


Abstract
MicroRNAs (miRNAs) are ∼22-nt long, non-coding RNAs that regulate gene silencing. It is known that many human miRNAs are deregulated in numerous types of tumors. Here we report the sequencing of small RNAs (17–25 nt) from 23 breast, bladder, colon and lung tumor samples using high throughput sequencing. We identified 49 novel miRNA and miR-sized small RNAs. We further validated the expression of 10 novel small RNAs in 31 different types of blood, normal and tumor tissue samples using two independent platforms, namely microarray and RT–PCR. Some of the novel sequences show a large difference in expression between tumor and tumor-adjacent tissues, between different tumor stages, or between different tumor types. We also report the identification of novel small RNA classes in human: highly expressed small RNA derived from Y-RNA and endogenous siRNA. Finally, we identified dozens of new miRNA sequence variants that demonstrate the existence of miRNA-related SNP or post-transcriptional modifications. Our work extends the current knowledge of the tumor small RNA transcriptome and provides novel candidates for molecular biomarkers and drug targets.
INTRODUCTION
MicroRNAs (miRNAs) are ∼22-nt long non-coding RNA species that negatively regulate gene expression. In recent years the role miRNAs play in cancer has been extensively explored and these non-coding genes were implicated in numerous types of cancer as either oncogenes or tumor-suppressor genes (1). miRNAs are already used as diagnostic biomarkers in clinical assays designed for several types of cancer, such as lung cancer and cancer of unknown primary (2,3).
Next generation sequencing methods, also known as ‘deep sequencing’, have been widely used in recent years. These high throughput and highly sensitive sequencing methods include Roche Applied Sciences (454) GS, Illumina’s Solexa 1G sequencer, and Applied Biosystem’s SOLiD system. Deep sequencing can be used for the discovery of novel miRNA species and other small RNAs that are missed by traditional sequencing of small RNA libraries. Human miRNAs were previously identified using deep sequencing (4–8). However, the miRNA content of solid human tumors has only been partially explored using these methods and yet-unknown miRNAs and other small RNAs may be part of the tumor transcriptome.
Here, we present deep sequencing analysis of miRNAs from 23 solid tumor specimens of four different types: breast, bladder, colon and lung. A computational approach was used to identify known miRNA sequences, miRNA sequence variants (isomiRs), and novel small RNA species in these tumors. Forty-nine novel miRNA and small RNA candidates were identified including several novel miRNA sized RNA sequences: a human endogenous siRNA candidate and highly abundant 22–25 bp small RNAs derived from Y RNA. We also provide sequencing evidence for the existence of the recently identified human miRNA–offset RNAs (MORs) in human tumors. Subsequently, 31 normal and tumor samples from various tissue types were hybridized to a miRNA-microarray containing the novel miRNAs. Some of the novel miRNAs are abundantly expressed in different types of tumors and others are expressed differently between tumor and non-tumor samples, between different tumor stages or between different types of tumors. In addition, using RT–PCR as a third platform we confirmed the expression of five of the novel small RNAs in normal human serum. We identified numerous, abundant, new isomiRs (sequence variants of miRNAs) that may be related to carcinogenesis and may be derived from DNA alterations (SNPs or cancer-related mutations) or post-transcriptional modifications. These new cancer miRNA candidates can potentially be used as diagnostic biomarkers or therapeutic targets in different types of cancer.
MATERIALS AND METHODS
RNA isolation and enrichment phase
Total RNA was extracted twice from each sample of 23 human formalin-fixed paraffin-embedded (FFPE) samples derived from cancerous tissue. RNA was isolated using 10 10-µm-thick tissue sections using the miRdicatorTM extraction protocol developed at Rosetta Genomics. Briefly, the sample was incubated repeatedly in xylene at 57°C to remove excess paraffin, followed by washing in ethanol. Proteins were degraded by incubation in proteinase K solution at 45°C for a few hours. The RNA was extracted with acid phenol:chloroform followed by ethanol precipitation and DNAse digestion. Total RNA quantity and quality were checked by spectrophotometry (Nanodrop ND-1000). Pools of samples of the small RNA fraction (∼200 nt and lower) within the total RNA were labeled and hybridized on arrays. After ensuring the presence and expression of more than 100 miRNAs per cancerous tissue pool, tissues were pooled together, resulting in a bladder+breast tumor pool and a colon+lung pool. Array expression (Supplementary Figure S1) revealed the presence of 157 miRNAs from bladder cancer FFPEs, 260 miRNAs from breast cancer FFPEs, 135 miRNAs from lung cancer FFPEs, and 239 miRNAs from colon cancer FFPEs. Total RNA (75 µg) of seven different colon cancer FFPEs were pooled together with 75 µg of six different lung cancer FFPEs, while 75 µg total RNA of five different bladder cancer FFPEs were pooled together with 75 µg of five different breast cancer FFPEs.
Cloning linker attachment phase
The 3′ and 5′ cloning linkers (3′ Linker: 5′-rAppCTGTAGGCACCATCAAT/3ddC/-3′; 5′ Linker: 5′-TGGAATrUrCrUrCrGrGrGrCrArCrCrArArGrGrU-3′) were ligated to purified small RNA species in preparation for cDNA synthesis and amplification, using miRCat™ miRNA Cloning Kit according to the user manual, as described below.
Amplification
Reverse transcription of the linkered RNA species was carried out followed by PCR amplification. Primer sequences were as follows:
RT: 5′-GATTGATGGTGCCTACAG -3′ (Tm: 50.2°C)
Fwd tag1 primer: (454 fwd1 –BL1 mm)
5′ GCCTCCCTCGCGCCATCAGcagtTGTAATTCTCGGTCACCAA 3′
Rev tag1 primer: (454-Rev1-BL1)
5′ GCCTTGCCAGCCCGCTCAGcatgATTGACGGTGCCTACAG 3′
Fwd tag2 primer: (454 fwd2 –BL1 mm)
5′ GCCTCCCTCaCGCCATCAGtagtTGTAATTCTCGGTCACCAA 3′
Rev tag2 primer: (454-Rev2-BL1)
5′ GCCTTGCCAGCCCGCTCAGtagtATTGACGGTGCCTACAG 3′
Small RNA enrichment
The libraries were built using the MirCat kit, with several modifications, as described below. Enrichment of small RNA was carried out by recovering the small RNA fraction (17–25 nt), identified by internal size markers, from a slice of a 12% denaturing (7 M Urea) polyacrylamide gel. The synthetic RNA size markers run in the lane adjacent to the cancer samples were 15 nt (5′-GCAAAGCACACGGCC-3′), 22 nt (5′-UAUGUAUCGAAUUUAAGCUCAA-3′) and 38 nt (5′-GCAAGGAUGACACGCAAAUUCGUGAAGCGUUCCAUAUU-3′).
