

Abstract
We have developed a thermoresponsive poly(N-isopropyl acrylamide)-based scaffold with degradability and controlled porosity. Biodegradable poly(N-isopropyl acrylamide) hydrogels were synthesized by photo-copolymerization of N-isopropylacrylamide with 2-methylene-1,3-dioxepane and polycaprolactone dimethacrylate. The hydrogels’ phase transition temperature, swelling and viscoelastic properties, as well as hydrolytic degradability at 25 and 37°C, were explored. A sphere-templating technique was applied to fabricate hydrogel scaffolds with controllable pore size and a highly interconnected porous structure. The scaffold pore diameter change as a function of temperature was evaluated and, as expected, pores decreased in diameter when the temperature was raised to 37°C. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) test results suggested neither the scaffolds nor their degradation products were cytotoxic to NIH3T3 cells. Scaffolds with 55±5 μm pore diameter were loaded with NIH3T3 cells and then were warmed to 37°C entrapping cells in pores approximately 39 μm in diameter, a size range we have found to be optimal for angiogenesis and biointegration. Cells showed uniform infiltration and an elongated morphology after 7 days of culture. Due to the controlled monodisperse pore diameter, highly interconnected architecture, fully degradable chemistry and thermoresponsive properties, the polyNIPAM-based scaffolds developed here are attractive for applications in tissue engineering.
Keywords: poly(N-isopropyl acrylamide), thermoresponsive polymer, hydrogel, degradable polymer, tissue engineering, scaffold
Introduction
Hydrogels are networks of hydrophilic cross-linked polymers which swell to equilibrium in the presence of water or physiological fluids. During the last two decades, naturally derived and synthetic hydrogels have been topics of extensive research in the field of tissue engineering for application as three-dimensional porous scaffolds for the repair and regeneration of various tissues and organs.1–6 Hydrogels are appealing for this goal due to their high water content and mechanical properties that mimic those of living tissues.5,7 The advantages of synthetic hydrogels compared to natural ones are reproduciblity, freedom from biological contamination and the ability to tune properties by synthetic chemistry modifications.1,5
So called “intelligent hydrogels” have been a subject of special interest due to their ability to respond with pronounced property changes to external stimuli.8–10 For example, thermosensitive degradable hydrogels have been used for cell entrapment 11 and protein release.12 Among the intelligent hydrogels, thermosensitive poly(N-isopropyl acrylamide) (polyNIPAM) is probably one of the most extensively studied. In aqueous media polyNIPAM hydrogels exhibit a volume phase transition temperature (VPTT) at around 32–34°C.13–15 The cross-linked polyNIPAM network undergoes an abrupt, reversible swelling-deswelling process below and above the VPTT.15 The VPTT of polyNIPAM hydrogels can be modulated by co-polymerization with hydrophilic or hydrophobic monomers to increase or decrease the transition temperature.9,16 Due to their thermosensitive nature, biostability and biocompatibility, polyNIPAM-based hydrogels are attractive candidates for biomedical applications including tissue engineering.17–20 Three-dimensional polymer scaffolds that mimic native extracellular matrices play a pivotal role in tissue engineering.16 The function of scaffolds is to direct growth of cells, either seeded within the porous structure or migrating from the surrounding tissue. Therefore, scaffold matrices must provide cell adhesion, proliferation, differentiation and infiltration.20–21 One of the criteria for the successful application of three-dimension scaffolds is high porosity.21 Various techniques have been used to fabricate interconnected porous scaffolds such as salt leaching, phase separation methods, gas foaming, fiber meshes and electrospinning.21 However, these methods do not provide precision control of pore size, structure or interconnectivity. Our research group has defined criteria for optimal porous scaffold biointegration and angiogenesis requiring that all pores are identical in size, well-interconnected and between 30 and 40 microns in diameter.40,46,48 Degradability is an additional important factor in designing scaffolds for applications in tissue and organ regeneration.1,21–22 Ideally, the scaffold should provide mechanical and biochemical support during the regeneration process and then, as new tissue forms, should be completely degraded and eliminated through metabolic processes.
The purpose of this study is to demonstrate the preparation of fully degradable, thermo-swellable scaffolds based on polyNIPAM hydrogels with controlled pore size and a highly interconnected porous structure that have potential for tissue engineering applications.
There are examples in the scientific literature of partially degradable polyNIPAM hydrogels where the degradable bonds are typically introduced into the cross-linking sites but not within the polymer backbone.13,19,23–32 Different biodegradable cross-linkers have been used for this purpose, such as amino-acid derivatives,23–25 degradable polyaspartic acid derivatives,13 as well as modified dextran,26–30 polylactic acid19,29 and poly(ε-caprolactone) (PCL).26,31 In all these cases, materials degrade through cleavage of crosslinkings leaving a non-degradable, high molecular weight polyNIPAM backbone. Such polyNIPAM chains have limited ability to be eliminated from the body by natural pathways and may induce the foreign body response.27,39
A non-degradable polymer backbone could be engineered for degradability by the introduction of ester linkages through radical ring-opening copolymerization (RROP)33 of cyclic monomers like 2-methylene-1,3-dioxepane (MDO)34–35 or benzo-2-methylene-1,3-dioxepane (BMDO).36 In the literature to date, a few studies on the preparation of degradable linear polyNIPAM by applying this technique have been reported37–39 and only one of them mentions preparation of fully degradable cross-linked polyNIPAM hydrogel by UV polymerization of NIPAM with BMDO in presence of poly(ethylene glycol-co-glycolic acid) diacrylate.39
Here we report a method for preparation of a thermoresponsive scaffolds based on a fully degradable polyNIPAM hydrogel with a controllable, monodisperse, highly interconnected porous structure for potential applications in tissue engineering. First, polyNIPAM hydrogels with degradable units within the polymer backbone and at the cross-linking sites were synthesized by photopolymerization of NIPAM with MDO and polycaprolactone dimethacrylate (PCLDMA). The VPTT of the hydrogels prepared at different [NIPAM]/[MDO] molar ratios were measured and their swelling and viscoelastic properties as well as hydrolytic degradability at 25 and 37 °C were explored. Second, a sphere-templating technique40 was applied to fabricate fully degradable scaffolds, with monodisperse controlled pore size and highly interconnected architecture, based on the aforementioned polyNIPAM hydrogels. The morphology of the scaffolds with different pore sizes was characterized and change in the pore size as a function of temperature was examined. The cytotoxicity of the scaffolds and their degradation products were analyzed using the MTT assay and the adhesion of the fibroblast cells to the material was explored. To demonstrate the potential of the developed material for tissue engineering, the scaffolds were loaded with NIH3T3 cells using the thermoresponsive properties of the material and cell distribution, infiltration and morphology were investigated.
