

Abstract
Selective inhibition of the neuronal isoform of nitric oxide synthase (nNOS) over endothelial nitric oxide synthase (eNOS) and inducible nitric oxide synthase (iNOS) has become a promising strategy for the discovery of new therapeutic agents for neurodegenerative diseases. However, because of the high sequence homology of different isozymes in the substrate binding pocket, developing inhibitors with both potency and excellent isoform selectivity remains a challenging problem. Herein we report the evaluation of a recently discovered peripheral hydrophobic pocket (Tyr706, Leu337, and Met336) that opens up upon inhibitor binding and its potential in designing potent and selective nNOS inhibitors using three compounds, 2a, 2b, and 3. Crystal structure results show that inhibitors 2a and 3 adopted the same binding mode as lead compound 1. We also found that hydrophobic interactions between the 4-methyl group of the aminopyridine ring of these compounds with the side chain of Met336, as well as the π-π stacking interaction between the pyridinyl motif and the side chain of Tyr706 are important for the high potency and selectivity of these nNOS inhibitors.
Keywords: nitric oxide synthase, selective inhibitor, crystal structure, peripheral hydrophobic pocket, pi-stacking
Overproduction of the small molecule nitric oxide (NO) by the neuronal isoform of nitric oxide synthase (NOS) in the brain is closely associated with numerous neurodegenerative diseases, including chronic pathologies such as Parkinson’s,1 Alzheimer’s,2 Huntington’s,3 headaches,4 as well as neuronal damage in stroke.5 Inhibition of nNOS, therefore, has become a compelling strategy for the treatment of neurodegeneration.6-8 In the past two decades, a large number of nNOS inhibitors have been reported, including derivatives of L-arginines, guanidines, isothioureas, isothioureidos, 2-iminopiperidines, amidines, aminopyridines, thioureas, thiazoles, imidazoles, and indazoles.9 None of these inhibitors, however, has entered clinical trials due to drawbacks with respect to their potency and isozyme selectivity.9 These inhibitors, when binding to nNOS, target the L-arginine binding pocket where nNOS has high sequence homology with the other two isozymes, eNOS and iNOS.6 This explains why most inhibitors suffer from poor isoform selectivity. At present, discovery of nNOS inhibitors with high potency and selectivity still represents a major challenge.
To overcome this limitation, a number of selective nNOS inhibitors have emerged from structure-based design.10-12 One of the more important is pyrrolidine-based inhibitor 1, which exhibits excellent potency (Ki = 5 nM) and selectivity for rat nNOS over bovine eNOS (3,800-fold) and murine macrophage iNOS (1,200-fold).13-15 Interestingly, crystal structures of nNOS and eNOS in complex with 1 revealed that this inhibitor adopts an unexpected “flipped” binding mode, shown in Figure 1, in which the 2-amino-4-methylpyridine motif extended beyond the edge of the heme distal site, hydrogen bonded to the heme propionate of pyrrole ring D, and fits nicely into a hydrophobic pocket (Tyr706, Leu337, and Met336) formed by an alternate rotamer of the aromatic side chain of Tyr706.13 A close look at this pocket indicates two potential beneficial interactions: (1) the hydrophobic interaction between the 4-methyl group of 1 and the side chains of Leu337 and Met336, and (2) the π-π stacking interaction between the aminopyridine motif of 1 and the aromatic side chain of Tyr706. More importantly, the specific residues around this pocket are significantly different in the 3 mammalian NOS isoforms. For example, instead of the three amino acid residues, Tyr706, Leu337, and Met336 for rat nNOS, for human nNOS the corresponding residues are Tyr710, His341, and Met340, while for bovine eNOS and murine iNOS, these are Tyr477, Leu107, and Val106, and Tyr487, Asn115, and Met114, respectively.16 Therefore, we speculate that this hydrophobic pocket may provide a new “hot spot” for developing isoform-selective inhibitors for NOSs.
Figure 1.
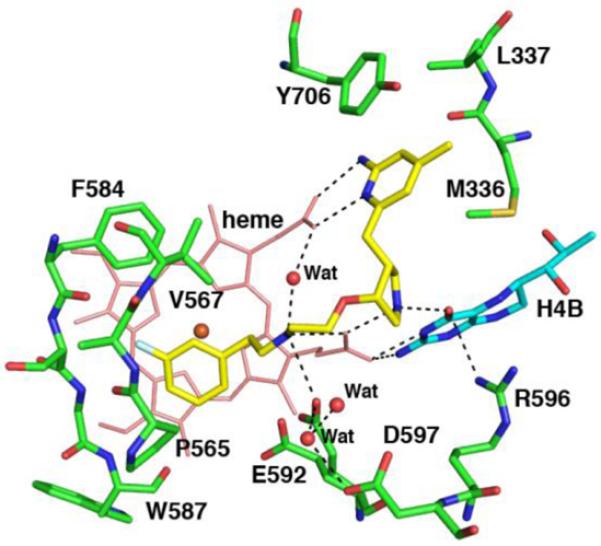
Binding of lead compound 1 to the rat nNOS active site. The major hydrogen bonds are drawn as dashed lines. The structural figures were prepared with PyMOL (www.pymol.org).
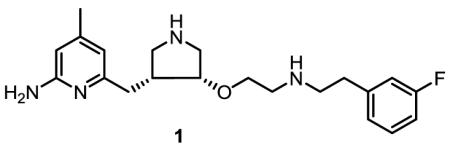
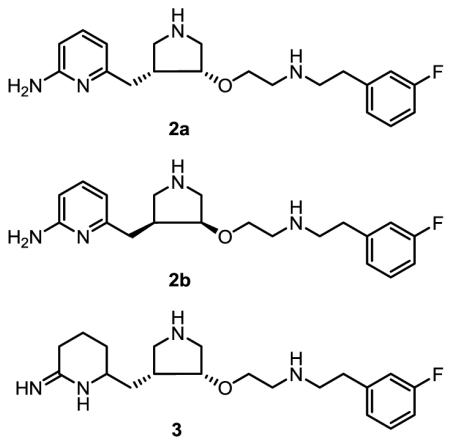
To further investigate the chemical environment of this peripheral site, and also to evaluate the possibility of utilizing it for the development of selective nNOS inhibitors, we report herein the synthesis and evaluation of three inhibitors, 2a, 2b, and 3. Compound 2a is an analog of 1, with the 4-methyl group removed from the 2-aminopyridine ring. The difference in potency between 1 and 2a for nNOS can demonstrate the importance of the methyl group to inhibitory activity. Compound 2b, the enantiomer of 2a, has opposite chirality at the pyrrolidine core. Therefore, 2b cannot adopt the “flipped” binding mode as with 2a, and thus will provide a direct comparison between the two different binding modes. Inhibitor 3 removes the aromaticity of the aminopyridine from 2a. A comparison of 3 and 2a demonstrates the role of the π-π stacking interaction between the pyridine ring of 1 and the side chain of Tyr706.
The syntheses of key intermediates 4a and 4b are shown in Scheme 1, using a recently reported method.13-15 The amino group of 2-amino-4,6-dimethylpyridine (5) was protected using (Boc)2O in the presence of triethylamine (TEA) to provide 6 in high yields. Compound 6 was treated with two equivalents of n-BuLi, and the resulting dianion was allowed to react with the epoxide (7)13 to generate the desired regioisomer (8) in modest yields. Then, the free NH group on the pyridine ring was further protected by a benzyl-protecting group using benzyl bromide to yield 9 in good yields. Next, trans alcohol 9 underwent a Mitsunobu reaction using acetic acid as a nucleophile to produce 10 in very high yields. Hydrolysis of 10 yielded cis-alcohol 11 in quantitative yields. Finally, the racemic mixture of 11 was successfully separated using a two step procedure. First, 11 was treated with (1S)-(−)-camphanic chloride in the presence of TEA and N,N-dimethylaminopyridine (DMAP) to yield two diastereomers (12a and 12b), which were successfully separated using silica chromatography. Then, each ester was subjected to aqueous Na2CO3 to produce 4a and 4b as single enantiomers in excellent yields.
Scheme 1.