Cleaning the desired RNA from the gel was carried out by GeBAflex-tube-midi column (GeBAflex-tube Gel ext & Dialysis Kit Midi, Manufactured by DNR http://www.dnr-is.com/Product.asp?Par=2.215.219&id=228) using an electric current of 300 V for 40 min until the nucleic acid exited from the gel slice, followed by applying reverse polarity of the current for 120 s. This step releases the nucleic acid from the membrane. Isolated RNA was precipitated by adding 8 µl of glycogen, a one-tenth volume of NaOAc 3 M, pH 5.2, and three volumes of cold 100% ETOH, with vortexing after each addition. The isolated RNA was precipitated overnight at −20°C, centrifuged for 1 h at 4°C at 14 000 rpm, followed by washing with 1 ml cold 85% ETOH and subsequent centrifugation for 5 min at 14 000 rpm.
RNA linkering
Following recovery of the enriched small RNA fraction from the acrylamide gel slice, the small RNAs were ligated with a 3′ and a 5′ linker in two separate reactions. First, 3′ ligation was performed in which the 3′ linker was ligated to the small RNAs using T4 RNA ligase in the absence of ATP in order to avoid circularization of the RNA fragments, as described in (9). The ligated product was purified by recovering the desired band, identified using size markers, from a slice of a 12% denaturing (7 M Urea) polyacrylamide gel. Two synthetic RNAs (24 and 38 nt, described previously) and two synthetic RNA transcripts (53 and 83 bp) were run adjacent to the cancer samples. Purification and precipitation were carried out as described previously.
The 5′ linker is ligated to the 3′ linkered small RNAs in the presence of 1.0 mM ATP, followed by recovering the desired band from a slice of a 12% denaturing (7 M Urea) polyacrylamide gel with the same size markers. Purification and precipitation done as described before.
Reverse transcription
The 5′ and 3′ ligated RNAs contained both RNA and DNA regions which were converted to DNA using reverse transcriptase with RT primer, according to MirCat protocol (http://eu.idtdna.com/CATALOG/smallRNAcloning/page1.aspx?display=mircatkit).
PCR amplification
The PCR amplification step was carried out using primers different from those provided by the MirCat kit since the primers provided cause strong self- and heterodimers. PCR was carried out using PfuUltra high fidelity DNA polymerase (Stratagene #600380) and pairs of longer PCR primers (40–42-mers) containing sequences complementary to the linkers, tag sequences (small letters) and sequences which were suitable for the 454 platform (underlined). Tag1-flagged colon and lung library and Tag2-flagged breast and bladder library followed by 454 sequences that will convert the small RNA libraries made to ones that can be directly sequenced on the 454 platform.
Samples from five PCR reactions were pooled, extracted with phenol:chloroform, followed by recovery of the desired band from slices of an 8% native polyacrylamide gel. The resulting library was sent for sequencing on the 454 platform.
Computational analysis of deep sequencing
The deep sequencing process yielded over 200 000 sequences from both libraries. Adaptors were removed using a Perl script allowing internal polyN sequences within the adaptors and one mismatch. About 1000 sequences were removed since they were too short after adaptor removal (<10 bp). The sequences were mapped to the human genome (UCSC hg18 build) using BLAST, allowing maximum 3 bp mismatched to the genome and maximum insertion/deletion (indels) of 3 bp. For each aligned sequence the highest scoring hit was retrieved. All sequences with position overlap were clustered together using a Perl script. We assigned each genomic cluster of sequences the most abundant sequence in this cluster and demanded that for candidate miRNAs, the most abundant sequence was mapped precisely to the genome (not allowing any mismatches/indels). The next step was to annotate known sequences. The following datasets were used for this task: RNA genes, sno/miRNA, RefSeq genes and RepeatMasker tables were downloaded from the UCSC table browser (10), and known miRNA precursors were downloaded from miRBase (11) in order to mark whether the sequence is part of a non-coding gene, a snoRNA, a protein-coding gene exon, a genomic repeat, or a known miRNA precursor, respectively. The sequences of the novel miRNA candidates were extended by several hundred base pairs within their chromosomes in order to predict possible miRNA precursors. An extended sequence was intended to predict the folding of a pri-miRNA that contains a hairpin-folded pre-miRNA. The candidate pri-miRNAs were folded using the Vienna package (12) or mfold (13) programs. All hairpin structures that had at least 6 bp, were at least 55 nt long and had a loop not longer than 20 nt were extracted from the minimum free energy fold of the predicted pri-miRNA (excluding overlapping hairpins). Each hairpin was assigned a hairpin score (Palgrade) and conservation score as described before (14). Predicted miRNA precursors have either Palgrade > 0 meaning it has structural and sequence characteristics of known miRNA) or have absolute value of conservation score > 0.9 (conserved in mammals). These criteria have a sensitivity of 86% for known miRNA precursors from miRBase 13.0. In addition, only sequences with 10 or less genomic copies, with a length of 17–25 bp and a GC content in known miRNA range (15–90%) were chosen as miRNA candidates. Most (75%) miRbase miRs detected in this analysis were detected three or more times. Therefore we consider sequences counted three times or more to have a good chance of being functional. All sequences were deposited in the GEO, accession GSE20418.
Microarray design
Custom microarrays (Biochips) were manufactured by Agilent Technologies (http://www.agilent.com) by in situ synthesizing DNA oligonucleotide probes to 949 known miRNAs and 876 sequences from deep sequencing, printed in triplicate. Out of 49, 44 novel miRNA and small RNAs were used in the microarray (five sequences were identified as novel miRNAs/small RNAs after the design of the microarray). Sequences from deep sequencing were characterized by:
Mapping to the human genome.
Being part of a predicted hairpin (folded by Vienna/MFold).
Not being part of an annotated sequence (known miRNA, small RNA, coding exon).
Having <10 genomic occurrences.
MiRNA-sized (17–25 bp).
10% < %GC content < 90%.
Each probe comprised an antisense sequence of the relevant sequence, followed by a tail sequence (GCAATGCTAGCTATTGCTTGCTATTAAAAA), and was trimmed to the length of 45 nt. Seventeen negative control probes were designed using the sense sequences of different miRNAs. Two groups of positive control probes were designed to hybridize to the array: (i) synthetic small RNA that were spiked to the sample RNA before labeling to verify the labeling efficiency, and (ii) probes for abundant small nuclear RNAs that were spotted on the array to verify RNA quality.
Microarray hybridization
The microarray was hybridized to 38 different samples (Supplementary Table S9). The samples were from 17 different tissue types and blood samples and were divided to normal (n = 8), tumor (n = 15), tumor adjacent (n = 5) and metastasis indications (n = 8).