Experimental Section
Materials
NIPAM (Aldrich, 97 %), MDO (Polysciences Inc.), tetraethylene glycol dimethacrylate (TEGDMA) (Polysciences Inc.), polycaprolactone diol (Mn ~530; Aldrich), methacryloyl chloride (Fluka, 97%), triethylamine (TEA) (Sigma, 99.5%), uncrosslinked poly(methyl methacrylate) (PMMA) microspheres (Microbeads® (sphere diameter ~ 38 μm); Kupa Inc. (sphere diameter ≤ 100 μm); Polyscience Inc. (sphere diameter ~ 200 μm, Mw 75, 000)), DMSO anhydrous (Sigma, 99.9%), ethylene glycol anhydrous (EG) (Sigma, 99.8%), 2,2-dimethoxy-2-phenylacetophenone (IRGACURE 651, Ciba-Geigy), dichloromethane (EMD Chemicals Inc., HPLC grade), acetone (EMD Chemicals Inc., HPLC grade), Vybrant MTT cell proliferation assay kit (Invitrogen), Dulbecco’s modified Eagle medium (DMEM) (Invitrogen).
Analytical Methods
1H-NMR spectra were obtained with a Bruker AV-300 spectrometer. For chloroform-d, chemical shifts are expressed in ppm downfield from tetramethylsilane used as an internal standard.
Photopolymerization was accomplished with a UV lamp (PC451050, Hanovia 450-W Hg lamp, HANOVIA Specialty Lighting LLC, NJ, USA. Distance of the lamp from polymerization mixture is 30 cm.)
The scaffold and cell morphology were determined by scanning electron microscopy (SEM) (FEI SEM XL Siron, Hillsboro, OR). Dry samples were Au/Pd sputter coated for 60 sec (SPI Supplies, West Chester, PA).
Details regarding additional equipment items that were used during this research are described later in pertinent paragraphs.
Synthesis of PCLDMA41
Polycaprolactone diol (Mn ~530, 25 g, 0.047 mol) and TEA (19.7 mL, 0.14 mol) were dissolved in anhydrous dichloromethane (150 mL) and the solution was cooled to 0°C. Methacryloyl chloride (13.8 mL, 0.14 mol) was added dropwise and the reaction mixture was stirred at room temperature (RT) for 18 h. The formed solid was filtered off and the dichloromethane solution was washed with 0.1N HCl, saturated NaHCO3 and H2O. The organic phase was dried over MgSO4, filtered and evaporated to produce an orange oil. Purified PCLDMA was obtained by precipitation into cold hexanes as a slightly yellow wax (yield 82%). The degree of methacrylation was determined with 1H NMR spectroscopy by ratio between the integral area under the peak at δ 6.12 (s, 2H, olefinic, cis) and 3.70 (m, 4H, -OCH2CH2OCH2CH2O-).
Synthesis of PolyNIPAM-20 and PolyNIPAM-40 Hydrogels
Fully degradable polyNIPAM-20 and polyNIPAM-40 hydrogels were prepared by photocopolymerization of NIPAM with a 20 and 40 molar percent of MDO monomer, respectively, as shown in Figure 1.
Figure 1.

Synthesis of fully degradable polyNIPAM-based hydrogel.
In a typical experiment, NIPAM (0.9 g, 0.008 mol), PCLDMA (0.158 g, 2.4×10−4 mol, 3% mol/mol), MDO (0.181 mL, 1.6×10−3 mol (20% mol/mol) or 0.362 ml, 3.2×10−3 mol (40% mol/mol)) and photoinitiator 2,2-dimethoxy-2-phenylacetophenone (25 mg, 8×10−5 mol (1% mol/mol)) were dissolved in anhydrous DMSO/EG solution (1.5 mL, 1:1 v/v). The reaction mixture was then cast into a mold (consisting of two microscope slides separated by 1 mm thick Teflon spacer) and photopolymerized for 3 min. The polymer sheet was then removed from the mold and washed with acetone and water to extract residues of unreacted reagents.
For NMR, linear polyNIPAM-40 was synthesized according the procedure described above but in the absence of PCLMDA crosslinker. After polymerization the crude linear material was dissolved in chloroform, precipitated from cold ether and dried under vacuum to obtain purified linear polyNIPAM-40 as a white powder.
Synthesis of Poly(NIPAM-TEGDMA)-20, Poly(NIPAM-TEGDMA)-40 and Poly(NIPAM-TEGDMA) Hydrogels
Partially degradable (NIPAM-TEGDMA)-20 and poly(NIPAM-TEGDMA)-40 hydrogels (with a degradable backbone but a non-degradable cross-linking sites), were prepared according the procedure described in the paragraph above, but in the presence of the non-degradable cross-linker TEGDMA (79 μL, 2.4×10−4 mol, 3% mol/mol) instead of PCLDMA. Non-degradable poly(NIPAM-TEGDMA) hydrogels (with non-degradable both backbone and cross-linking site), were prepared as described herein, but in the absence of MDO monomer.
Synthesis of PolyNIPAM Hydrogel
Partially degradable polyNIPAM hydrogel with a non-degradable backbone and degradable cross-linking sites was prepared according the procedure for synthesis of fully degradable hydrogels but in absence of MDO monomer.
Hydrogel Compositions Synthesized in this Work
Table I summarizes compositions and anticipated degradability of the hydrogels prepared in the present work.
Table I.
Summary of the Hydrogels Compositions Synthesized in this Work
| Hydrogel Name | MDO %, mol/mol | Degradable |
|
|---|---|---|---|
| Backbone | Cross-linking Site | ||
| PolyNIPAM-20 | 20 | √ | √ |
| PolyNIPAM-40 | 40 | √ | √ |
| PolyNIPAM | ---- | ---- | √ |
| Poly(NIPAM-TEGDMA)-20 | 20 | √ | ---- |
| Poly(NIPAM-TEGDMA)-40 | 40 | √ | ---- |
| Poly(NIPAM-TEGDMA) | ---- | ---- | ---- |
Degradation Studies
1. Impact of NIPAM Copolymerization with MDO on the Degradation of the PolyNIPAM Backbone
Non-degradable poly(NIPAM-TEGDMA) and partially degradable poly(NIPAM/TEGDMA)-20 and poly(NIPAM/TEGDMA)-40 hydrogel sheets were punched into 8 mm disks. The disks were lyophilized to determine the original dry weight (W1). The dry disks were then placed in 0.1 N NaOH solution and the samples were shaken at RT. Poly(NIPAM-TEGDMA), poly(NIPAM/TEGDMA)-20 and poly(NIPAM/TEGDMA)-40 samples (triplicates) were taken at different points in time and washed with H2O to remove residual NaOH. The samples were then lyophilized to determine dry weight after degradation (W2). The degradation was evaluated by calculating the percent of weight loss using equation (1):
| (1) |
2. Hydrolytic Degradation of Fully Degradable PolyNIPAM-Based Hydrogels
Fully degradable polyNIPAM-20 and polyNIPAM-40 hydrogel sheets were punched into 8 mm disks. The disks were lyophilized to determine the original dry weight (W1). Then, the dry disks were placed in 0.007 N NaOH solution and the samples were shaken at 25 and 37°C. For both temperatures, polyNIPAM-20 and polyNIPAM-40 samples (triplicates) were taken at different points in time, washed with H2O and lyophilized to determinate dry weight after degradation (W2). The degradation was evaluated by calculating the percent of weight loss using equation (1).