Synthesis of 4a and 4b.
a Reagents and conditions: (a) (Boc)2O, TEA, t-BuOH, 50 °C, 24 h, 90%; (b) (i) n-BuLi (2 equiv.), −78 °C to r.t., 30 min, (ii) 7, −78 °C to r.t., 3 h, 55%; (c) NaH, 0 °C to r.t., 10 min, benzyl bromide, r.t., 8 h, 92%; (d) PPh3, DIAD, acetic acid, r.t., 12 h, 95%; (e) NaOH (2N)/MeOH, 50 °C, 4 h, 90%; (f) (1S)-(−)-camphanic chloride, TEA, DMAP, r.t. 4 h, 49% for each diastereomer; (g) Na2CO3, H2O/MeOH, r.t., 3 h, 99%.
Next, allylation of 4a/b gave 13a/b in excellent yields (Scheme 2).13-15 Alkenes 13a/b were subjected to ozonolysis using Zn as the reducing reagent to provide aldehydes (14a/b), which underwent reductive aminations with 2-(3-fluorophenyl)ethanamine to provide secondary amines 15a/b. The secondary amino group was protected with a Boc-protecting group, and then the benzyl-protecting groups of 16a/b were removed by catalytic hydrogenation at 60 °C to give 17a/b. Finally, the three Boc-protecting groups were removed in a 2:1 mixture of 6 N HCl and MeOH to generate inhibitors 2a and 2b. Inhibitor 3 can be synthesized from 15a using high pressure catalytic hydrogenation conditions in very high yields.
Scheme 2.

Syntheses of 2a, 2b, and 3.
a Reagents and conditions: (a) NaH, allyl bromide, 0 °C to r.t. 30 min, 99%; (b) (i) O3, −78 °C, (ii) Zn, acetic acid, −78 °C to r.t., 70-75%; (c) 2-(3-fluorophenyl)ethanamine, NaHB(OAc)3, r.t., 3 h, 52-55%; (d) Pd(OH)2/C, H2, 2:1 EtOH/HCl (12 N), r.t., 575 psi, 40 h, 100%; (e) (Boc)2O, Et3N, MeOH, r.t., 0.5 h, 99%; (f) Pd(OH)2/C, H2, 60 °C, 24 h, 35-50%; (g) 6 N HCl/MeOH (2:1), r.t., 4 h, 80-85%.
In the crystal structure of the active site of rat nNOS, 2a adopts the same binding mode as lead compound 1 (Figure 2A). The aminopyridine motif extends to the same peripheral hydrophobic pocket containing Tyr706, Leu337, and Met336, forming a charge-charge interaction with the heme propionate D, as well as a π-π stacking interaction with the aromatic side chain of Tyr706. However, removal of the 4-methyl group from the 2-aminopyridine motif dramatically impaired the potency (7-fold) of the inhibitor (2a vs 1). This result highlights the crucial role of the 4-methyl group for retaining the high inhibitory activity of 1 for rat nNOS. Importantly, the selectivity of 2a for rat nNOS over bovine eNOS also dropped significantly (2.3-fold). This was mainly the result of the lower sensitivity of eNOS to the presence of the 4-methyl group, only a 3-fold difference when comparing 2a to 1. This methyl group might make less favorable contacts with the smaller side chain of Val106 in eNOS. Inhibitor 2b, the corresponding enantiomer of 2a, adopts the normal binding mode with its 2-aminopyridine hydrogen bonded to the side chain of Glu592 (Figure 2B), resulting in a 4-fold lower potency for rat nNOS (2b vs 2a). However, eNOS has no preference for the two binding modes with 2b and 2a showing comparable affinities. Inhibitor 3, with the 2-aminopyridine of 2a reduced to a cyclic amidine, showed diminished potency for rat nNOS (3 vs 2a). This result indicates that the π-π stacking interaction between the pyridine ring and Tyr706 is an important factor for tight binding of 2a to nNOS. The stacking interaction provides less contribution to the binding affinity of 2a to eNOS, as its Ki values are similar for both 3 and 2a. This is probably because the Tyr477 side chain in eNOS does not interact as closely with the 2-aminopyridine ring of the inhibitors, as demonstrated previously in crystal structures for other pyrrolidine inhibitors complexed to eNOS and nNOS.13
Figure 2.