A total of 2–2.5 µg of total RNA was labeled by ligation of an RNA-linker, p-rCrU-Cy/dye (Eurogentec S.A.; Cy3 or Cy5), to the 3′ end. Synthetic small RNA was spiked into the RNA before labeling to verify the labeling efficiency. Slides were incubated with the labeled RNA for 12–16 h at 55°C and then washed according to Agilent GE washes. Arrays were scanned using Agilent DNA Microarray Scanner Bundle (Agilent Technologies, Santa Clara, CA) at a resolution of 5 µm, dual pass at 100 and 10% PMT power. Array images were analyzed using Agilent Feature Extraction software.
Array signal calculation and normalization
Array images were analyzed using the Feature Extraction software (FE) 9.5.1 (Agilent, Santa Clara, CA). Triplicate spots were combined to produce one signal for each probe by taking the logarithmic mean of reliable spots. All data were log-transformed (natural base) and the analysis was performed in log-space. A reference data vector for normalization R was calculated by taking the median expression level of a subset of all probes (all miRNAs in miRBase 10) across samples. For each sample data vector S, a second degree polynomial F was found so as to provide the best fit between the sample data and the reference data, such that R ≈ F(S). For each probe in the sample (element Si in the vector S), the normalized value (in log-space) Mi was calculated from the initial value Si by transforming it with the polynomial function F, so that Mi = F(Si). P-values were calculated using a two-sided t-test on the log-transformed normalized fluorescence signal. The fold-difference (ratio of the median normalized fluorescence) was calculated for each miRNA. The signal of a sequence is defined as differential between sample ‘A’ and sample ‘B’ if the fold change between the signal in sample A and sample B is either larger than the 95th percentile of fold changes of all sequences expressed in both samples, or >8. Results of all microarray experiments were deposited in Gene Expression Omnibus GSE20418.
Expression detection by qRT–PCR
RNA was subjected to a polyadenylation reaction as described previously (15). Briefly, RNA was incubated in the presence of poly (A) polymerase (PAP; Takara-2180A), MnCl2 and ATP for 1 h at 37°C. Then, using an oligodT primer harboring a consensus sequence, reverse transcription was performed on total RNA using SuperScript II RT (Invitrogen). Next, the cDNA was amplified by real time PCR; this reaction contained a miRNA-specific forward primer, and universal TaqMan probe complementary to the 3′ end of the oligodT plus part of the tail, and a universal reverse primer complementary to the consensus 3′ sequence of the oligodT tail. For each miR, expression signals were calculated by the formula 42 – Ct(miR-X).
Dual-luciferase reporter assay
Dual-luciferase assay was conducted using psiCHECK-2 dual luciferase plasmid (Promega) harboring the relevant sequencing in its 3′UTR (GeneScript). Hep3B cells were plated at a density of 4000 cells per well, on white collagen coated plates (with a transparent bottom). Cells were transfected the next day with plasmid baring the relevant 3′UTR, and with or without antisense oligo for the relevant small RNA sequence. Transfection was carried out using 0.3 µl Lipofectamine 2000/well (Invitrogen, Cat# 11668027). Luminescence was assayed 24 h after Transfection, using the Dual Luciferase reporter assay kit (Promega, Cat#E1961). Luminescence was read on Luminoskan Ascent (Thermo). Firefly luciferase from the same plasmid was used for normalization of transfection efficiency. A plasmid vector without 3′UTR alteration in the renilla 3′UTR was used as a reference for constitutive luciferase expression. Results were shown as the ratio between the various treatments and cells transfected with an empty vector.
RESULTS
Deep sequencing of small RNAs from solid tumor samples
In order to identify new cancer-related miRNAs, 23 formalin-fixed paraffin-embedded (FFPE) samples of primary solid tumors were obtained from the following tumor tissues: breast (n = 5), bladder (n = 5), colon (n = 7) and lung (n = 6) (samples are described in Supplementary Table S1). Total RNA enriched of small RNA was extracted, RNA quality was examined by hybridization to a custom miRNA microarray. Tissue-specific miRNAs of the tissues sampled were clearly expressed in the microarray experiment, supporting the high quality of the RNA (Supplementary Figure S1). Next, the RNA samples from breast and bladder were pooled together, and RNA samples from colon and lung were pooled together. Small RNA (17–25 nt) was separated and small RNA libraries were prepared and sequenced using 454 Life Sciences technology.
The sequencing process yielded 141 023 sequences from the bladder+breast tumor pool and 90 986 sequences from the colon+lung pool. After combining identical sequences, 27 968 unique sequences remained, 81% of which are 18- to 26-bp long, demonstrating that the libraries are highly enriched in small RNAs. This small RNA size fraction accounts for 93% of all redundant sequences. Sequences were mapped to the human genome using the Blast program (16), allowing up to three mismatched base pairs and indels of up to 3 bp. This process yielded 723 485 genomic loci of mapped sequences. Eighty-three percent of the unique sequences were mapped to the human genome using these criteria and 59% of the unique sequences were mapped with maximum 1 nt mismatch. We postulate that some mismatches, mainly in the 5′ and 3′ edges of the sequences, could result from inaccurate removal of the sequence-flanking adaptors. Subsequently, 565 224 clusters of sequences with genomic position overlap were created. For example, if sequence X was mapped to positions 1-20 within the plus strand of chromosome 1 and a sequence Y was mapped to positions 15–35 on the same chromosome and strand, then the two sequences were unified in the same genomic cluster of chromosome 1, plus strand, positions 1–35. The clusters of sequences represent segments of expressed genes. The statistics of the sequencing and genome mapping procedures are summarized in Table 1.
Table 1.
Statistics of sequencing and genome mapping procedures
| Total reads (redundant sequences): | 141 023 (bladder+breast), 90 986 (colon+lung) |
|---|---|
| Unique sequences in both libraries: | 27 986 |
| Unique short (18–26 bp) sequences in both libraries: | 22 648 |
| Unique sequences in both libraries mapped to the genome with up to three mismatches/indels (% of total unique sequences) | 23 271 (83%) |
| Unique sequences in both libraries mapped to the genome with up to one mismatch and no indels (% of total unique sequences) | 16 564 (59%) |
| Unique sequences in both libraries mapped to the genome in exact match (% of total unique sequences) | 10 182 (36%) |
| Genomic loci of mapped sequences (up to 3 mismatches/indels) | 723 485 |
| Genomic loci clusters of mapped sequences (up to 3 mismatches/indels) | 565 224 |
Expression and sequence variability of known miRNAs
We first mapped the sequenced reads to known miRNAs from miRBase database (11) according to genomic position overlap, inclusion of sequenced reads in mature miRNA sequences or inclusion of mature miRNA sequences in the sequenced reads. The small RNA libraries were found to be enriched with human miRNAs. Known miRNAs occupy 61% (140 255/230 740) of the total small RNA reads. Out of 885, 387 (44%) human miRNAs were sequenced in at least one read in the different tumor libraries. However, the expression of known miRNAs is very heterogeneous, ranging from 1 read to 25 780 reads. The 10 most abundantly expressed miRNAs in both tumor libraries, are presented in Table 2. Most of these miRNAs were indeed expected to be highly expressed in the tumor tissues screened. hsa-miR-21 is a well known oncogene that is expected to be abundant in solid tumors. We also observed high expression of the miR-141-200 family that is known to be expressed in epithelial tumors similarly to the tumors we screened (3). In addition, we sequenced dozens of rare miRNAs that were only found to be lowly expressed in specific tissues and lack independent validation in other tissues or by other methods. These rare miRNAs include miRNAs that were identified specifically in chronic lymphocytic leukemia cells (17), embryonic stem cells (5,7), colorectal cancer cells (18), or cervical cancer cells (19), detailed in Supplementary Table S2. The expression of these rare miRNAs in tumor tissues enhances their reliability as true miRNAs.
