

Abstract
Allergen-induced respiratory inflammation facilitates and/or elicits the extravasation of proinflammatory leukocytes by well understood mechanisms that mediate the movement of multiple cell types. The non-specific character of these pathways led us to hypothesize that circulating cancer cells use similar mechanisms, promoting secondary tumor formation at distal sites. To test this hypothesis, the frequency of metastasis to the lung as a function of allergic pulmonary inflammation was assessed following the intravenous injection of B16-F10 melanoma cells in mice. These studies demonstrated that allergen-induced pulmonary inflammation resulted in a >3-fold increase in lung metastases. This increase was dependent on CD4+ T cell activities; however, it occurred independent of the induced eosinophilia associated with allergen provocation. Interventional strategies showed that existing therapeutic modalities for asthma, such as inhaled corticosteroids, were sufficient to block the enhanced pulmonary recruitment of cancer cells from circulation. Additional mechanistic studies further suggested that the ability of circulating cancer cells to extravasate to surrounding lung tissues was linked to the activation of the vascular endothelium via one or more Gαi-coupled receptors. Interestingly, a survey of a clinical breast cancer surgical database showed that the incidence of asthma was higher among patients with lung metastases. Thus, our data demonstrate that allergic respiratory inflammation may represent a risk factor for the development of lung metastases and suggests that amelioration of the pulmonary inflammation associated with asthma will have a direct and immediate benefit to the 7–8% of breast cancer patients with this lung disease.
Keywords: Metastasis, Asthma, Endothelial Cell, Diapedesis, Breast Cancer
INTRODUCTION
Disseminated metastases remains the main cause of malignancy-related death in cancer patients (1) and as such are the focus of intensive investigations. Nonetheless, despite these efforts the mechanisms that facilitate metastasis remain the subject of debate (2, 3). It appears likely that cells shed from primary tumors enter the peripheral and/or lymphatic circulation and subsequent cell-cell interactions with the vascular endothelium lead to adhesion and extravasation from circulation (see for example (4, 5)). In many respects, hypotheses describing these processes share a great deal of similarity with the well characterized recruitment of leukocytes in response to inflammation (i.e., the rolling of circulating leukocytes, tethering of these cells, and their firm adherence to activated vascular endothelium (reviewed in (6)). Moreover, adherent leukocytes subsequently elicit signaling events within the vascular endothelial cells that are necessary for the enhanced permeability of the endothelium and diapedesis of these leukocytes into adjacent tissues (7, 8). Though it is now well accepted that the tumor microenvironment is an important component in the neoplastic process (9), the specific mechanisms mediating seeding and development of metastases remain unresolved. Inflammation and tumor onset/growth have been linked in various animal models of cancer and human neoplastic conditions (10, 11). Interestingly, several studies have also demonstrated that sites of chronic inflammation are often associated with the establishment and growth of malignancies (e.g., mesothelioma, melanoma, oral squamous cell carcinoma); these observations suggest that inflammation may be supportive of tumor onset/growth (12, 13). Similar to leukocyte recruitment, circulating cancer cells also appear to respond to the same rich milieu of localized chemokines and cell adhesion molecules engaged during inflammatory processes (see for example (14)). Moreover, pro-inflammatory leukocytes themselves have also been shown to be a significant source of these same inflammatory mediators, suggesting that the presence of these leukocytes alone may facilitate tumor cell extravasation (14, 15). Thus, while localized tissue inflammation is necessary for the appropriate recruitment of pro-inflammatory leukocytes, this response itself may also create a favorable environment facilitating the recruitment and growth of circulating tumor cells.
Our recent studies utilizing mouse models of pulmonary inflammation mediated by either endotoxin or allergen have demonstrated the importance of signaling events occurring within endothelial cells for leukocyte extravasation (16). In addition, other studies suggest that changes in vascular permeability, cell adhesion, and the creation of a tumor-favorable microenvironment are all likely events contributing to metastasis (12, 17). Collectively, these observations suggest a link between inflammation and metastasis highlighted by common mechanisms by which both leukocytes and circulating cancer cells are recruited to tissue compartments. That is, the cadre of inflammatory mediators (i.e., cytokines, growth factors, adhesion receptor-ligand interactions, and molecular/cellular signaling events) that lead to directed leukocyte recruitment may, in turn, be exploited by circulating cancer cells to facilitate distant organ metastasis and the spread of cancer.
This study utilizes mouse models of allergic pulmonary inflammation and a well established model of metastasis in mice to show that allergic respiratory inflammation not only promotes pro-inflammatory leukocyte recruitment to the lung but also facilitates the recruitment of circulating cancer cells leading to enhanced metastasis. Significantly, the activation of the pulmonary vascular endothelium and the concomitant extravasation of cancer cells were shown to be required events for the increased rate of metastasis observed in mice following allergen provocation. More importantly, this relationship between allergic respiratory inflammation and elevated levels of metastasis may also exist in humans. In particular, the frequency of asthma appears to be higher than expected among breast cancer patients who have experienced a recurrence of their malignancy in the lung, suggesting that treatments targeting lung inflammation may have value as therapeutic strategies suppressing/preventing metastasis.
MATERIALS AND METHODS
Animals
C57BL/6J mice were either purchased from The Jackson Laboratory (Bar Harbor, ME) or bred within the Mayo Clinic Arizona animal facility. Transgenic mice devoid of eosinophils (line: PHIL (backcrossed on a C57BL/6J background >10 generations) were developed in our laboratory as previously described (18). Gαi2−/− mice were generated as previously described (8) and backcrossed >10 generations onto the 129/SvJ background. All procedures were conducted on female mice 8–16 weeks of age. Protocols and studies involving animals were conducted in accordance with National Institutes of Health and Mayo Clinic institutional guidelines.