Active site structures of rat nNOS in complex with 2a (A, PDB code 3NNY) and 2b (B, PDB code 3NNZ). Shown also the 2Fo–Fc electron density for inhibitor at 1σ contour level. The major hydrogen bonds are drawn as dashed lines.
We also tested the inhibitory activity of 1, 2a, 2b, and 3 against the human isoform of nNOS (Table 2). Human nNOS shows very high sequence homology to rat nNOS in the active site;16 the only different residue in the peripheral site is His341 in place of Leu337, which makes the hydrophobic pocket in human nNOS smaller than that in rat nNOS. As a result, the hydrophobic pocket of human nNOS should prefer smaller fragments. Even though inhibitors 1, 2a, 2b, and 3 are less potent with human nNOS compared to rat nNOS, the selectivity (Ki-human/Ki-rat) dropped 1.2-fold and 1.6-fold for 2a and 3, respectively, compared to 1. These results indicate that the relatively smaller hydrophobic pocket in human nNOS prefers to bind the non-substituted aminopyridine fragment.
Table 2. Ki values of 1, 2a, 2b, and 3 for rat nNOS and human nNOS.
| Kia(nM) | selectivity | ||
|---|---|---|---|
| inhibitor | |||
| rat nNOS | human nNOS | rat/human | |
| 1 | 7 | 50 | 7.1 |
| 2a | 50 | 290 | 5.8 |
| 2b | 220 | 1,800 | 8.2 |
| 3 | 110 | 490 | 4.5 |
The Ki values represent at least duplicate measurements with standard deviations of ±10%.
In summary, we demonstrate the feasibility and potential of using the peripheral site (Tyr706, Leu337, and Met336 for rat nNOS) as a “hot spot” for designing potent and selective nNOS inhibitors. Three new inhibitors, 2a, 2b, and 3, have been synthesized. The crystal structures show that 2a shares the same flipped binding mode as lead compound 1, while 2b binds in a normal mode as expected by their stereochemistry of the pyrrolidine ring. The inhibitory activities were tested against various NOS isoforms. Our results highlight the importance of the hydrophobic interaction between the 4-methyl group of the 2-aminopyridine motif and the side chain of Met336 for tight binding and high isoform-selectivity of the inhibitors. We also show that the π-π stacking interaction between the aminopyridine ring and the aromatic side chain of Tyr706 is important for good potency. The insensitivity of eNOS to the binding mode of pyrrolidine-based inhibitors and the weaker π-π stacking interaction from its Tyr477 to the 2-aminopyridine ring of the inhibitors are isoform specific properties valuable to future inhibitor design.
Supplementary Material
Table 1. Ki values of 1, 2a, 2b, and 3 for rat nNOS, bovine eNOS and murine iNOS.
| Kia(nM) | selectivity | ||||
|---|---|---|---|---|---|
| inhibitor | |||||
| rat nNOS | eNOS | iNOS | n/e | n/i | |
| 1 | 7 | 19,000 | 5,800 | 2,700 | 830 |
| 2a | 50 | 58,000 | 10,000 | 1,200 | 200 |
| 2b | 220 | 59,000 | 13,200 | 270 | 60 |
| 3 | 110 | 58,000 | 21,800 | 530 | 200 |
The Ki values represent at least duplicate measurements with standard deviations of ±10%
Acknowledgments
The authors are grateful to the National Institutes of Health for financial support to R.B.S. (GM49725), T.L.P (GM57353), and Dr. Bettie Sue Masters (GM52419, with whose laboratory P.M. and L.J.R. are affiliated). B.S.S.M. is the Robert A. Welch Distinguished Professor in Chemistry (AQ0012). P.M. is supported by grants 0021620806 and 1M0520 from MSMT of the Czech Republic.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zhang L, Dawson VL, Dawson TM. Pharmacol. Ther. 2006;109:33. doi: 10.1016/j.pharmthera.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 2.Dorheim M-A, Tracey WR, Pollock JS, Grammas P. Biochem. Biophys. Res. Commun. 1994;205:659. doi: 10.1006/bbrc.1994.2716. [DOI] [PubMed] [Google Scholar]
- 3.Norris PJ, Waldvogel HJ, Faull RLM, Love DR, Emson PC. Neuronsicence. 1996;72:1037. doi: 10.1016/0306-4522(95)00596-x. [DOI] [PubMed] [Google Scholar]
- 4.Ashina M. Exp. Opin. Pharmacother. 2002;3:395. doi: 10.1517/14656566.3.4.395. [DOI] [PubMed] [Google Scholar]
- 5.Sims NR, Anderson MF. Neurochem. Int. 2002;40:511. doi: 10.1016/s0197-0186(01)00122-x. [DOI] [PubMed] [Google Scholar]
- 6.Alderton WK, Cooper CE, Knowles RG. Biochem. J. 2001;357:593. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Southan GJ, Szabo C. Biochem. Pharmacol. 1996;51:383. doi: 10.1016/0006-2952(95)02099-3. [DOI] [PubMed] [Google Scholar]
- 8.Babu BR, Griffith OW. Curr. Opin. Chem. Biol. 1998;2:491. doi: 10.1016/s1367-5931(98)80125-7. [DOI] [PubMed] [Google Scholar]
- 9.Ji H, Erdal EP, Litzinger EA, Seo J, Zhu Y, Xue F, Fang J, Huang J, Silverman RB. In: Frontiers in Medicinal Chemistry. 4. Reitz AB, Choudhary MI, Atta-ur-Rahman, editors. Vol. 5. Bentham Science Publishers; 2009. p. 842. [Google Scholar]
- 10.Silverman RB. Acc. Chem. Res. 2009;42:439. doi: 10.1021/ar800201v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fedorov R, Vasan R, Ghosh DK, Schlichting I. Proc. Natl. Acad. Sci. U.S.A. 2004;101:5892. doi: 10.1073/pnas.0306588101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garcin ED, Arvai AS, Rosenfeld RJ, Kroeger MD, Crane BR, Andersson G, Andrews G, Hamley PJ, Mallinder PR, Nicholls DJ, St-Gallay SA, Tinker AC, Gensmantel NP, Mete A, Cheshire DR, Connolly S, Stuehr DJ, Aberg A, Wallace AV, Tainer JA, Getzoff ED. Nature Chem. Biol. 2008;4:700. doi: 10.1038/nchembio.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delker DL, Ji H, Li H, Jamal J, Fang J, Xue F, Silverman RB, Poulos TL. J. Am. Chem. Soc. 2010;132:5437. doi: 10.1021/ja910228a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lawton GR, Ranaivo HR, Wing LK, Ji H, Xue F, Martesek P, Roman LJ, Watterson DM. Bioorg. Med. Chem. 2009;17:2371. doi: 10.1016/j.bmc.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xue F, Fang J, Lewis WW, Martasek P, Roman LJ, Silverman RB. Bioorg. Med. Chem. Lett. 2010;20:554. doi: 10.1016/j.bmcl.2009.11.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ji H, Li H, Flinspach M, Poulos TL, Silverman RB. J. Med. Chem. 2003;46:5700. doi: 10.1021/jm030301u. [DOI] [PubMed] [Google Scholar]
- 17.Hevel JM, Marletta MA. Method Enzymol. 1994;233:250. doi: 10.1016/s0076-6879(94)33028-x. [DOI] [PubMed] [Google Scholar]
- 18.Li H, Shimizu H, Flinspach M, Jamal J, Yang W, Xian M, Cai T, Wen EZ, Jia Q, Wang PG, Poulos TL. Biochemistry. 2002;41:13868. doi: 10.1021/bi020417c. [DOI] [PubMed] [Google Scholar]
- 19.McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, Soltis SM, Kuhn P. J Synchrotron Radiat. 2002;9:401. doi: 10.1107/s0909049502015170. [DOI] [PubMed] [Google Scholar]
- 20.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 21.Murshudov GN, Vagin AA, Dodson EJ. Acta Cryst. 1997;D53:240. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 22.Jones TA, Zou J-Y, Cowan SW, Kjeldgaarrd M. Acta Cryst. 1991;A47:110. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 23.Emsley P, Cowtan K. Acta Cryst. 2004;D60:2126. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 24.Winn MD, Isupov MN, Murshudov GN. Acta Cryst. 2001;D57:122. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.