Table 2.
Highly expressed miRNAs in the two deep sequencing libraries
| miRNA name | No. of reads in bladder+breast tumor library | No. of reads in colon+lung tumor library |
|---|---|---|
| hsa-miR-200c | 20 259 | 5480 |
| hsa-miR-200b | 9493 | 6059 |
| hsa-miR-143 | 4642 | 3961 |
| hsa-miR-21 | 3337 | 3835 |
| hsa-miR-200a | 4246 | 2845 |
| hsa-miR-23a | 3545 | 3010 |
| hsa-miR-26a | 3328 | 2456 |
| hsa-miR-29b | 1867 | 1369 |
| hsa-miR-23b | 2030 | 1075 |
| hsa-miR-141 | 2207 | 715 |
| Total | 54 954 | 30 805 |
Most miRNAs were sequenced in several sequence variants that were previously referred to as isomiRs (7). The different isomiRs were predominantly variable in the 3′ end of the mature miRNA sequence, a region which is less precisely defined than the miRNA 5′ end (20). Although we expected to find different isomiRs for most miRNAs, we found two surprising phenomena. First, for 74 known miRNAs the most abundant isomiR in our cancer tissue survey was much more abundant (at least 20%) than the reference miRNA sequence from miRBase database (Supplementary Table S3). This suggests that the relative abundance of isomiRs may be inherently different between normal tissue and tumors. Second, 59 known miRNAs had an abundant isomiR with at least one mismatch to the human genome sequence, suggestive of the discovery of novel miRNA-related SNPs/cancer mutations or post-transcriptional modification of the miRNAs (Supplementary Table S4). All these isomiRs were expressed in at least the same number of reads as the miRBase isomiR and seven of these miRNAs were supported by at least 10 reads. Examples are shown in Figure 1. Most of the sequence modifications (69%) occurred in the 3′-end of the miRNA and involved either DNA base modification, 3′ uridylation (Figure 1A and B) or 3′ adenylation (Figure 1C and D). 3′ additions of G or C were completely absent in our data. The high abundance and the specificity of the 3′ terminal single nucleotide insertions suggest that these are regulated post-transcriptional modifications and not DNA-level changes (SNPs/mutations), which are expected to occur in a more random manner. Interestingly, the common 3′ uridylation of hsa-miR-143 (Figure 1A) and the common 3′ adenylation of hsa-miR-100 (Figure 1C) were also identified by other independent methods of small RNA library preparation and sequencing (7,18,21) supporting the idea that these modifications are the result of meaningful cellular processes and not merely a technical artifact. The 3′ adenylation/uridylation described is unlikely to be the result of incorrect adaptor removal, as the 3′ adaptors that were used start with cytosine. We also noted several sequence modifications that occur internally within the miRNA sequence (Figure 1E). These isomiRs demonstrated primarily (77%) C → T or A → T nucleotide modifications, again suggesting involvement of post-transcriptional RNA editing by cytidine deaminase or ADAR enzymes, respectively, contrary to DNA level changes. We cannot exclude the possibility that the rare sequence modifications, sequenced only in few reads, are sequencing errors.
Figure 1.
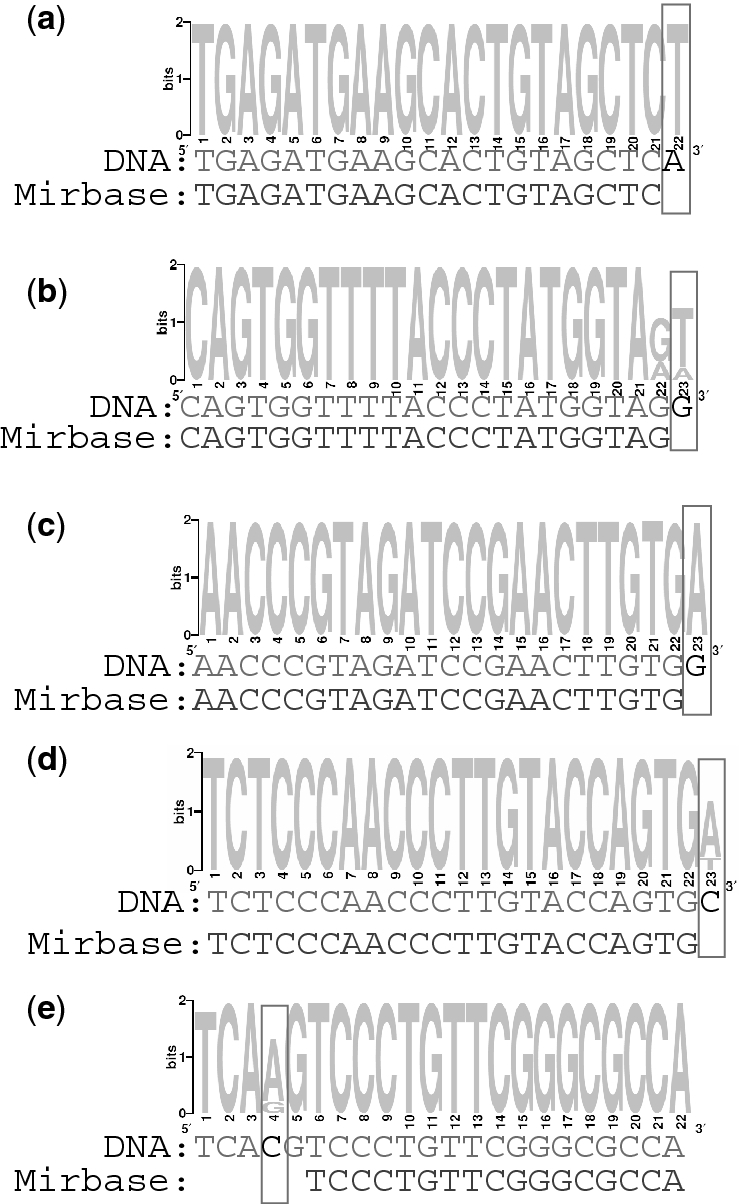
IsomiRs that demonstrate existence of SNPs or post-transcriptional modifications. For each presented miRNA, its main isomiRs, responsible for at least 5% of all sequences in the tumor libraries, were retrieved. These were aligned using ClustalW (51) and the resulting alignment is visualized as position-specific scoring matrix using WebLogo (52). The sequenced isomiRs of each miRNA are compared to the human genome and the reference miRNA sequence from miRBase database. (A) hsa-miR-143, (B) hsa-miR-140-5p, (C) hsa-miR-100, (D) hsa-miR-150 and (E) hsa-miR-1274b.
Identification of novel miRNAs and miRNA sized small RNAs
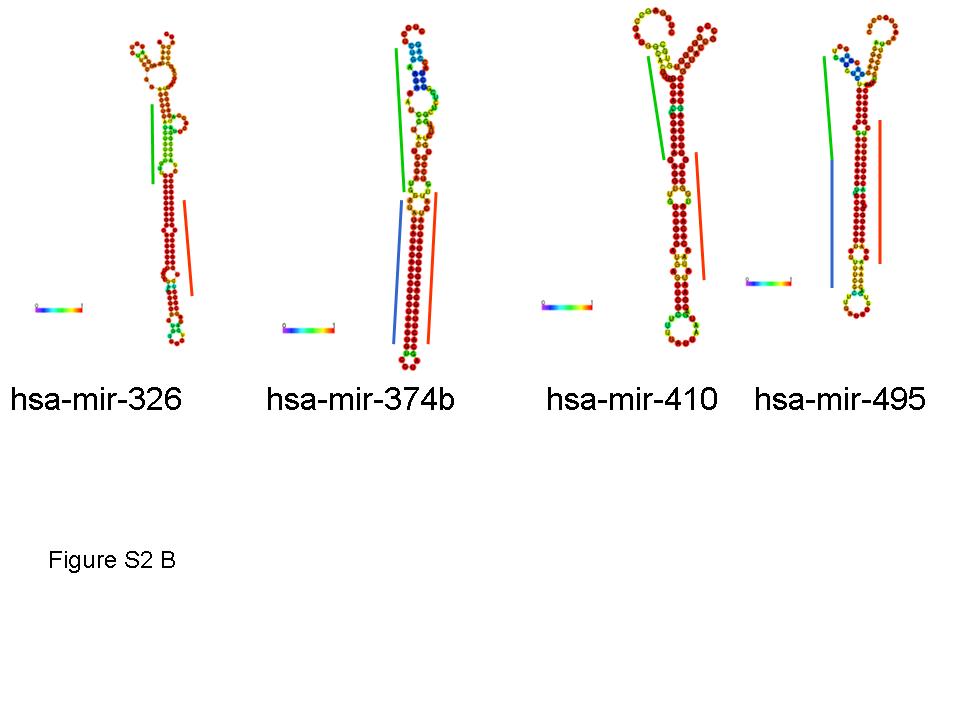
Next, we set out to identify novel miRNAs expressed in the tested cancer tissues. Our miRNA discovery pipeline (Figure 2) led to the identification of four different groups of novel miRNAs and miRNA sized small RNAs: (i) miRNAs derived from known miRNA precursors, (ii) miRNAs derived from novel miRNA precursors, (iii) miRNA sized small RNAs derived from annotated small RNA and genomic repeats, (iv) Endogenous siRNA sequences. The first group (Supplementary Table S5) mainly consisted of the complementary miRNA (miRNA star) sequences of known human miRNAs. These are ∼22 nt RNA species nearly complementary to a known miRNA, which are located within the miRNA precursor and which may have an inhibitory activity (22). We identified 18 such novel miRNA star species. In several cases, e.g. hsa-miR-1307-5p and hsa-miR-412-5p, the novel complementary mature miRNA was more abundant than the known miRNA, suggesting that the miRNA identified here is the major active product of the miRNA precursor, at least in the tested tumor samples. In addition we identified seven cases of miRNA-offset RNAs (MORs), a miRNA-like group that was recently characterized (23). MORs are part of the miRNA precursor and are processed from a ∼22 bp dsRNA region directly upstream to the miRNA-miRNA star dsRNA region (an example of a sequenced 5′-MOR can be seen in Figure 3A). All MORs sequenced in the human tumors are highly conserved, derived exclusively from the 5′ stem of the miRNA precursor directly upstream to the 5′ miRNA, and lowly expressed relative to the main miRNA product of the precursor. These findings are in accordance with previous results (24). The MORs identified here tend to be located in a region of lower dsRNA stability than the main miRNA-miRNA star pair of the miRNA precursor (Supplementary Figure S2). Therefore, the miRNA precursor of a MOR may switch between different folded RNA structures, only part of which accommodates the MOR in a dsRNA region that would be processed by the canonical miRNA pathway. This may explain the relatively low expression of MORs in comparison to the main mature miRNAs of the precursors. In the case of hsa-miR-410 and hsa-miR-326 we sequenced the 5′ MORs, whereas the miRNA stars of the precursors were not sequenced. Several of the new miRNA star and MOR species identified here were recently identified by an independent study (24), however they do not yet appear in miRBase.
Figure 2.

miRNA and small RNA discovery pipeline. Four different groups of novel miRNA and small RNAs were identified: novel miRNAs/ miRNA-offset RNAs (MORs) derived from known miRNA precursors, novel miRNAs derived from novel miRNA precursors, miRNA sized small RNA derived from annotated repeats/small RNAs, and endogenous siRNAs.
Figure 3.

Novel types of small RNA identified in deep sequencing of tumors. (A) 5′ miRNA-offset RNA from hsa-mir-20a. In parenthesis are numbers of sequenced reads. (B) Antisense miRNA and miRNA star of hsa-miR-338. Red segments denote hsa-miR-338 precursor and mature miRNAs (long segments are precursors); blue segments denote the antisense miRNA-338 precursor and mature miRNAs. Numbers beside the short segments (mature miRNA) denote number of sequenced reads. The predicted folded hairpin of the antisense miRNA precursor is shown on the left with the positions of the mature miRNAs marked by black lines. Nucleotides colored in red have a high probability to occupy the depicted fold, green colored nucleotides have a low probability to be in the given fold. (C) miRNA sized Small RNA derived from Y-RNA. The entire Y RNA is folded as a hairpin, similar to miRNA precursor (left). The sequenced small RNA is derived from the 5′ arm of the hairpin. (D) Putative endogenous siRNA derived from mitochondrial-derived pseudogene. Segments in blue derive from plus strand and start in the transcription start site of the tRNA pseudogene. Segments in red derive from the minus strand. Numbers beside the segments denote number of sequenced reads. This figure is based on the UCSC genome browser (53).
The second group contained completely novel miRNAs from novel miRNA precursors. In this case we used only reads that were exactly mapped to the genome. Reads that were mapped to more than 10 loci were filtered out, since human miRNAs rarely map to more than a few genomic loci. Other reasons for which sequences were discarded include their rare occurrence (i.e. very few reads), length exceeding normal miRNA length and GC content (%GC) higher than the %GC of known miRNAs. After filtering out by these criteria, as well as filtering sequences located within already annotated sequences (known miRNAs, other small RNAs, transposons, coding exons), we predicted miRNA precursors by folding several hundred base pairs flanking the final miRNA candidates using RNAfold (25). In order to reduce the number of false positive predictions, we kept only predicted miRNA precursors that were either evolutionarily conserved or had structural features of known miRNAs. Such structural features include limited lengths of bulges and loops and low folding energy. A miRNA precursor score was computed by integrating these parameters (14). This process resulted in the identification of 20 novel miRNAs (Supplementary Table S6). An example of a novel miRNA precursor, which yields the mature miRNA MID-20989, can be seen in Figure 3B. miRNA MID-20989 is more abundant (16 reads) than hsa-miR-338 (five reads), the product of hsa-mir-338, which is located antisence to the MID-20989 precursor. Although we filtered out sequences within annotated small RNAs, we noticed that two new miRNAs that correspond to a miRNA–miRNA star pair of the same miRNA precursor (MID-16049 and MID-18078) are located within a HBII-82B snoRNA. Several miRNA have previously been identified by other groups as having been derived from snoRNAs (26–28), which makes it plausible that these two sequences indeed function as miRNAs.
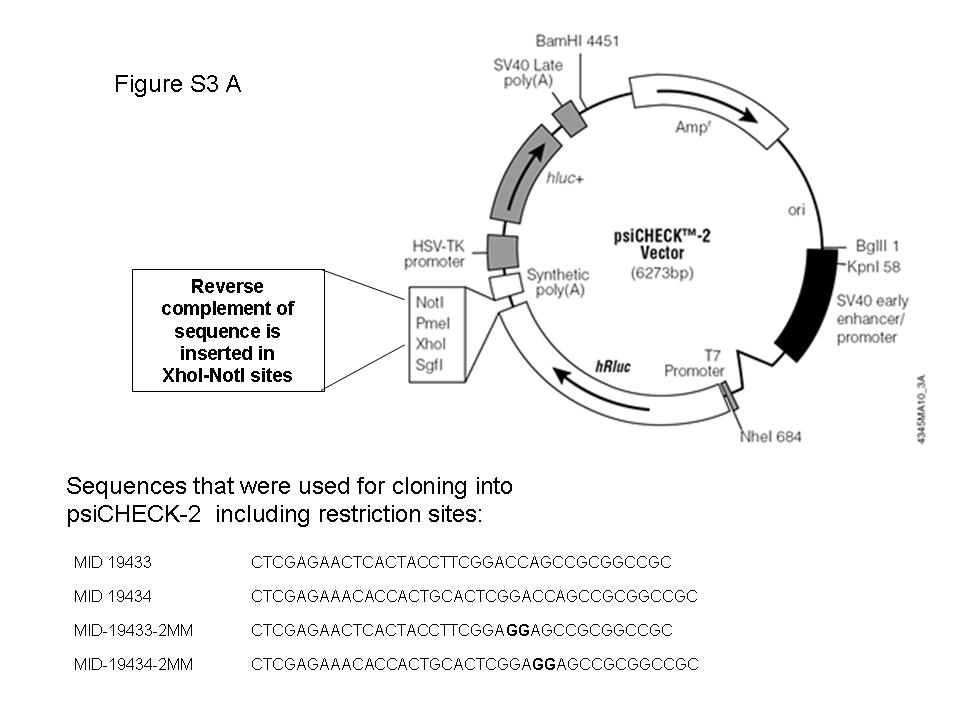
The third group contained miRNA sized sequences derived from annotated small RNAs and genomic repeats. Several miRNAs (e.g. hsa-miR-28, hsa-miR-548 family) were previously described as having been derived from such genetic elements (26–30). Sequences whose length exceeded the conventional size of miRNAs (17–25 bp) were discarded. MiRNA precursors were predicted using RNAfold and mFold and the precursor score described above. Finally, only sequences with at least 10 reads were taken, in order to ensure that the identified novel miRNAs were likely to be consistent products of enzymatic excision and not rare degradation products. This strict criterion was used for this group only as these derive from known RNA species that are often highly expressed and their degradation products are expected to be found in the cell, therefore their re-annotation as miRNAs needs stronger evidence. This process revealed three novel miRNA sized sequences (Supplementary Table S7). One of the candidates in this group, MID-24078, is derived from a local hairpin-fold of an Alu repeat. The other two (MID-19433, MID-19434) are, interestingly, derived from Y RNAs. Y RNAs are relatively unexplored non-coding RNA species that are implicated in chromosomal DNA replication (31) and RNA quality control (32). MID-19434 is a 25-nt long RNA derived from a ∼100-nt long hY3 RNA like sequence. MID-19434 was highly expressed, with 200 sequenced reads, which is more abundant than over 300 known miRNAs sequenced in the tumor samples analyzed here. The predicted well-folded precursor of this miRNA was precisely aligned to the hY3 RNA (Figure 3C), suggesting that the Y RNA is processed, possibly by Dicer, to yield a 25-bp mature miRNA. MID-19433 is derived from hairpin-folded hY1 Y RNA. Following this finding, we went back to miRBase and found that two relatively newly known miRNAs, hsa-miR-1975 and hsa-miR-1979, which were also sequenced in this study, are actually Y RNA-derived miRNAs. We next set out to explore whether these Y RNA-derived miRNA candidates had gene silencing activity, similarly to known miRNAs. Therefore, we designed a Luciferase assay experiment. This experiment aimed to verify reduced Luciferase activity of a transfected Luciferase gene carrying a complete complementary sequence to the Y RNA-derived miRNA candidates within its 3′ UTR. However, this experiment failed to show gene silencing activity of the miRNA candidates tested, at least in Hep3B cells that were endogenously expressing these sequences (Supplementary Figure S3). This result challenges our hypothesis that the Y RNA derived miRNA sized sequences (including hsa-miR-1975 and hsa-miR-1979) are bona fide miRNAs. A plausible explanation for the high abundance of these miRNA sized RNA species is that these are the specific 20–25 bp Y RNA apoptotic degradation products that remain intact since they are protected by the Ro60 protein (33). However, the high abundance and sequence stability of these small RNAs may suggest that these small RNAs may have a yet unknown function.
Finally, we have also identified a candidate human siRNA. Endogenous siRNA were recently described in mouse oocytes (34), but have not yet been identified in the human transcriptome. These are ∼21-bp long RNA species that are processed from a dsRNA by Dicer and assembled in the RNA induced silencing complex (RISC). Figure 3D depicts the candidate human endogenous siRNA we identified in the studied cancer libraries (see also Supplementary Table S8). This is a ∼20-nt dsRNA that could be derived from bi-directional transcription of the same locus. Six sequenced reads are transcribed in the same orientation as a mitochondrial tRNA as well as a tRNA-derived pseudogene in several chromosomes, which is the more likely source of the siRNA sequences. Their transcription starts in the transcription start site of the tRNA, suggesting that these sequences are processed from the tRNA transcripts. Two antisense reads create a dsRNA with a short 5′ overhang, as opposed to common siRNA which are characterized by a 3′ overhang. The sense and antisense reads are mapped to nine different genomic loci. Therefore, it is also possible that the complementary sequences were derived from independent single-stranded RNAs and not from a hybridized dsRNA.
Cross platform validation and differential expression of novel miRNAs
In order to verify the expression of the novel miRNAs in another independent platform, and to identify differentially expressed miRNAs in specific tissues or tumors, we designed a custom microarray. The microarray contained probes designed for deep sequencing reads that passed minimal criteria (see Materials and methods section), including 44 of the novel miRNA and small RNAs identified above. The microarray also contained probes for known miRNAs. The microarray was hybridized to 38 different samples (Supplementary Table S9) from 17 different tissue types that included tumor, tumor adjacent, normal and metastasis samples, as well as blood samples. A total of 684 probes were expressed in at least one sample, out of which 584 had %GC<0.75, which would ensure more reliable hybridization, and 244 had a deep sequencing count of 2 or above (in the two libraries combined). Table 3 shows the distribution of miRNAs by each of the above criterion, for known miRNAs, and miRNAs detected by deep sequencing. Only 23% of the known miRNAs were detected in both platforms, i.e. by the microarray and the deep sequencing. Thus, although only a small fraction of the newly defined miRNAs were detected by both platforms, a larger fraction (roughly four times this fraction) could nonetheless be identified as true miRNAs.
Table 3.
Statistics for the different types of microarray probes
| New miRNAs | miRBase miRNAs | Deep sequencing sequences | |
|---|---|---|---|
| Total | 44 | 852 | 815 |
| Expressed (% of total) | 12 (29%) | 315 (37%) | 114 (14%) |
| And GC<0.75 (% of total) | 10 (23%) | 301 (35%) | 86 (10%) |
| And DS count ≥2 (% of total) | 9 (20%) | 194 (23%) | 43 (3%) |
The correlation between deep sequencing and microarray signals is significant for all sequence types, but is much higher for known miRNAs (Table 4). Sequences identified as novel miRNAs have a higher correlation than the overall correlation of deep sequencing reads. This strengthens the notion that many of these are indeed true miRNAs.
Table 4.
Correlation between deep sequencing and microarray
| Pearson’s correlation coefficienta | P-value | |
|---|---|---|
| Deep sequencing reads | 0.39 | <10−6 |
| Novel miRNAs | 0.44 | 0.00272 |
| Known (miRBase) miRNAs | 0.61 | <10−6 |
aMedian microarray signal of tumor samples.
The expression pattern of the newly identified miRNAs in different tissues is shown in Figure 4. Some of the new miRNAs are expressed in similar levels as miRNAs with a known role in cancer, suggesting that some of the new miRNAs may also play an important role in cancer. For example, comparison of colon tumor versus adjacent normal colon tissue (Figure 4A) supports the known upregulation of hsa-miR-20a (35–37) and hsa-miR-17 (37) in colon cancer. In addition, nine new miRNAs were expressed in both colon tumor and tumor-adjacent colon tissues in high and comparable levels to known miRNAs. Four of these were expressed at least 1.5-fold higher in colon tumor. Figure 4B displays a comparison between lung cancers versus nine non-lung tumor types. hsa-miR-200c, hsa-miR-141 and hsa-miR-205 are miRNA that are known biomarkers of epithelial tumors (3). A new miRNA, MID-19667, which is the star sequence of hsa-miR-663, demonstrates lung tumor tissue-specificity, together with six additional novel miRNAs. Figure 4C depicts a comparison between primary breast tumor and breast metastasis to the lymph node. hsa-mir-130b is known to be upregulated in breast metastasis (38). Three novel miRNAs were expressed higher in non-metastatic breast tumor. Finally, several novel miRNAs were abundantly expressed across many tumor types (Figure 5). MID-19433 and MID-19434 (the Y RNA derived small RNAs) are, notably, expressed in comparable levels to the expression of hsa-miR-21, a well-established oncomiR, supporting their potential role as oncogenic small RNAs. Five of the new miRNAs were also expressed in the serum of healthy people (Figure 6) using a third platform (RT–PCR). Four of the novel miRNAs and miRNA sized small RNAs (MID-19433, MID-19434, MID-17356 and MID-16489) were expressed in all three platforms used.
Figure 4.

Microarray expression in different tissues. Normalized log-scale expression of various sequences printed on a custom made microarray. Yellow circles indicate newly identified miRNAs, black dots indicate miRbase miRs, and plus signs (+) indicate all other deep sequencing detected sequences. (A) Colon tumor versus colon tumor adjacent tissue from the same patient. Labeled miRNAs are known to be expressed or deregulated in colon tumors (3,35–37,54). The newly identified miRNAs, marked in yellow, are expressed in levels similar to those of known relevant miRNAs. (B) Median of two lung tumor samples versus median expression of other tumors, from the following tissues: bile duct, bladder, breast, colon, kidney, liver, lung, ovary, pancreas and prostate. One lung tumor sample is from type non-small squamous, the other is a mix of various lung tumor types. Labeled miRNAs are known to be expressed in epithelial tumors (3). A novel miRNA, MID-19667, is preferentially expressed in lung tumors. (C) Breast primary tumor versus breast metastasis to the lymph node. Labeled miRNAs are known to be high in breast cancer or differential between breast metastasis and primary tumors (38,55).
Figure 5.

Microarray expression of novel miRNAs and small RNAs, as well as the known oncomiR hsa-miR-21 in several tumor tissues. Expression is normalized, and shown in log2 scale. Red and green denotes high and low expression level, respectively. Expression varies between different tumors, some new miRNAs expression levels compare with hsa-miR-21, whereas others are expressed in lower levels.
Figure 6.

RT–PCR expression of novel miRNAs and small RNAs, in human serum. RNA was measured in sera of 19 normal humans and in negative controls not containing RNA. Shown is the median of expression signals for each miRNA in all tested samples. Black bars show expression in experimental samples and white bars show expression in negative controls. hsa-miR-200b is given as reference since it is known to be expressed in blood (56,57).
Finally, we set out to identify the target genes of several of the novel miRNAs. We used the TargetScan program (39) in order to predict target genes. Since the novel miRNAs were identified in tumor tissues, we hypothesized that these may have a potential role in carcinogenesis. Therefore, we crossed the predicted target genes with cancer-related genes from the cancer gene census (40). This analysis yielded a list of 16 402 predicted targets for the 49 candidate miRNA and miRNA sized small RNAs from deep sequencing (Supplementary Table S10). We identified that some novel miRNAs have multiple binding sites in known tumor suppressor genes or oncogenes, e.g. TP53 (MID-22124, MID-16049, MID-18078 and MID-20830), NRAS (MID-16770) and KRAS (MID-16489, MID-17963, MID-24263 and MID-32019). All predicted targets, not only cancer related genes, are shown in Supplementary Table S10.
DISCUSSION
In this study a large set of 23 human tissue samples from four different tumor types was screened for small RNAs using deep sequencing. Nearly 400 known miRNAs were detected and 49 novel miRNAs and miRNA sized small RNA sequences were identified. Further support is provided for the expression of 10 novel sequences in a different platform (10 by microarray and five also by RT–PCR) and in a broader range of blood, normal and cancer tissues that were not surveyed by deep sequencing. Some of the novel sequences are expressed differently between different tissues, such as tumor and adjacent normal tissue. Novel types of miRNA sized sequences are reported here, revealing new small RNA groups, including Y-RNA-derived small RNAs, and putative endogenous siRNAs. In addition we identified a large variety of new isomiRs, many of which demonstrate DNA change (SNP/cancer-related mutation) or post-transcriptional modification.
Deep sequencing is a useful tool that was previously used to uncover unknown small RNA groups, such as piRNAs, murine endogenous siRNAs, and mirtrons (34,41–43). Here we report the identification of two novel small RNA groups from human solid tumors. We estimate that these novel sequences were ignored in former deep sequencing analyses due to the following reasons: (i) Low expression—the putative endogenous siRNA were sequenced in <10 reads each. (ii) Location within annotated sequences—as part of deep sequencing analysis, annotated reads are as a rule filtered out in order to identify new sequences. The endo-siRNA is located within tRNA-derived pseudogene; Y-RNA derived miRNAs are located within annotated small cytoplasmic Y-RNA. This former annotation may have caused the overlooking of these sequences when analyzed by others before. (iii) Deep sequencing of unexplored tumor tissues. Relatively few human tumor tissues were deeply sequenced till now. Specific expression of the identified sequences in these tissues may suggest that the described sequence novelties are related to carcinogenesis of solid tissues, e.g. Y-RNA derived small RNAs may be the result of aberrant processing of Y-RNA in cancerous tissue. In addition we identified other recently identified human miRNA sized species such as: MORs (24), antisense transcribed miRNA (4), and snoRNA-derived miRNA (26–28).
The two novel small RNAs that are most abundantly expressed in different tumors in all platforms (high throughput sequencing, microarray, and RT–PCR), MID-19433 and MID-19434, are derived from small cytoplasmic Y RNA. However, the microarray and RT–PCR cannot differentiate between intact Y RNA and small RNA derived from Y RNA. Therefore, further validation is needed in order to rule out the possibility that the bulk of the signals recorded in these platforms are due to unprocessed Y RNAs. The potential importance of these two novel miRNA sized sequences in tumorgenesis is supported by a recent work reporting that the Y RNAs hY1 and hY3, that are the unprocessed Y RNAs of MID-19433 and MID-19434, respectively, are overexpressed in carcinomas of the bladder, cervix, colon, kidney, lung and prostate (44). Therefore, measuring the highly expressed small RNAs from these Y RNAs can be used for molecular diagnostics of these cancers. The fact that these were also detected in serum confers their potential usage in non-invasive assays.
In this survey, we report the identification of dozens of new highly abundant isomiRs, including some isomiRs that demonstrate either existence of novel SNP or clear RNA modification. Several small-scale variations in miRNA genes were implicated before with carcinogenesis of various tissues, such as breast cancer (45,46), pancreatic cancer (47), lung cancer (48) and thyroid cancer (49). Additional data of isomiRs expression in normal tissues is needed in order to gain insight whether the novel isomiRs are related to the carcinogenesis of solid tumors. Some of the novel isomiRs were found to be significantly more abundant in this study than their reference miRNA sequences in miRBase database. Therefore, we suggest that in these cases the reference miRNA sequence should be considered for revision to the most abundantly expressed isomiR described here.
Although the number of novel miRNAs identified in this study is relatively high, it is important to note that the novel miRNAs are expressed on average in much lower levels than known miRNAs, with the exception of the Y-RNA derived small RNAs that are abundantly expressed in both tissue pools. Our work joins recently published works that reported a similar result regarding scarcity of newly identified miRNAs, which calls for additional verification in order to establish functional relevance (50). The fact that miRNAs discovered in the last two years using very sensitive methods are generally of low expression or are tissue-specific, suggests that the more abundant and non-specific miRNAs have mostly been identified already. However, as this work demonstrates, using a sensitive method of next generation sequencing on RNA extracted from tumors, in addition to careful computational analysis and followed by verification experiments can identify yet unknown sequences such as the new miRNAs, MORs, Y-RNA derived sequences and endogenous siRNAs presented in this analysis. The identification of such tumor-specific small RNAs, could lead to the development of new therapeutic targets, which may be utilized as a treatment more specific than the set of tools currently available.
ACCESSION NUMBER
GEO accession number: GSE20418.
FUNDING
Funding for open access charge: Internal funding.
Conflict of interest statement. Authors affiliated with Rosetta Genomics are full-time employees and/or hold equity in the company, which develops microRNA based diagnostic products and may stand to gain by publications of these findings.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
The authors thank the technicians and researchers at Rosetta Genomics for their assistance and contributions, and Ranit Aharonov, Moshe Hoshen and Shai Rosenwald from Rosetta Genomics for scientific discussions and comments on the manuscript.
REFERENCES
- 1.Visone R, Croce CM. MiRNAs and cancer. Am. J. Pathol. 2009;174:1131–1138. doi: 10.2353/ajpath.2009.080794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lebanony D, Benjamin H, Gilad S, Ezagouri M, Dov A, Ashkenazi K, Gefen N, Izraeli S, Rechavi G, Pass H, et al. Diagnostic assay based on hsa-miR-205 expression distinguishes squamous from nonsquamous non-small-cell lung carcinoma. J. Clin. