell. 2008;30:203–213. doi: 10.1016/j.molcel.2008.02.024. [DOI] [PubMed] [Google Scholar]
- 13.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the Rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 14.Alessi DR, Andjelkovic M, Caudwell FB, Cron P, Morrice N, Cohen P, et al. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- 15.Ugi S, Imamura T, Maegawa H, Egawa K, Yoshizaki T, Shi K, et al. Protein phosphatase 2A negatively regulates Akt (protein kinase B) activity in 3T3-L1 adipocytes. Mol Cell Biol. 2004;24:8778–8789. doi: 10.1128/MCB.24.19.8778-8789.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu W, Yauan X, Jung YJ, Yang Y, Basso AD, Rosen N, et al. The heat shock protein 90 inhibitor geldanamycin and the ErbB inhibitor ZD1839 promote rapid PP1 phosphatase-dependent inactivation of AKT in ErbB2 overexpressing breast cancer cells. Cancer Res. 2003;63:7777–7784. [PubMed] [Google Scholar]
- 17.Brognard J, Newton AC. PHLiPPing the switch on Akt and protein kinase C signaling. Trends Endocrinol Metab. 2008;19:223–230. doi: 10.1016/j.tem.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dillon RL, White DE, Muller WJ. The phosphatidyl inositol 3-kinase signaling network: Implications for human breast cancer. Oncogene. 2007;26:1338–1345. doi: 10.1038/sj.onc.1210202. [DOI] [PubMed] [Google Scholar]
- 19.Liu W, Bagaitkar J, Watabe K. Roles of AKT in breast cancer. Front Biosci. 2007;12:4011–4019. doi: 10.2741/2367. [DOI] [PubMed] [Google Scholar]
- 20.Sun M, Wang J, Pacigga JE, Feldman RI, Yuan Z-Q, Ma X-L, et al. Akt1/PKBα kinase is frequently elevated in human cancers and its constitutive activation is required for oncogenic transformation in NIH3T3 cells. Am J Pathol. 2001;159:431–437. doi: 10.1016/s0002-9440(10)61714-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun M, Paciga JE, Feldman RI, Yuan Z-q, Coppola D, Lu YY, et al. Phosphatidylinositol-3-OH kinase (PI3K)/AKT2, activated in breast cancer, regulates and is induced by estrogen receptor α (ERα) via interaction between ERα and PI3K. Cancer Res. 2001;61:5985–5991. [PubMed] [Google Scholar]
- 22.Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–445. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 23.Liu X, Shi Y, Han EK, Chen Z, Rosenberg SH, Giranda VL, et al. Downregulation of Akt1 inhibits anchorage-independent cell growth and induces apoptosis in cancer cells. Neoplasia. 2001;3:278–286. doi: 10.1038/sj.neo.7900163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arboleda MJ, Lyons JF, Kabbinavar FF, Bray MR, Snow BE, Auyala R, et al. Overexpression of AKT2/protein kinase Bβ leads to up-regulation of β1 integrins, increased invasion, and metastasis of human breast and ovarian cancer cells.Cancer Res 200363196–206. [PubMed] [Google Scholar]
- 25.Stahl JM, Sharma A, Cheung M, Zimmermann M, Cheng JQ, Bosenberg MW, et al. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res. 2004;64:7002–7010. doi: 10.1158/0008-5472.CAN-04-1399. [DOI] [PubMed] [Google Scholar]
- 26.Liang J, Slingerland JM. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle. 2003;2:339–345. [PubMed] [Google Scholar]
- 27.Bozulic L, Hemmings BA. PIKKing on PKB: regulation of PKB activity by phosphorylation. Curr Opin Cell Biol. 2009;21:256–261. doi: 10.1016/j.ceb.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 28.Gonzalez E, McGraw TE. The Akt kinases: Isoform specificity in metabolism and cancer. Cell Cycle. 2009;8:2502–2508. doi: 10.4161/cc.8.16.9335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng JQ, Lindsley CW, Cheng GZ, Yang H, Nicosia SV. The Akt/PKB pathway: Molecular target for cancer drug discovery. Oncogene. 2005;24:7482–7492. doi: 10.1038/sj.onc.1209088. [DOI] [PubMed] [Google Scholar]
- 30.Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/Akt pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- 31.Yang L, Dan HC, Sun M, Liu QY, Sun XM, Feldman RI, et al. Akt/protein kinase B signaling inhibitor-2, a selective small molecule inhibitor of Akt signaling with antitumor activity in cancer cells overexpressing Akt. Cancer Res. 2004;64:4394–4399. doi: 10.1158/0008-5472.CAN-04-0343. [DOI] [PubMed] [Google Scholar]
- 32.Schweinsberg PD, Smith RG, Loo TL. Identification of the metabolites of an antitumor tricyclic nucleoside (NSC-154020) Biochem Pharmacol. 1981;30:2521–2526. doi: 10.1016/0006-2952(81)90577-3. [DOI] [PubMed] [Google Scholar]
- 33.Wotring LL, Crabtree GW, Edwards L, Parks RE., jr Mechanism of activation of triciribine phosphate (TCN-P) as a prodrug form of TCN. Cancer Treat Rep. 1986;70:491–497. [PubMed] [Google Scholar]
- 34.Garrett CR, Coppola D, Wenham RM, Cubitt CL, Neuger AM, Frost TJ, et al. Phase I pharmacodynamic study of triciribine phosphate, a small molecule inhibitor of Akt phosphorylation, in adult subjects with solid tumors containing phosphorylated Akt. 10th Annual AACR Meeting; Denver, CO. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hiromura M, Okada F, Obata T, Auguin D, Shibata T, Roumestand C, et al. Inhibition of Akt kinase activity by a peptide spanning the βA strand of the proto-oncogene TCL1. J Biol Chem. 2004;279:53407–53418. doi: 10.1074/jbc.M403775200. [DOI] [PubMed] [Google Scholar]
- 36.Thomas CC, Deak M, Alessi DR, van Aalten DM. High-resolution structure of the pleckstrin homology domain of protein kinase b/Akt bound to phosphatidylinositol (3,4,5)-trisphosphate. Curr Biol. 2002;12:1256–1262. doi: 10.1016/s0960-9822(02)00972-7. [DOI] [PubMed] [Google Scholar]
- 37.Auguin D, Barthe P, Auge-Senegas MT, Stern MH, Noguchi M, Roumestand C. Solution structure and backbone dynamics of the pleckstrin homology domain of the human protein kinase B (PKB/Akt). Interaction with inositol phosphates. J Biomol NMR. 2004;28:137–155. doi: 10.1023/B:JNMR.0000013836.62154.c2. [DOI] [PubMed] [Google Scholar]
- 38.Bellacosa A, Chan TO, Ahmed NN, Datta K, Malstrom S, Stokoe D, et al. Akt activation by growth factors is a multiple-step process: the role of the PH domain. Oncogene. 1998;17:313–325. doi: 10.1038/sj.onc.1201947. [DOI] [PubMed] [Google Scholar]
- 39.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 40.Fukuda T, Guo L, Shi X, Wu C. CH-ILKBP regulates cell survival by facilitating the membrane translocation of protein kinase B/Akt. J Cell Biol. 2003;160:1001–1008. doi: 10.1083/jcb.200212113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kazi A, Lawrence H, Guida WC, McLaughlin ML, Springett GM, Berndt N, et al. Discovery of a novel proteasome inhibitor selective for cancer cells over non-transformed cells. Cell Cycle. 2009;8:1940–1951. doi: 10.4161/cc.8.12.8798. [DOI] [PMC free article] [PubMed] [Google Scholar]