VPTT Determination
VPTT of the polyNIPAM-20, polyNIPAM-40 and polyNIPAM hydrogels was determined by differential scanning calorimetery (DSC) (Netzsch DSC 200). The hydrogel sheets, swollen to equilibrium, were punched into 3 mm diameter disk and the excess water on the disk surface was removed with wet filter paper. Then, the samples were placed in an aluminum pan and sealed with an aluminum lid. The samples were scanned from 25 to 50°C at a heating rate of 3 °C/min under dry nitrogen. The VPTT was defined as the onset temperature of the endotherm.
Swelling Study
PolyNIPAM-20, polyNIPAM-40 and polyNIPAM hydrogel sheets were punched into 8 mm disks. The disks were lyophilized to determinate the dry weight (Wdry). The samples were then swollen in distilled water at 4, 25 and 37°C for 24 h to reach the equilibrium state. For each temperature, the excess water on the swollen hydrogel surface was removed with wet filter paper and the weight of the swollen sample (Wswollen) was determined (in triplicate). The swelling percent was calculated using equation (2):
| (2) |
Rheology
Viscoelastic properties of polyNIPAM-20 and polyNIPAM-40 were characterized by dynamic shear oscillation measurements. The rheology study was performed on swollen hydrogel disks (25 mm diameter, 0.5 cm thickness) using a rheometer (AR-G2, TA Instruments, New Castle, DE, USA) with a 20 mm diameter parallel plate. The effect of the temperature on the storage modulus (G′) was determined from 25 to 45°C with a heating rate of 1°C/min, at constant frequency and shear strain of 1 Hz and 10%, respectively. The temperature of the plate was controlled by connection to a recirculating water bath.
Sphere-Templated Scaffold Fabrication
Step 1. Template Preparation
PMMA microspheres were fractionated to the following size cuts: 35 ± 1μm; 49 ± 8μm; 81 ± 8μm and 188 ± 26 μm, with an ATM model L3P Sonic Sifter. The beads were transferred to a mold (composed of two microscope slides separated by a 1 mm thick Teflon spacer) and sonicated for 10 min for optimal packing. The beads were then sintered for 20 h at 140°C to obtain PMMA templates with neck sizes (interconnects between the beads) of 30% of the bead diameter.
Step 2. Scaffold Fabrication
PMMA templates were infiltrated with the reaction mixture composed of NIPAM (0.9 g, 0.008 mol), PCLDMA (0.158 g, 2.4×10−4 mol, 3% mol/mol), MDO (0.181 mL, 1.6×10−3 mol (20% mol/mol) or 0.362 ml, 3.2×10−3 mol (40% mol/mol)) and photoiniator 2,2-dimethoxy-2-phenylacetophenone (25 mg, 8×10−5 mol (1% mol/mol)) dissolved in anhydrous DMSO/EG solution (1.5 mL, 1:1 v/v). The mixture was photopolymerized for 5 min and the PMMA template infused with polymerized hydrogel was removed from the mold and placed in dichloromethane to dissolve the PMMA beads. The scaffold obtained, composed of a fully degradable polyNIPAM gel, was washed in acetone and then hydrated in distilled water.
Shrinkage
Swollen polyNIPAM-40-based scaffolds disks (10 mm diameter with different pore diameters) were embedded in a gel (Tissue Tek® O.C.T. Compound), frozen in liquid nitrogen and then cut to 25 μm thick sections with a Leica ultramicrotome CM1850. The sections were immersed in water and observed under an inverted optical microscope equipped with an incubation warmer (Nikon eclipse TE200). Pore diameter was measured with MetaMorph® software (version 6.0, Molecular Devises, PA, USA) at 25°C (D25) and 37°C (D37) and shrinkage percent was calculated by using equation (3):
| (3) |
Cytotoxicity
Cytotoxicity of the scaffolds and their degradation products were evaluated by using an MTT assay.
1. Scaffold Cytotoxicity
Scaffolds were sterilized with 70% ethanol, washed with sterile PBS and placed in DMEM for 24 h. At the same time, 0.5 mL of 3T3 mouse fibroblasts (1×104 cells/well) were seeded in 24-well culture plates for 24 h. The media then was removed from the wells and replaced with 0.5 mL of scaffold eluent. The cells were incubated with media eluted from the scaffold for an additional 24 h, then washed with phenol red-free DMEM media and afterwards exposed for 4 h to 0.5 mL of phenol red-free DMEM media containing 10% of MTT solution (5 mg/mL MTT reagent in PBS). 1 mL of DMSO was then added to extract a blue product by vigorous pipetting. Tissue culture polystyrene (TCPS) and latex were used as the negative and positive controls, respectively, and were treated similarly as above. 200 μL aliquot from each well was transferred into a 96-well plate and absorbance was measured at 550 nm with a microplate reader (tunable VERSAmax microplate reader, Molecular Devices, CA, USA).
2. Degradation Products Cytotoxicity
Scaffolds were hydrolyzed in 0.1 N NaOH. The solution was neutralized and degradation products were extracted with chloroform. The organic phase was dried over MgSO4, filtered, and chloroform was evaporated to obtain a white powder. The degradation products were dissolved into media at concentrations of 5, 10 and 15 mg/mL and cytotoxicity at different concentrations was evaluated according the procedure for the scaffold eluent, described in the paragraph above.
Cell Adhesion and Morphology
PolyNIPAM-40-based scaffold was punched into 8 mm disks, sterilized with 70% ethanol, washed with sterile PBS and soaked in culture media at 4°C overnight prior to seeding. Afterwards, disks immersed in DMEM media were placed at 37°C for 24 h to allow shrinkage of the material. The shrunken disks then were removed from the media and placed into a 96-well plate to fit exactly the diameter of the well and to serve as a surface for cell culture. 180 μL of cell suspension (1×104cells/well) was added and cells were cultured on the surface of the scaffold for 2 and 5 days. At each point in time the media was removed, the scaffold’s surface was washed with warm sterile PBS and then fixed with 2% glutaraldehyde solution at 37°C. At the end of fixation the sample was immediately frozen in liquid nitrogen and lyophilized. The samples were observed under SEM to explore cell adhesion and morphology.
Cell Loading Study
The polyNIPAM-40-based scaffold with 55 ± 5 μm pore diameter was punched into 10 mm disks, sterilized with 70% ethanol, washed with sterile PBS and soaked in culture media at 4°C overnight. Prior to loading with cells, the excess of media was removed from the scaffold’s surface with wet filter paper. Afterward, the disks were placed on dry filter paper and 50 μL of the 3T3 cell suspension (1×107 cells/mL) was added 4 times to the top of each disk (triplicates). The scaffolds were then placed into a 24-well plate and 2 mL of media was carefully added. The cells were cultured for 7 days and the media was changed every other day. The scaffolds were then fixed with a methanol-glacial acetic acid solution (9:1 v/v) for 30 min at 37°C. For histological analysis, samples were dehydrated through graded ethanol/xylene at 37°C and then embedded in paraffin. Paraffin blocks were sectioned (5 μm thick) onto charged Superfrost® plus slides (VWR International, West Chester, PA, USA), deparaffinized, rehydrated and stained with hematoxylin and eosin. Images were obtained with a Nikon E800 upright microscope equipped with MetaMorph® software (version 6.0, Molecular Devises, PA, USA). For observation under SEM the samples’ cross-sections of approximately 1mm thick were deparaffinized, rehydrated, frozen in liquid nitrogen and lyophilized.
Results and Discussion
Fully Degradable PolyNIPAM-Based Hydrogels
Homopolymerization of cyclic MDO monomer results in PCL formation through the RROP mechanism34 and in the present work this feature was utilized to engineer degradable polyNIPAM backbone. Figure 1 illustrates the synthesis of a polyNIPAM-based hydrogel by photo-copolymerization of NIPAM with MDO and PCLDMA degradable cross-linker that completely degrades to oligomeric units that can cleared from the body.
Copolymerization of NIPAM with MDO contributes ester linkages to the polymer backbone chains through incorporation of CL units and therefore leads to the formation of a degradable polyNIPAM backbone. Thus, by polymerization of NIPAM in presence of MDO and PCLDMA, one can obtain degradable sites throughout the polymer network (backbone and the cross-links).
In order to demonstrate the impact of NIPAM copolymerization with MDO on the degradable properties of the polymer backbone, polyNIPAM-based hydrogels with the non-degradable cross-linker TEGDMA were synthesized. Non-degradable control, poly(NIPAM-TEGDMA), was prepared in absence of MDO; partially degradable poly(NIPAM-TEGDMA)-20 and poly(NIPAM-TEGDMA)-40 were synthesized in presence of 20 and 40% mol/mol of MDO, respectively. The accelerated degradation of the hydrogels in 0.1N NaOH solution was evaluated by measuring weight loss (%) vs. time and is summarized in Figure 2.
Figure 2.

Degradation of the poly(NIPAM-TEGDMA), poly(NIPAM-TEGDMA)-20 and poly(NIPAM-TEGDMA)-40 hydrogels in 0.1N NaOH.
Figure 2 demonstrates that control, poly(NIPAM-TEGDMA) does not exhibit degradation while both poly(NIPAM-TEGDMA)-20 and poly(NIPAM-TEGDMA)-40 show significant weight loss of 48 and 65%, respectively, after 10 hours of exposure to alkaline solution. The hydrogels were synthesized with non-degradable cross-linker and the modification was applied only in the polymer chain and therefore this experiment is a proof for incorporation of ester linkages into the polyNIPAM backbone by the presence of MDO during copolymerization. 1H-NMR of linear polyNIPAM-40 also confirms the presence of the CL units in the polyNIPAM backbone: 1H-NMR (CHCl3) δ 1.14 (-CONCH(CH3)2), 1.26–1.93 (-CO2CH2CH2CH2CH2-) and (-CH2-CH(CONCH(CH3)2), 2.13 (-CH2-CH(CONCH(CH3)2), 2.85 (-CH2CO2CH2CH2CH2CH2-), 3.54 (-CH2CO2CH2CH2CH2CH2-), 4.03 (-CONCH(CH3)2).
VPTT of fully degradable hydrogels, polyNIPAM-20 and polyNIPAM-40 (prepared in presence of 20 and 40% mol/mol MDO, respectively) and partially degradable polyNIPAM (prepared in presence of PCLDMA but in absence of MDO) were determined by DSC and defined as the onset temperature of the endotherms, as shown in Figure 3.
Figure 3.
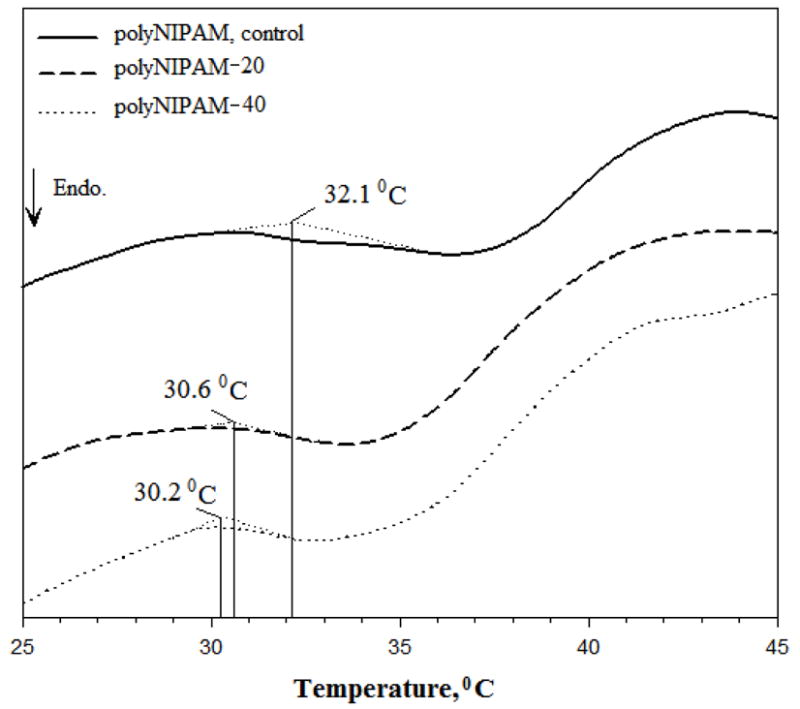
DSC thermograms of the polyNIPAM, polyNIPAM-20 and polyNIPAM-40 hydrogels.