Induction of allergic pulmonary inflammation
Allergic pulmonary inflammation was induced in mice using an established ovalbumin (OVA) sensitization/aerosol challenge model of asthma as described earlier (19). Briefly, mice (20–30 grams) were sensitized by intraperitoneal (i.p.) injections (100μl) of 20μg chicken ovalbumin (OVA; Sigma, St. Louis, MO) emulsified in 2 mg of Imject® Alum (Al(OH)3/Mg(OH)2; Pierce, Rockfield, IL) on days −29 and −15 of a protocol in which the day of cancer cell administration is day 0 (Figure 1). Sensitized mice were challenged with an aerosol, derived by nebulizing a 1% OVA solution in saline (OVA), on protocol days −5, −4, and −3 and then provoked with a 5% OVA nebulant on protocol days −1, +4, and +9; control animals (OVA-sensitized) were challenged/provoked with saline alone (SAL).
Figure 1. Schematic time-line representing the merger of an established model of allergic respiratory inflammation and the metastasis of circulating cancer cells to the lung.

Experimental animals were sensitized to ovalbumin (OVA) by i.p. injections on days −29 and −15 of this protocol followed by a series of aerosol challenges (protocol days −5, −4, and −3) derived from a 1% OVA solution in saline (control animals received saline alone) focusing allergic immune responses to the lung. These inflammatory responses in the lung were maintained throughout the remaining 12 days of the protocol by provoking the mice on days 0, +4, and +9 with a nebulant derived from a 5% OVA solution in saline (again control animals received saline alone). During the aerosol provocation phase of this protocol, the mice received an intravenous (i.v.) injection of B16-F10 melanoma cells (2 ×105) on day 0 of the time-line and lung metastases were assessed in these animals twelve days later (tumor harvest). Variations of this protocol representing the described experiments of this report are shown below the main time-line. Depletion of CD4+ T cells (T cell depletion) was achieved via i.p. administration of the depleting monoclonal antibody GK1.5 on protocol days −7, −1, and +5. Additional groups of mice were treated with the corticosteroid budesonide through intranasal (i.n.) challenges of mice (0.5mg/kg of body weight) during the OVA challenge and provocation phases of the protocol (days −6, −5, −4, −3, −1, +4, and +9).
Generation of lung metastasis using B16F10 melanoma and MC38 colon carcinoma cell lines
B16-F10 melanoma cells (ATCC, Manassas, VA) and MC38 colon sarcoma (a kind gift from Dr. Lotze (University of Pittsburgh, Pittsburgh, PA)), both of which are derived from C57BL/6J mice, were cultured (humidified atmosphere containing 5% CO2 at 37°C) in DMEM medium, supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 1% Penicillin/streptomycin. Trypsinized cells were recovered and washed with 1x PBS prior to inoculation into recipient animals as a cell suspension in 1x PBS pH 7.4. All tissue culture reagents were purchased from Invitrogen (Carlsbad, CA).
C57BL/6J wild type, PHIL, or Gαi2−/− mice undergoing the OVA sensitization/aerosol challenge protocol outlined above were injected (400μl total volume) with 2 × 105 B16-F10 melanoma cells (in some studies MC38 colon sarcoma cells were used) into a lateral tail vein (protocol day 0). Injected animals were euthanized on day 12 for assessment of the number of lung metastases. Specifically, harvested lungs were inflated with a fixed volume (1ml) of 10% formalin and the numbers of metastatic colonies on the surface were assessed using a low power microscope. All evaluations of metastases were performed in duplicate as independent observer-blinded assessments.
Budesonide treatment
Corticosteriod (i.e., budesonide) treatment of mice was performed as previously described (20). Briefly, 0.5mg/kg body weight of budesonide (Sigma, St. Louis, MO) in 0.9% saline with 1% carboxymethylcellulose (Sigma, St. Louis, MO) and 0.1% polyoxyethylenesorbitan monooleate (Tween 80 (Sigma, St. Louis, MO)) was administered by i.p. injection on protocol days −5, −4, −3, −1, +4, and +9 one hour prior to the aerosol OVA challenges on those days (Figure 1).
Antibody mediated depletion of CD4+ T cells
The anti-CD4 monoclonal antibody (mAb) GK1.5 was used to deplete CD4+ T cells using a modification of a previously described protocol (21). Specifically, CD4+ T cells were depleted from mice by administration (i.p.) of GK1.5 mAb to mice (0.5mg/100μl) on protocol days −8, −1, and 6 in which the day of cancer cell administration is day 0 (Figure 1); control groups of mice were administered nonspecific rat IgG.
Histology and immunohistochemical staining
Lung tissue for histological analyses was obtained by instilling 1 ml of 10% neutral-buffered formalin (30 cm H2O constant pressure) through a cannula inserted into the trachea. The excised lung was immersed in 10% formalin for 24 hours (at 4°C) and then paraffin-embedded. Four (4) μm sagittal sections were stained with hematoxylin and eosin and analyzed by bright field microscopy (n = ≥ 5 animals/group). Eosinophil recruitment to lung tissue was determined by immunohistochemistry using a rabbit polyclonal anti-mouse major basic protein (MBP) antisera (22).
Ex vivo assessments of melanoma cell adhesion, and migration
Direct measures of cell adhesion between B16-F10 melanoma cells and an endothelial cell monolayer (i.e., mHEVa endothelial cells) were assessed in static cultures using a “cell association assay” (23). Rat anti-mouse VCAM-1 (clone MVCAM.A, BD PharMingen (San Diego, CA)) and anti-α4 integrin (clone PS/2, BioDesign International (Kennebunk, ME)) were used as blocking agents in the “cell association” assays to demonstrate the requirement of these cell adhesion molecules for B16-F10 melanoma cell transendothelial cell migration. Endothelial cell viability after pertussis toxin (Sigma, St. Louis, MO) treatment was determined either by assessment of GADPH release into the medium using the Molecular Probes Vybrant Cytotoxicity Assay (V23111; Carlsbad, CA) or by microscopic assessment of cells following disassociation with 0.3% EDTA using trypan blue exclusion.
A parallel plate flow chamber was used to examine migration under conditions of laminar flow (24). B16-F10 melanoma cell migration across an endothelial cell monolayer (i.e., mHEVa cells) was demonstrated earlier to occur as a consequence of endothelial cell constitutive production of the chemokines/growth factors (see for example (25)). The mHEVa cells have been spontaneously immortalized but not transformed (26). mHEVa cell monolayers were grown to 95% confluence on slides and treated overnight with media only or media containing pertussis toxin (endothelial cells grew to confluence in all groups as observed by microscopy) prior to inserting the slide in the parallel plate flow chamber and performing migration assays (16, 24).
Asthma occurrence among patients in the Mayo Clinic Surgical Breast Cancer Database
The Mayo Clinic Surgical Breast Cancer Database contains information on all patients at Mayo Clinic Rochester who had a primary diagnosis of breast cancer and have undergone surgery beginning in 1988. Patients diagnosed with ductal carcinoma in situ (DCIS) or Stage I – Stage III breast cancer were identified through the Mayo Clinic Surgical Index and reviewed by a trained medical abstractor. Information abstracted included date of breast cancer surgery, clinical characteristics, first course of treatment, pathology, and recurrence data. Recurrence data were updated by patient visit information as well as linkage to the Mayo Clinic Tumor Registry that is maintained for reporting to the statewide tumor registry and the American College of Surgeons. Through August, 2006, there were 769 recurrences identified in this database, of which 571 were distant metastases. The cohort of patients used in our current study were the 176 patients with distant metastases that were to the lung. A medical abstractor screened all charts for physician or self-reported diagnosis of asthma. An experienced pulmonologist next reviewed all charts with a diagnosis of asthma for corticosteroid use and to obtain the age of asthma onset, or if not available, time of asthma diagnosis relative to breast cancer diagnosis and lung metastases. Protocols and studies involving human subject information were conducted in accordance with National Institutes of Health and Mayo Clinic institutional guidelines.
Statistical analysis
Data were analyzed and graphed using GraphPad Prism statistics program (GraphPad Prism Software, San Diego, CA). Results are presented as means ± SE. Statistical analysis was performed using t-tests with differences between means considered significant when p<0.05.
RESULTS
Allergic respiratory inflammation in mice elicited a significant increase in the number of metastases to the lung
The similarities between pro-inflammatory cell recruitment to the lung following allergen provocation and mechanisms by which circulating cancer cells seed outlying tissue compartments prompted us to examine if allergic respiratory inflammation facilitated cancer metastasis to the lung. This potential relationship was assessed in mice by overlaying an established model of asthma and an established model of lung metastasis. The details of this protocol are presented in Figure 1. Specifically, we adopted and extended an established model of acute allergen exposure to include the time period necessary for the development of lung metastases using a melanoma cell injection model system (19). An established model of lung metastasis was overlaid onto this model of allergic respiratory inflammation through the intravenous (i.v.) injection of 2 × 105 B16-F10 melanoma cells (protocol day 0) and the subsequent development of metastases to the lung (protocol days 0 through +12).
The induced pulmonary inflammation associated with both asthma and mouse models of allergic respiratory inflammation is characterized by the appearance of perivascular/peribronchial leukocyte infiltrates dominated by eosinophils ((27, 28)). Immunohistochemistry of lung sections (Figure 2(A)) using a rabbit polyclonal antibody targeting the eosinophil-specific granule protein eosinophil major basic protein (MBP (22)), provides a representative example of the pulmonary tissue eosinophilia induced in OVA sensitized/aerosol challenged mice (i.e., relative to saline-treated control animals). Significantly, this allergen-mediated inflammation is also accompanied by a corresponding increase in the number of lung metastases following i.v. administration of melanoma cells (Figure 3(A)). Quantitative assessments of the lungs of these mice show that the induction of allergic pulmonary inflammation led to a >3-fold increase in lung metastases relative to non-allergic saline control animals (Figure 3(B)). This increase in lung metastasis was not a function of the cell line employed in these studies. That is, in addition to B16-F10 melanoma cells, our studies showed that i.v. injection of other unrelated tumor cell lines (e.g., a colon sarcoma cell line (MC38)) also led to an increase in lung metastases among allergen sensitized/aerosol challenged mice relative to control animals (data not shown).
Figure 2. Allergen sensitization/aerosol challenge promotes a CD4+ T cell dependent recruitment of eosinophils to the lungs of mice.

(A) Lung sections from wild type saline control (WT Saline) and ovalbumin-treated (WT OVA) mice were stained with hematoxylin-eosin (left panels) for generalized histopathological assessments. Immunohistochemistry using a rabbit polyclonal antibody specific for mouse MBP demonstrated the spatial localization of the pulmonary eosinophils (greyish/black staining cells) uniquely occurring in OVA sensitized/aerosol challenged wild type mice. (B) The tissue eosinophilia associated with OVA sensitization/aerosol challenge is eliminated in mice depleted of CD4+ T cells using the GK1.5 monoclonal antibody (GK1.5 OVA) and is absent in mice congenitally devoid of eosinophils (PHIL OVA). Scale bars = 100μm.
Figure 3. T cell mediated allergic respiratory inflammation is associated with an increase in the number of lung metastases following i.v. administration of B16-F10 melanoma cells.

The increase in lung metastasis was associated with a CD4+ T cell dependent allergic inflammation but occurred independent of the allergen-induced accumulation of pulmonary eosinophils. (A) Representative photographs of lungs ressected from mice following the protocol outlined in Figure 1 showed that allergen-treated mice (wild type, OVA) experience a significant elevation in the number of metastases relative to control mice (wild type, Saline). These data also show that the allergen-induced increase in the number of metastases was lost in mice depleted of CD4+ T cells (OVA + GK1.5) but nonetheless occurred in mice devoid of eosinophils (OVA + PHIL). (B) Lung metastases were assessed by gross morphological determinations using a low power (4-10x) stereo-dissecting microscope. A quantitative summary of the mean number of lung metastases (n = 7–16 mice per group). *P<0.05.
The increase in lung metastasis associated with allergic pulmonary inflammation required CD4+ T cell responses but occurred independent of accumulating eosinophils
Previous studies have demonstrated that allergen-induced pulmonary pathologies (including the recruitment/accumulation of pulmonary eosinophils) are mediated by CD4+ T cell activities (29). We investigated whether the allergen-induced increase in lung metastasis was similarly dependent on CD4+ T cell activities through the administration of a cell-depleting antibody (GK1.5 (21)) using an administration protocol and time-line described in the Materials and Methods and Figure 1, respectively. Flow cytometric assessment of splenocytes derived from mice at the time of tumor harvest (protocol day +12) demonstrated that this cell depletion strategy is both effective (i.e., no CD4+ T cells are detectable in GK1.5-treated mice) and specific (i.e., no effects were observed on any other T cell population). Immunohistochemical assessment of lung sections using anti-MBP antibodies demonstrated the absolute dependence of the allergen-induced eosinophilia on CD4+ T cells with the complete absence of these granulocytes in GK1.5-treated mice (Figure 2(B)). This phenomenon also extended to circulating cancer cells as the depletion of CD4+ T cells resulted in a concomitant decrease in the number of lung metastases following administration of melanoma cells (Figure 3(A)). That is, similar to the complete abrogation of allergen-induced pulmonary pathologies occurring in GK1.5-treated mice, the loss of CD4+ T cells reduced the number of lung metastases in OVA-treated mice to levels observed in allergen-naive control animals (Figure 3(B)).
In contrast to the apparent requirement of CD4+ T cell activities, the singular absence of eosinophils had no effect on the increased metastasis observed in OVA-treated mice. Specifically, these studies exploited the availability of our transgenic mice (line: PHIL) that are congenitally devoid of eosinophils (18). PHIL mice subjected to the OVA provocation/metastasis protocol outlined in Figure 1 failed to recruit any eosinophils following OVA challenges/provocations (Figure 2(B)). However, unlike the depletion of CD4+ T cells, the loss of OVA-mediated recruitment/accumulation of pulmonary eosinophils in PHIL mice did not prevent an increase in lung metastases following administration of melanoma cells (Figure 3(A)). Significantly, the lack of an effect on lung metastasis in OVA-treated PHIL mice was unambiguous as no quantitative difference was observed in the number of lung metastases relative to OVA-treated wild type control mice (Figure 3(B)).
Attenuation of allergic pulmonary pathologies by administration of corticosteroid blocks the induced increase in the number of lung metastases
Administration of inhaled corticosteroids, a therapeutic regimen commonly used to curtail the pulmonary inflammation occurring in asthma patients, was assessed in our mouse model of asthma/metastasis for its ability not only to block the allergen-induced inflammation but also for its ability to attenuate the observed increase in metastasis to the lung. In these studies cohorts of OVA-treated mice were administered the corticosteroid budesonide (control animals received PBS vehicle alone) during the OVA challenge/provocation phase of the protocol outlined in Figure 1. Administration of budesonide significantly decreased the level of lung histopathologies (e.g., extracellular matrix collagen deposition, airway goblet cell metaplasia and mucin accumulation) and airway Th2 (e.g., IL-4, -5, and -13) cytokine expression (20, 30), as well as the recruitment and tissue accumulation of both T cells (31) and pro-inflammatory eosinophils (Figure 4(A)). More importantly, administration of budesonide to OVA-treated wild type mice also resulted in a significant decrease in metastasis to the lung (Figure 4(B)). Interestingly, no difference was observed in the number of metastases occurring between saline control groups receiving budesonide or vehicle controls, suggesting that non-specific effects of steroid administration were not responsible for the observed decrease in metastasis.
Figure 4. Administration of budesonide prevented the OVA-induced pulmonary inflammation leading to eosinophil accumulation in the lungs and concomitantly abolished the increased number of metastases occurring in the lung.

(A) Lung sections from control OVA-treated mice receiving PBS (Vehicle) and OVA-treated mice administered i.p. with corticosteroid (budesonide). Immunohistochemistry identifying MBP+ eosinophils infiltrating the lung (greyish/black staining cells) demonstrates the effectiveness of budesonide treatment to block this inflammatory response. (B) A quantitative summary of the mean number of lung metastases (n = 8–10 mice per group) showed that budesonide treatment of mice effectively abolished the OVA-induced increase in metastasis occurring in OVA-treated wild type mice receiving vehicle. Interestingly, budesonide treatment only reduced the number of observable metastases to saline control levels, suggesting that corticosteroid treatment was not having a larger, more profound, effect on the level of metastasis occurring in these mice. Scale bar = 100μm. *P<0.05.
Blockade of endothelial cell activation prevents allergen-induced recruitment of both pro-inflammatory leukocytes and circulating cancer cells
The trafficking of leukocytes from the blood to sites of inflammation is the cumulative result of receptor-ligand mediated signaling events associated with the leukocytes themselves as well as with the underlying vascular endothelium. Our recently published studies have shown that receptors coupled to intracellular heterotrimeric G-proteins containing a Gαi2 subfamily member elicit signaling pathway(s) in the vascular endothelium that regulate(s) a critical step required for transmigration (i.e., diapedesis) of leukocytes through the endothelium and into surrounding lung tissues (16). Specifically, these data showed that the loss of a Gαi2 signaling event(s) had no effects on allergen-mediated Th2 immune responses. However, the loss of Gαi2 signaling in the vascular endothelium of the lung prevented the accumulation of pro-inflammatory leukocytes in both an allergic model of asthma and an endotoxin-mediated model of pulmonary inflammation. The generalized mechanisms described by these data also suggested that other invasive cells may utilize a similar trafficking pathway. We subjected Gαi2 knockout mice to the concurrent asthma and metastasis model to define the role of endothelial cell activation in the firm adhesion and diapedesis of circulating cancer cells. Significantly, the loss of these signaling events in knock out mice prevented the accumulation of eosinophils in the perivascular/peribronchial regions (Figure 5(A)). We have previously demonstrated that this blockade is at the level of transmigration across the vascular endothelium (16) and, significantly, our current studies show that metastasis formation in OVA-treated Gαi2 knockout mice displays a similar decrease relative to OVA-treated wild type animals (Figure 5(B)).
Figure 5. Allergen treatment of Gαi2−/− mice demonstrated that Gαi2 signaling was required for both the allergen-induced pulmonary inflammation leading to eosinophil accumulation and the increased number of pulmonary metastases.
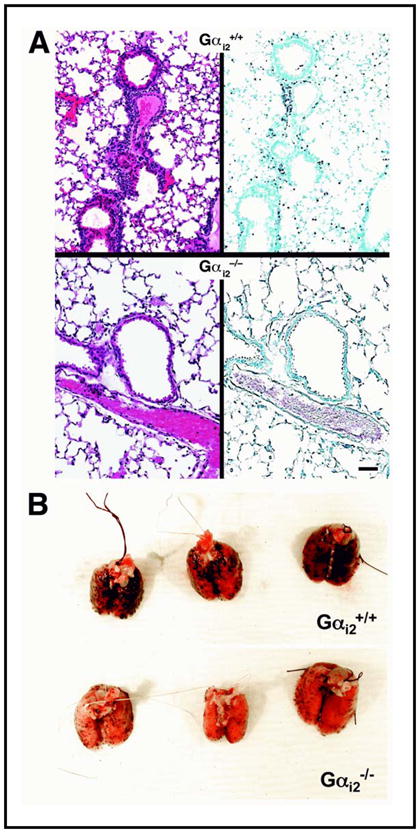
(A) In the absence of Gαi2 signaling, immunohistochemistry identifying MBP+ eosinophils infiltrating the lung (greyish/black staining cells) revealed that the loss of these signaling events effectively abolished the accumulation of eosinophils in the lungs of OVA-treated knockout mice (Gαi2−/−) relative to wild type animals (Gαi2+/+). (B) Representative photographs of lungs ressected from Gαi2+/+ vs. Gαi2−/− mice following the OVA/metastasis protocol showed that OVA-treatment of Gαi2−/− mice fails to elicit the significant elevation in the number of metastases observed in OVA-treated Gαi2+/+ animals. Scale bar = 100μm.
Ex vivo studies of adhesion and transmigration were performed to demonstrate and ultimately define a potential mechanism of tissue invasion by cancer cells. Specifically, ex vivo laminar flow assays (Figure 6) were used to assess VCAM-1/α4 integrin-dependent melanoma cell adherence and migration through an endothelial cell monolayer (32, 33). In these studies, Gαi-signaling events were abolished in the endothelial cell monolayer by pre-treatment with pertussis toxin (PTX (16)). This pre-treatment of the endothelial cells had no effect on cytotoxicity/viability or surface expression levels of the adhesion ligand VCAM-1 (16). The laminar flow study showed that α4-integrin/VCAM-1 dependent firm adhesion of melanoma cells was unaffected in the absence of endothelial cell Gαi-signaling (Figure 6(B)), suggesting that effects on adhesion mediated by endothelial Gαi2 signaling were not responsible for the blockade of metastasis in OVA-treated knockout mice. However, the subsequent migration of firmly adherent melanoma cells through the endothelial cell monolayer (i.e., endothelial transmigration) in the laminar flow assay showed that this process was a Gαi-dependent event and was thus significantly inhibited following PTX-treatment of the monolayer (Figure 6(C)). This result suggests that a signaling event(s), absent in the venule endothelium of Gαi2−/− mice, is required specifically for efficient transmigration/diapedesis.
Figure 6. Melanoma cell migration through an endothelial cell monolayer was dependent on one or more Gαi signaling events.

(A) Melanoma cell binding and transmigration was assessed using an ex vivo parallel plate flow chamber under conditions of laminar flow comparable to pressures observed in post-capillary venules (i.e., 2 dynes/cm2). (B) VCAM-1 dependent firm adhesion in a static cell association assay was blocked by antibodies against endothelial VCAM-1, but was unaffected by the blockade of Gαi signaling events using pertussis toxin (PTX) pre-treatment of the endothelial cell monolayer. *Significantly different (P< 0.05) from no-antibody control group. (C) Melanoma cell migration through an endothelial cell monolayer using a parallel plate flow chamber was blocked by pre-treatment of the endothelial cells with pertussis toxin (PTX). “No treatment”, endothelial cells treated with PBS. *Significantly different (P< 0.05) from PBS-treated control group.
The prevalence of asthma is disproportionately higher among human breast cancer patients with recurrence of the disease as pulmonary metastasis
In an attempt to provide initial insights regarding the translatability of our mouse model studies to human subjects, we examined patient records from a Mayo Clinic Surgical Breast Cancer Database (MCSBCD) assessing the frequency of asthma in patients whose disease recurrence was evidenced by pulmonary metastases. That is, we determined the prevalence of an asthma diagnosis among the 176 breast cancer patients in the MCSBCD with recurrence of disease as lung metastases. A review of these 176 breast cancer cases revealed that 30 patients had a diagnosis of asthma, although it could not be determined in one case when asthma was diagnosed. Thus, this survey of the MCSBCD revealed 29 patients with a diagnosis of asthma of which 23 were identified at least one year prior to the diagnosis of distant metastases. It is noteworthy that further examination of the patient medical records showed that none of these 23 patients had received oral and/or intravenous administration of corticosteroids. Indeed, these records demonstrated that only two of these patients had any indication of inhaled corticosteroid use. Remarkably, these 23 patients alone represent >13% of the breast cancer patients who developed distant metastases in the lung (23/176). That is, despite being a conservative assessment, this percentage is nearly twice the predicted frequency of asthma among a random population of women in the United States (i.e., 7–8% (Surveillance for Asthma, National Center for Health Statistics (NCHS), U.S. CDC, 2003), suggesting that the asthma of these cancer patients may have contributed to the appearance of lung metastases.
DISCUSSION
The lung is the second most common site for metastasis of various tumors, including breast, colorectal, and skin (34–36). The progression of disease by metastasis to this largely vascularized organ reduces the chance for long term survival of these patients and presents a significant clinical obstacle to their successful treatment. The present study identifies allergic pulmonary inflammation as a potential contributing factor in the process by which the lung is a selected target of circulating cancer cells. Our underlying hypothesis is that the non-specific character of mechanisms leading to pro-inflammatory cell recruitment to the lung following allergen provocation represent pathways that circulating cancer cells may exploit to extravasate from peripheral/lymphatic circulation. These pathways include common cell adhesion molecule interactions as well as the expression of inflammatory cytokines/chemokines leading to cancer cell recruitment, localized proliferation, and/or the increased survival of accumulating cells.
The parallels between allergen-induced pro-inflammatory cell recruitment to the lung and the selective recruitment of circulating cancer cells to this organ were explored through the unique strategy of combining established mouse models of asthma and lung metastasis. Collectively, these studies showed that allergen-induced pulmonary inflammation is linked to a significant increase in the appearance of lung metastases following i.v. administration of cancer cells. It is noteworthy that allergen-treatment of eosinophil-less PHIL mice showed that the recruitment of eosinophils to the lungs is not required for enhanced metastasis. Moreover, enhanced metastasis occurred in OVA-treated PHIL mice despite the fact that the loss of eosinophils resulted in a >50% reduction in allergen-induced end point histopathologies (18, 37). These results suggest that the enhanced metastasis was not a linear function of either leukocyte recruitment/accumulation or a downstream consequence of induced inflammation to structural components of the lung.
Similar to the ablation of CD4+ T cells, administration of an immunosuppressing corticosteroid (budesonide) to the lungs of allergen-treated mice abolished both the induced pulmonary pathologies (including lung-specific cytokine/chemokine expression and the allergen-induced eosinophil infiltrate (20)) and, more importantly, eliminated the increased lung metastasis associated with allergen provocation. Although budesonide was shown to modulate gene expression of lung tumors and, in turn, primary tumorigenesis in the lung (38), the lack of any effect on the number of lung metastases in baseline saline control animals with and without budesonide treatment suggests that the corticosteroid treatment itself was not a regulator of pulmonary metastasis. The importance of this observation has implications beyond studies of mouse models as inhaled corticosteroids represent the prominent therapeutic modality used in the treatment of the inflammation associated with asthma patients.
Our earlier studies of pro-inflammatory cell recruitment in mouse models of allergen challenge demonstrated that T cell mediated activation of the vascular endothelium via one or more Gαi2-coupled receptors, and not Gαi2-mediated elaboration of Th2 immune responses, was critical to the extravasation of pro-inflammatory leukocytes from circulation (16). The data presented in this report demonstrate that circulating cancer cells utilize this same mechanism as both allergen-induced eosinophil recruitment and enhanced lung metastasis were lost in OVA-treated Gαi2−/− mice. Moreover, our ex vivo studies using a parallel plate flow chamber system showed that whereas adhesion of melanoma cells to endothelial cells was not Gαi2-dependent, the ability of these cancer cells to invade and transmigrate through the endothelial cell monolayer was a Gαi-dependent phenomenon. In addition, our ex vivo adhesion data showed that adhesion of the melanoma cells to endothelial cells was nonetheless dependent on interactions with endothelial cell expressed VCAM-1 (39, 40). This observation is particularly important because allergen provocation of the lung is associated with a T cell dependent activation of the vascular endothelium, including the enhanced endothelial cell expression of VCAM-1 (41, 42). Thus, the increased metastasis observed in mice following allergen provocation may be a consequence of both a Gαi2-dependent ability to extravasate from circulation and the availability of increased VCAM-1:α4 integrin interactions. That is, cancer cell avidity to the vascular endothelium of the pulmonary bed is increased following allergen provocation and, together with immune responses that lead to endothelial cell activation, these events promote transmigration in the lung parenchyma.
The significance of these mouse model studies was greatly enhanced by our preliminary examination of breast cancer patients from a surgical database showing that the frequency of asthma among patients with reoccurrence of their disease as lung metastases is elevated relative to that expected from cross-sectional assessments of the general female population. Clearly, the provocative character of these observations will require additional and more detailed studies of the patients in this database. In particular, future studies are necessary to evaluate the importance of patient age and ethnicity, the time of asthma diagnosis as well as disease severity, and whether the apparent effects of allergic respiratory inflammation on pulmonary metastasis are lung specific versus a phenomenon affecting metastasis to other distal sites. Moreover, what effects on lung metastasis, if any, occur as a consequence of specific asthma therapies. For example, the medical records of the patients in our breast cancer database indicated that only two were receiving corticosteroids in any form, however, will larger studies reveal that guideline use of inhaled corticosteroids for asthma control reduces the occurrence of pulmonary metastases? These are but a few questions that will be answerable from continued studies attempting to link asthma and lung metastasis. The importance of establishing a link between increased metastasis to the lung among breast cancer patients who have asthma would be difficult to overestimate given that 7–8% of adult women in the United States are diagnosed with this respiratory disease and 1 in 8 women (by age 90) are diagnosed with breast cancer. Our demonstration of such a link has the potential to affect significantly the way cancer patients are treated. In particular, aggressive treatment of the underlying inflammation associated with asthma may have a considerable impact on the quality of life and/or survival of breast cancer patients with this respiratory disease.
Acknowledgments
We wish to acknowledge the invaluable contributions of Drs. Grzegorz Cieslewicz and Mark Inman as well as Katie O’Neill, Dana Colbert, and Ralph Pero. In addition, we wish to thank Linda Mardel and Shirley “Charlie” Kern for their excellent administrative support. The work presented was supported in part by the Mayo Foundation, the Intramural Research Program of the NIH (LB), and grants from the NIH to JJL (R01CA112442 and K26-RR019709), NAL (HL058723), and AGT (F32HL83718).
References
- 1.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–72. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 2.Mehlen P, Puisieux A. Metastasis: A question of life or death. Nat Rev Cancer. 2006;6:449–58. doi: 10.1038/nrc1886. [DOI] [PubMed] [Google Scholar]
- 3.Tse JC, Kalluri R. Mechanisms of metastasis: Epithelial-to-mesenchymal transition and contribution of tumor microenvironment. J Cell Biochem. 2007;101:816–29. doi: 10.1002/jcb.21215. [DOI] [PubMed] [Google Scholar]
- 4.Kobayashi H, Boelte KC, Lin PC. Endothelial cell adhesion molecules and cancer progression. Curr Med Chem. 2007;14:377–86. doi: 10.2174/092986707779941032. [DOI] [PubMed] [Google Scholar]
- 5.Langley RR, Fidler IJ. Tumor cell-organ microenvironment interactions in the pathogenesis of cancer metastasis. Endocr Rev. 2007 doi: 10.1210/er.2006-0027. [DOI] [PubMed] [Google Scholar]
- 6.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: The multistep paradigm. [review] Cell. 1994;76:301–14. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 7.Barreiro O, Yanez-Mo M, Sala-Valdes M, et al. Endothelial tetraspanin microdomains regulate leukocyte firm adhesion during extravasation. Blood. 2005;105:2852–61. doi: 10.1182/blood-2004-09-3606. [DOI] [PubMed] [Google Scholar]
- 8.van Buul JD, Anthony EC, Fernandez-Borja M, Burridge K, Hordijk PL. Proline-rich tyrosine kinase 2 (pyk2) mediates vascular endothelial-cadherin-based cell-cell adhesion by regulating {beta}-catenin tyrosine phosphorylation. J Biol Chem. 2005;280:21129–36. doi: 10.1074/jbc.M500898200. [DOI] [PubMed] [Google Scholar]
- 9.van Kempen LC, de Visser KE, Coussens LM. Inflammation, proteases and cancer. Eur J Cancer. 2006;42:728–34. doi: 10.1016/j.ejca.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 10.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krishnamoorthy S, Honn KV. Inflammation and disease progression. Cancer Metastasis Rev. 2006;25:481–91. doi: 10.1007/s10555-006-9016-0. [DOI] [PubMed] [Google Scholar]
- 12.de Visser KE, Coussens LM. The inflammatory tumor microenvironment and its impact on cancer development. Contrib Microbiol. 2006;13:118–37. doi: 10.1159/000092969. [DOI] [PubMed] [Google Scholar]
- 13.Vakkila J, Lotze MT. Inflammation and necrosis promote tumour growth. Nat Rev Immunol. 2004;4:641–8. doi: 10.1038/nri1415. [DOI] [PubMed] [Google Scholar]
- 14.Peng HH, Liang S, Henderson AJ, Dong C. Regulation of interleukin-8 expression in melanoma-stimulated neutrophil inflammatory response. Exp Cell Res. 2006 doi: 10.1016/j.yexcr.2006.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Slattery MJ, Liang S, Dong C. Distinct role of hydrodynamic shear in leukocyte-facilitated tumor cell extravasation. Am J Physiol Cell Physiol. 2005;288:C831–9. doi: 10.1152/ajpcell.00439.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pero RS, Borchers MT, Spicher K, et al. G alpha i2 mediated signaling events in the endothelium are involved in controlling leukocyte extravasation. Proc Natl Acad Sci U S A. 2007;104:4371–76. doi: 10.1073/pnas.0700185104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kopfstein L, Christofori G. Metastasis: Cell-autonomous mechanisms versus contributions by the tumor microenvironment. Cell Mol Life Sci. 2006;63:449–68. doi: 10.1007/s00018-005-5296-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee JJ, Dimina D, Macias MP, et al. Defining a link with asthma in mice congenitally deficient in eosinophils. Science. 2004;305:1773–6. doi: 10.1126/science.1099472. [DOI] [PubMed] [Google Scholar]
- 19.Cieslewicz G, Tomkinson A, Adler A, et al. The late, but not early, asthmatic response is dependent on il-5 and correlates with eosinophil infiltration. J Clin Invest. 1999;104:301–8. doi: 10.1172/JCI7010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen H, O’Byrne PM, Ellis R, Wattie J, Tang C, Inman MD. The effects of intranasal budesonide on allergen-induced production of interleukin-5 and eotaxin, airways, blood, and bone marrow eosinophilia, and eosinophil progenitor expansion in sensitized mice. Am J Respir Crit Care Med. 2002;166:146–53. doi: 10.1164/rccm.2008161. [DOI] [PubMed] [Google Scholar]
- 21.Gavett SH, Chen X, Finkelman F, Wills-Karp M. Depletion of murine cd4+ t lymphocytes prevents antigen-induced airway hyperreactivity and pulmonary eosinophilia. Am J Respir Cell Mol Biol. 1994;10:587–93. doi: 10.1165/ajrcmb.10.6.8003337. [DOI] [PubMed] [Google Scholar]
- 22.Denzler KL, Farmer SC, Crosby JR, et al. Eosinophil major basic protein-1 does not contribute to allergen-induced airway pathologies in mouse models of asthma. J Immunol. 2000;165:5509–17. doi: 10.4049/jimmunol.165.10.5509. [DOI] [PubMed] [Google Scholar]
- 23.Tudor KS, Deem TL, Cook-Mills JM. Novel alpha 4-integrin ligands on an endothelial cell line. Biochem Cell Biol. 2000;78:99–113. [PubMed] [Google Scholar]
- 24.Ager A, Mistry S. Interaction between lymphocytes and cultured high endothelial cells: An in vitro model of lymphocyte migration across high endothelial venule endothelium. Eur J Immunol. 1988;18:1265–74. doi: 10.1002/eji.1830180818. [DOI] [PubMed] [Google Scholar]
- 25.Matheny HE, Deem TL, Cook-Mills JM. Lymphocyte migration through monolayers of endothelial cell lines involves vcam-1 signaling via endothelial cell nadph oxidase. J Immunol. 2000;164:6550–9. doi: 10.4049/jimmunol.164.12.6550. [DOI] [PubMed] [Google Scholar]
- 26.Cook-Mills JM, Gallagher JS, Feldbush TL. Isolation and characterization of high endothelial cell lines derived from mouse lymph nodes. In Vitro Cell Dev Biol Anim. 1996;32:167–77. doi: 10.1007/BF02723682. [DOI] [PubMed] [Google Scholar]
- 27.Kips JC, Anderson GP, Fredberg JJ, et al. Murine models of asthma. Eur Respir J. 2003;22:374–82. doi: 10.1183/09031936.03.00026403. [DOI] [PubMed] [Google Scholar]
- 28.Hamid Q. Eosinophils in allergic inflammation. J Allergy Clin Immunol. 2004;113:182–4. doi: 10.1016/j.jaci.2003.10.058. [DOI] [PubMed] [Google Scholar]
- 29.Gonzalo JA, Lloyd CM, Kremer L, et al. Eosinophil recruitment to the lung in a murine model of allergic inflammation. The role of t cells, chemokines, and adhesion receptors. J Clin Invest. 1996;98:2332–45. doi: 10.1172/JCI119045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McMillan SJ, Xanthou G, Lloyd CM. Therapeutic administration of budesonide ameliorates allergen-induced airway remodelling. Clin Exp Allergy. 2005;35:388–96. doi: 10.1111/j.1365-2222.02193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loza MJ, Foster S, Peters SP, Penn RB. Interactive effects of steroids and beta-agonists on accumulation of type 2 t cells. J Allergy Clin Immunol. 2008;121:750, e1–5, e3. doi: 10.1016/j.jaci.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 32.Simiantonaki N, Jayasinghe C, Kirkpatrick CJ. Effect of pro-inflammatory stimuli on tumor cell-mediated induction of endothelial cell adhesion molecules in vitro. Exp Mol Pathol. 2002;73:46–53. doi: 10.1006/exmp.2002.2440. [DOI] [PubMed] [Google Scholar]
- 33.Okahara H, Yagita H, Miyake K, Okumura K. Involvement of very late activation antigen 4 (vla-4) and vascular cell adhesion molecule 1 (vcam-1) in tumor necrosis factor alpha enhancement of experimental metastasis. Cancer Res. 1994;54:3233–6. [PubMed] [Google Scholar]
- 34.Abrams HL, Spiro R, Goldstein N. Metastases in carcinoma; analysis of 1000 autopsied cases. Cancer. 1950;3:74–85. doi: 10.1002/1097-0142(1950)3:1<74::aid-cncr2820030111>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 35.Crow J, Slavin G, Kreel L. Pulmonary metastasis: A pathologic and radiologic study. Cancer. 1981;47:2595–602. doi: 10.1002/1097-0142(19810601)47:11<2595::aid-cncr2820471114>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 36.Weiss L. Comments on hematogenous metastatic patterns in humans as revealed by autopsy. Clin Exp Metastasis. 1992;10:191–9. doi: 10.1007/BF00132751. [DOI] [PubMed] [Google Scholar]
- 37.Jacobsen EA, Ochkur SI, Pero RS, et al. Allergic pulmonary inflammation in mice is dependent on eosinophil-induced recruitment of effector t cells. J Exp Med. 2008;205:699–710. doi: 10.1084/jem.20071840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yao R, Wang Y, Lemon WJ, Lubet RA, You M. Budesonide exerts its chemopreventive efficacy during mouse lung tumorigenesis by modulating gene expressions. Oncogene. 2004;23:7746–52. doi: 10.1038/sj.onc.1207985. [DOI] [PubMed] [Google Scholar]
- 39.Delneste Y, Jeannin P, Gosset P, et al. Allergen-stimulated t lymphocytes from allergic patients induce vascular cell adhesion molecule-1 (vcam-1) expression and il-6 production by endothelial cells. Clin Exp Immunol. 1995;101:164–71. doi: 10.1111/j.1365-2249.1995.tb02293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wardlaw AJ, Brightling C, Green R, Woltmann G, Pavord I. Eosinophils in asthma and other allergic diseases. Br Med Bull. 2000;56:985–1003. doi: 10.1258/0007142001903490. [DOI] [PubMed] [Google Scholar]
- 41.Elices MJ, Osborn L, Takada Y, et al. Vcam-1 on activated endothelium interacts with the leukocyte integrin vla-4 at a site distinct from the vla-4/fibronectin binding site. Cell. 1990;60:577–84. doi: 10.1016/0092-8674(90)90661-w. [DOI] [PubMed] [Google Scholar]
- 42.Nagata M, Yamamoto H, Tabe K, Sakamoto Y. Eosinophil transmigration across vcam-1-expressing endothelial cells is upregulated by antigen-stimulated mononuclear cells. Int Arch Allergy Immunol. 2001;125 (Suppl 1):7–11. doi: 10.1159/000053844. [DOI] [PubMed] [Google Scholar]