Both polyNIPAM-20 and polyNIPAM-40 exhibit a VPTT around 30°C compared to polyNIPAM homopolymer with a VPTT of 32.1°C. In general, VPTT of themosensitive polyNIPAM-based hydrogels is usually governed by the relative hydrophobicity of the bulk.9,16 As a result of the copolymerization of NIPAM with MDO, the hydrophobic/hydrophilic balance of the polyNIPAM hydrogel is changed due to incorporation of CL units into the polyNIPAM backbone.37 The presence of the hydrophobic CL fragments increases the hydrophobicity of the hydrogel network and therefore decreases the VPTT of fully degradable polyNIPAM-20 and polyNIPAM-40 compared to partially degradable polyNIPAM. The polyNIPAM-40 has been suggested to have more hydrophobic character then polyNIPAM-20 since it was prepared with a higher molar percent of MDO and consequently contains more CL fragments. This assumption can explain the slight difference in the VPTT values of polyNIPAM-20 and polyNIPAM-40 hydrogels: 30.2 and 30.6°C, respectively. The relatively large breadth of the endotherms, which is observed for polyNIPAM-20, polyNIPAM-40 and polyNIPAM samples, probably can be related to gradual, not sharp, deswelling of the hydrogels.47
Figure 4 shows the swelling of polyNIPAM-20, polyNIPAM-40 and polyNIPAM hydrogels at temperatures below and above the VPPT.
Figure 4.

Swelling (%) of polyNIPAM, polyNIPAM-20 and polyNIPAM-40 hydrogels as a function of temperature.
As seen in Figure 4, polyNIPAM-20, polyNIPAM-40 and polyNIPAM hydrogels decrease in swelling as temperature increases, which is the same tendency in thermoresponsive behavior consistent with previously reported data regarding polyNIPAM-based materials.19,25,29,37 A significant decrease in swelling occurs at 37°C, the temperature above the VPTT, due to intensive dehydration of the polymer network. Figure 4 also demonstrates that at each temperature, the hydrogels exhibit slightly different swelling ability that depends on their copolymer composition. As a result of copolymerization with MDO, both polyNIPAM-20 and polyNIPAM-40 possess increased hydrophobic nature compared to polyNIPAM and therefore show lower swelling. The polyNIPAM-40 exhibits lower swelling at each temperature comparing to polyNIPAM-20, as shown in Figure 4, which can be explained by the increased hydrophobicity.
Figure 5 shows polyNIPAM-40 hydrogel disks that were swollen in water at 4, 25 and 37 °C to visualize the difference in swelling at temperatures below and above the VPTT.
Figure 5.

PolyNIPAM-40 hydrogel disks swollen in water at different temperatures.
Figure 5 demonstrates a decrease in the diameter of the swollen disks with increasing temperature (the initial diameter of the three dry disks before swelling was exactly the same). Visual inspection of the poly-NIPAM-40-based hydrogel indicates a transparent appearance at the temperatures below VPTT and an opaque appearance at 37 °C due to dehydration of the bulk. Thus, the picture presented in Figure 5 supports the results summarized in Figure 4.
To explore the temperature-dependent viscoelastic properties of polyNIPAM-20 and polyNIPAM-40, the storage modulus, G′, was measured under oscillatory shear conditions. Figure 6 demonstrates the storage modulus of fully degradable polyNIPAM- 20 and polyNIPAM-40 hydrogels as a function of temperature.
Figure 6.

Storage modulus, G′, of polyNIPAM-20 and polyNIPAM-40 hydrogels as a function of temperature at a frequency of 1 Hz and 10% shear strain.
Both polyNIPAM-20 and polyNIPAM-40 exhibit the typical rheological behavior of thermosensitive polyNIPAM-based materials.23,26,47 As shown in Figure 6, below the VPTT there are non-significant changes in values of the storage modulus with increase in temperature. At approximately 30°C, the temperature corresponding to the VPTT, there is an increase in G′, which becomes more dramatic with increasing temperature. This behavior indicates the deswelling for both hydrogels leading to the formation of more rigid materials.
Figure 7 demonstrates the degradation of polyNIPAM-20 and polyNIPAM-40 hydrogels at 25 and 37°C that was monitored by measuring weight loss of the hydrogels in 0.007 N NaOH as a function of time.
Figure 7.

Degradation of polyNIPAM-20 and polyNIPAM-40 in 0.007N NaOH at 25°C (A) and 37°C (B).
Figure 7 shows that the degradation rate of both hydrogels at 37°C is significantly slower than at 25°C. This behavior is reasonable, since at 25°C the hydrogels are more swollen by alkaline solution and degradable sites are available and accessible for accelerated hydrolysis. On the other hand, at 37°C, the temperature above the VPTT, the hydrogels had expelled much water. This increases the hydrogels’ bulk hydrophobicity and affects chain conformation by collapse due to dehydration. Now degradable sites are less accessible for hydrolysis and polyNIPAM-20 and polyNIPAM-40 probably degrade through surface erosion, which can explain the decreased degradation rate. Figure 7 also demonstrates that at both temperatures, polyNIPAM-40 exhibits higher weight loss as compared to polyNIPAM-20. For example, after 24 h of degradation at 25°C polyNIPAM-20 and polyNIPAM-40 lost 29 and 51% of the initial weight, respectively, and after 5 days of degradation at 37°C polyNIPAM-20 and polyNIPAM-40 exhibit decreases of 6 and 18% of their initial weight, respectively. It can be assumed that polyNIPAM-40 has more degradable sites since it was prepared in the presence of a higher concentration of MDO and therefore degrades faster than polyNIPAM-20.
In this study, degradation of the polyNIPAM-20 and polyNIPAM-40 hydrogels was explored in an alkaline environment rather than at physiological pH in order to accelerate the degradation process of the PCL blocks. PCL is a well known biodegradable polyester used in biomedical applications with degradation kinetics considerably slower than other aliphatic polyesters due to its hydrophobicity and crystallinity.42,43 PCL-containing materials degrade in vitro (under physiological conditions) and in vivo on time scales of 3–6 month to 2 years, depending on composition, PCL molecular weight, etc.41–43 In the present work we structured our experiments to demonstrate that our new polymers can indeed degrade to soluble products. Since PCL polymers have often been shown to appropriately degrade upon in vivo implantation, these new polymers should behave similarly under in vitro accelerated conditions. When we focus this polymer system on a specific application with defined degradation time requirements, we will modify our material accordingly and explore degradation under the appropriate physiological conditions.
Fully Degradable PolyNIPAM Scaffolds
Figure 8 illustrates the process for the sphere-templated fabrication of fully degradable, highly interconnected polyNIPAM-based scaffolds with thermally controllable pore size. This specific approach to sphere-templating was developed by Ratner and Marshall and involves forming a crosslinked polymer scaffold around a template of sinter-fused, monodisperse-sized porogens and removing the template to produce a porous biomaterial.48
Figure 8.

Sphere-templated fabrication of a fully degradable polyNIPAM-based scaffold.
In this study, monodispersed PMMA microspheres of the desired diameter are introduced into the mold, sintered to fuse particles, infiltrated with a monomer mixture of NIPAM, MDO, PCLDMA and photoinitiator dissolved in DMSO, polymerized in situ to obtain a composite of polyNIPAM hydrogel and PMMA particles and finally exposed to solvent to dissolve the PMMA particles. This yields a fully degradable, porous highly interconnected scaffold. In summary, the pore size of the scaffold is defined by the initial diameter of PMMA particles and the interconnectivity is obtained through sintering for longer or shorter times.
This sphere-templating protocol differs from previous versions in the use of DMSO as the monomer solvent. Methods described by Marshall et al.40, Bryant et al.44 and Linnes et al.45 used water to dissolve monomers for poly(2-hydroxyethyl methacrylate) (polyHEMA) or fibrin scaffold fabrication. In the present work, water could not be used as a solvent since MDO is moisture sensitive and the PCL-based crosslinker is not soluble in water. Thus, we needed to develop a system based on an organic solvent that, on the one hand, led to a homogenous solution of the monomers and reagents but, on the other hand, will not dissolve the PMMA templates during the infiltration. DMSO was found to meet these requirements and the sphere-templated procedure was adjusted to the present reagents composition.
Fully degradable scaffolds based on the polyNIPAM-40 hydrogel with pore diameters of 36 ± 2, 55 ± 5, 90 ± 8 and 204 ± 26 μm (at 25°C) were fabricated by using templates composed of PMMA beads of 35 ± 1, 49 ± 8, 81 ± 8 and 188 ± 26 μm, respectively. Figure 9 shows a representative SEM image of the polyNIPAM-40-based scaffold with a 55 ± 5 μm pore diameter.
Figure 9.

SEM image of the polyNIPAM-40-based scaffold with 55 ± 5 μm pore diameter.
The image demonstrates a monodispersed highly interconnected porous morphology that is typical for the scaffolds of all pore sizes.
Our objective was to achieve a change in pore size of the polyNIPAM-40-based scaffolds going from RT to body temperature. Figure 10 shows light microscope images of the scaffold’s cross-section with pore diameter of 36 ± 2 μm at 25 (A) that was reduced to 29 ± 1 μm at 37°C (B).
Figure 10.

Light microscope images of polyNIPAM-40-based scaffold’s cross-section at 25 (A) and 37°C (B) with pore size of 36 ± 2 μm and 29 ± 1 μm, respectively. Scale bar is 100 μm.
Figures 10-A and 10-B represent the same area of the section at the same magnification and it can be qualitatively seen that at 37°C (Figure 10-B) the pores shrank. The shrinkage can be attributed to volume phase transition since the temperature was raised above the hydrogel’s VPTT.
Table II quantifies the percent shrinkage at 37°C in polyNPAM-40-based scaffolds with different pore sizes.
Table II.
Shrinkage at 37°C in PolyNIPAM-40-based Scaffolds with Different Pore Diametersa
| Pore diameter, μm |
Shrinkage, % |
||
|---|---|---|---|
| 25 °C | 37 °C | Linear | Volumetric |
| 36 ± 2 | 29 ± 1 | 20 | 48 |
| 55 ± 5 | 39 ± 3 | 29 | 64 |
| 90 ± 8 | 67 ± 4 | 25 | 59 |
| 204 ± 26 | 138 ± 19 | 32 | 69 |
The shrinkage study was performed as described in the Experimental Section
Table 2 demonstrates the decrease in pore diameter at 37°C for polyNIPAM-40-based scaffolds of all explored pore sizes. For example, the scaffolds with pore diameter of 36 and 90 μm exhibit linear shrinkage in a pore size of about 20 and 25 %, respectively. Based on the aforementioned results one can assume that the shrinkage might affect not only the pore size but also the pore throat diameter (which is about 30% of the pore size for the sintering conditions used). This must be taken into consideration when planning scaffolding based on thermosensitive polyNIPAM because at body temperature the throat size should be large enough for efficient cell distribution throughout the scaffold. For example, at 25°C the throat size of the scaffold with 36 μm diameter is about 11 μm but at body temperature it’s probably will be reduced by approximately 20% and could be too small for cell penetration. Therefore, polyNIPAM-40-based scaffolds that have potential for tissue engineering should have a pore size starting at 55 μm at room temperature for sufficient throat size at body temperature.
The fact that the polyNIPAM-40-based scaffold can be engineered to have larger pores at room temperature than at 37°C makes the scaffold attractive for cell loading and delivery. We have a special interest in scaffolds with pore diameters of 30–40 μm because in previous studies we found that monodisperse pores of approximately 35–40 μm are optimal for vascularized, relatively non-fibrotic healing. This was seen with both silicone elastomer and polyHEMA based scaffolds.40,46 Cells have been found to show an increased adhesion to the polyNIPAM-based materials at 37°C due to protein adsorption to the more hydrophobic polymer.49 Thus, we hypothesized that due to the relatively large pore size and low cell-adhesiveness, the polyNIPAM-40-based scaffolds with pore diameter of 55 μm may efficiently be loaded with cells at RT and then at 37°C the cells will be locked, both geometrically and with cell-polymer adhesive interactions, within the scaffold with optimal pore size of 39 μm. Thus, upon implantation, the cell-loaded sphere-templated scaffold could sustain the loaded cells through enhanced angiogenesis and decreased fibrosis, and, after providing mechanical and biochemical support, may degrade leaving behind regenerated, vascularized tissue. Results regarding cell loading will be presented later in the article.
An additional benefit of the poly-NIPAM scaffolds is a unique synthetic polymer composition allowing physicochemical properties to be tuned for optimal performance in tissue engineering. For example, degradation rate of the scaffold can be controlled by the molar ratio between NIPAM and MDO monomers, as well as by cross-linker nature and network density.
The in vitro cytotoxicity of the fully degradable scaffolds and their degradation products were evaluated by a colorimetric MTT test. MTT is a yellow tetrazolium salt that is reduced to form violet formazan only in living, metabolically active cell mitochondria and thus, the number of living cells can be spectrophotometrically quantitated. In the present study, the absorbance values of the samples were normalized to the negative control (TCPS). Figure 11 shows representative results of the MTT test for the scaffold with 55 ± 5 μm pore diameter and its degradation products compared to negative and positive (latex) controls.
Figure 11.

Cytotoxicity of the polyNIPAM-40-based scaffold with 55 ± 5 μm pore diameter and its degradation products at 5, 10 and 15 mg/mL. TCPS and latex were used as negative and positive controls, respectively. Absorbance of the samples has been normalized to the TCPS’s value.
From data presented in Figure 11 it can be concluded that neither the scaffold itself nor its degradation products in the concentration range between 5–15 mg/mL are cytotoxic toward fibroblast cells. The same results were obtained for polyNIPAM-20 and polyNIPAM-40-based scaffolds of different pore sizes (data not shown).
In order to explore the adhesion of the cells to the polyNIPAM-40-based scaffold, the 55 ± 5 μm pore diameter was used as a surface for culturing fibroblast for 2 and 5 days. Representative SEM images of the scaffold’s surface at different magnifications after 2 days of cell culture are seen in Fig. 12-A. The cells are properly attached to the surface and show appropriate morphology. After 5 days of culture (Figure 12-B) the formation of a cell sheet on the surface of the scaffold as well as cell infiltration within the pores was observed. This behavior indicates the ability of the polyNIPAM-40-based scaffold to support cell attachment and growth and suggests cell compatibility with the material.
Figure 12.

SEM images of the scaffold surface with fibroblast cells after two days of culture (A, scale bar from top to bottom is 50, 10 and 10 μm) and five days of culture (B, scale bar from top to bottom is 50, 20 and 10 μm).
To demonstrate the potential of the polyNIPAM-40-based scaffold material for tissue engineering, the scaffold with a 55 ± 5 μm pore diameter was loaded with a model system for many cell types, NIH3T3 cells. The scaffold with this particular diameter was chosen since its diameter decreases at 37°C to 39 μm (Table 2). As was mentioned, this pore size was found to be optimal for vascularized, relatively non-fibrotic integration into tissue.40,46
Figure 13 shows relatively uniform cell distribution within the scaffold from the top to the bottom. The denser cell population on the one of the edges indicates the direction of the cell seeding. The magnified images show cell infiltration through the interconnecting throats and their appropriate elongated morphology.
Figure 13.

Light microscope images of histological section of NIH3T3 cells loaded into a polyNIPAM-40 scaffold with 55 ± 5 μm pore diameter.
Representative SEM images of the cell-loaded polyNIPAM-40-based scaffold with a pore diameter of 55 ± 5 μm (at room temperature) are shown in Figure 14. The cross-section images demonstrate cell attachment and infiltration throughout the scaffold and support the histological data.
Figure 14.

Representative SEM images (4 different areas of the scaffold) of cell loaded polyNIPAM-40-based scaffold cross-sections with a pore diameter of 55 ± 5 μm (at 25°C).
Conclusions
Thermosensitive scaffolds with two important criteria, full degradability and controlled porosity, were developed. First, fully degradable polyNIPAM-20 and polyNIPAM-40 hydrogels with ester linkages within both the backbone and the cross-linking sites were synthesized and characterized. Second, porous, highly interconnected polyNIPAM-40-based scaffolds with defined pore sizes were fabricated by using the sphere-templating technique. The scaffold with a pore diameter of 55 ± 5 μm was efficiently loaded with fibroblast cell at 25°C. The potential to use the scaffolds for tissue engineering was demonstrated by increasing the temperature to 37°C, thereby locking cells in the proangiogenic 39 μm pores. Future studies on the scaffolds will focus on loading the scaffold with 55 ± 5 μm pore diameter with cells and following their proliferation in vitro, as well as implantation of the scaffold in an animal model and the exploration of its behavior in vivo. Due to controlled monodisperse pore diameter, highly interconnected morphology, fully degradable nature and thermoresponsive properties, this novel polyNIPAM-based scaffold is an attractive candidate for applications in tissue engineering.
Acknowledgments
This work was supported by NIH grant R01 HL64387 and funding from the Coulter Foundation.
References
- 1.Drury JL, Mooney DJ. Biomaterials. 2003;24:4337–4351. doi: 10.1016/s0142-9612(03)00340-5. [DOI] [PubMed] [Google Scholar]
- 2.Lutolf MP. Nature Mater. 2009;8:451–453. doi: 10.1038/nmat2458. [DOI] [PubMed] [Google Scholar]
- 3.Langer R, Peppas NA. AIChE J. 2003;49:2990–3006. [Google Scholar]
- 4.Hoffman AS. Adv Drug Deliv Rev. 2002;43:3–12. doi: 10.1016/s0169-409x(01)00239-3. [DOI] [PubMed] [Google Scholar]
- 5.Karande TS, Agrawal CM. In: Nanotechnology and Tissue Engineering: the Scaffold. Laurencin CT, Nair LS, editors. CRC press; Boca Raton, FL: 2008. p. 54. [Google Scholar]
- 6.Williams CG, Elisseeff JH. In: Scaffolding in Tissue Engineeing. Ma PX, Elisseeff JH, editors. CRC press; Boca Raton, FL: 2005. p. 169. [Google Scholar]
- 7.Peppas NA, Huang Y, Torres-Lugo M, Ward JH, Zang J. Annu Rev Biomed Eng. 2000;2:9–29. doi: 10.1146/annurev.bioeng.2.1.9. [DOI] [PubMed] [Google Scholar]
- 8.Hasirci V, Yucel D. In: Encyclopedia of biomaterials and biomedical engineering. Wnek EG, Bowlin GL, editors. Informa Health Care; New York, NY: 2008. p. 228. [Google Scholar]
- 9.Kong J, Mu L. In: Smart polymers: applications in biotechnology and biomedicine. Galaev I, Mattiassin B, editors. CRC press; Boca Raton, FL: 2007. p. 247. [Google Scholar]
- 10.Kopecek J, Yang J. Polym Inter. 2007;56:1078–1098. [Google Scholar]
- 11.Vermonden T, Fedorovich NE, van Geemen D, Alblas J, van Nostrum CF, Dhert WJA, Hennink WE. Biomacromolecules. 2008;9:919–926. doi: 10.1021/bm7013075. [DOI] [PubMed] [Google Scholar]
- 12.Censi R, Vermonden T, van Steenbergen MJ, Deschout H, Braeckmans K, De Smedt SC, van Nostrum CF, di Martino P, Hennink WE. J Cont Rel. 2009;140:230–236. doi: 10.1016/j.jconrel.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 13.Yu Y, Xu Y, Ning H, Zhang S. Colloid Polym Sci. 2008;286:1165–1171. [Google Scholar]
- 14.Grinberg VY, Dubovik AS, Kuznetsov DV, Grinberg NV, Grosberg AY, Tanaka T. Macromolecules. 2000;33:8685–8692. [Google Scholar]
- 15.Cho EC, Lee J, Cho K. Macromolecules. 1990;23:283–289. [Google Scholar]
- 16.Feil HF, Bae YH, Feijen J, Kim SW. Macromolecules. 1993;26:2496–2500. [Google Scholar]
- 17.Klouda L, Mikos AG. Eur J Pharm Biopharm. 2008;68:34–45. doi: 10.1016/j.ejpb.2007.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roy I, Gupta MN. Chem Biol. 2003;10:1161–1171. doi: 10.1016/j.chembiol.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 19.Huang X, Zhang Y, Donahue HJ, Lowe TL. Tissue Eng. 2007;13:2645–2651. doi: 10.1089/ten.2007.0084. [DOI] [PubMed] [Google Scholar]
- 20.Lee KY, Mooney DJ. Chem Rev. 2001;101:1869–1879. doi: 10.1021/cr000108x. [DOI] [PubMed] [Google Scholar]
- 21.Khang G, Kim MS, Lee HB. In: A Manual for Biomaterials/Scaffold Fabrication Technology. Khang G, Kim MS, Lee HB, editors. World Scientific; Hackensack, NJ: 2007. p. 1. [Google Scholar]
- 22.Nair LS, Laurencin CT. Prog Polym Sci. 2007;32:762–798. [Google Scholar]
- 23.Kim S, Chung EH, Gilbert M, Healy KH. J Biomed Mater Res. 2005;75:73–88. doi: 10.1002/jbm.a.30375. [DOI] [PubMed] [Google Scholar]
- 24.Kim S, Healy E. Biomacromolecules. 2003;4:1214–1223. doi: 10.1021/bm0340467. [DOI] [PubMed] [Google Scholar]
- 25.Perez P, Gallardo A, Corrigan OI, Roman SJ. J Biomater Sci Polym Edn. 2008;19:769–783. doi: 10.1163/156856208784522074. [DOI] [PubMed] [Google Scholar]
- 26.Wu D, Qiu F, Wang T, Jiang X, Zhang X, Zhuo R. ACS Appl Mater Interfaces. 2009;1:319–327. doi: 10.1021/am8000456. [DOI] [PubMed] [Google Scholar]
- 27.Zang X, Sun G, Wu D, Chu C. J Mater Sci-Mater M. 2004;15:865–875. doi: 10.1023/B:JMSM.0000036274.83104.fe. [DOI] [PubMed] [Google Scholar]
- 28.Namkung S, Chu C. J Biomater Sci Polymer Edn. 2007;18:901–924. doi: 10.1163/156856207781367701. [DOI] [PubMed] [Google Scholar]
- 29.Huang X, Nayak BR, Lowe TL. J Polym Sci Polym Chem. 2004;42:5054–5066. [Google Scholar]
- 30.Kumashiro Y, Huh KM, Ooya T, Yui N. Biomacromolecules. 2001;2:874–879. doi: 10.1021/bm015527y. [DOI] [PubMed] [Google Scholar]
- 31.Paris R, Quijada-Garrido I. Polym Int. 2009;58:362–367. [Google Scholar]
- 32.Ooya T, Akutsu M, Kumashiro Y, Yui N. Sci Technol Adv Mat. 2005;6:447–451. [Google Scholar]
- 33.Sanda F, Endo T. J Polym Sci Polym Chem. 2001;39:265–276. [Google Scholar]
- 34.Jin S, Gonsalves KE. Macromolecules. 1997;30:3104–3106. [Google Scholar]
- 35.Jin S, Gonsalves KE. Macromolecules. 1998;31:1010–1015. [Google Scholar]
- 36.Bailey WJ, Ni Z, Wu S. Macromolecules. 1982;15:711–714. [Google Scholar]
- 37.Sun L, Zhuo R, Liu Z. Macromol Biosci. 2003;3:725–728. [Google Scholar]
- 38.Ren L, Agarwal S. Macromol Chem Phys. 2007;208:245–253. [Google Scholar]
- 39.Siegwart DJ, Bencherif SA, Srinivasan A, Hollinger JO, Matyjaszewski K. J Biomed Mater Res. 2008;87:345–358. doi: 10.1002/jbm.a.31708. [DOI] [PubMed] [Google Scholar]
- 40.Marshall AJ, Irvin CA, Barker T, Sage HE, Hauch KD, Ratner BD. ACS Polym Prep. 2004;45:100–101. [Google Scholar]
- 41.Atzet S, Curtin S, Trinh P, Bryant S, Ratner B. Biomacromolecules. 2008;9:3370–3377. doi: 10.1021/bm800686h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kweon HY, Yoo MK, Park IK, Kim TH, Lee HC, Lee HS, Oh JS, Akaike T, Cho CS. Biomaterials. 2003;24:801–808. doi: 10.1016/s0142-9612(02)00370-8. [DOI] [PubMed] [Google Scholar]
- 43.Hongfan S, Lin M, Cunxian S, Xiumin C, Pengyan W. Biomaterials. 2006;27:1735–1740. [Google Scholar]
- 44.Linnes MP, Ratner BD, Giachelli CM. Biomaterials. 2007;28:5298–5306. doi: 10.1016/j.biomaterials.2007.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bryant SJ, Cuy JL, Hauch KD, Ratner BD. Biomaterials. 2007;28:2978–2986. doi: 10.1016/j.biomaterials.2006.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ratner BD. Polym Inter. 2007;56:1183–1185. [Google Scholar]
- 47.Geever LM, Minguez CM, Devine DM, Nugent MJD, Kennedy JE, Lyons JG, Hanley A, Devery S, Tomkins PT, Higginbotham CL. J Mater Sci. 2007;42:4136–4148. [Google Scholar]
- 48.Ratner BD, Marshal AJ. 20080075752. US Appl. 2008
- 49.Okano T, Yamada N, Okuhara M, Sakai H, Sakurai Y. Biomaterial. 1995;16:297–303. doi: 10.1016/0142-9612(95)93257-e. [DOI] [PubMed] [Google Scholar]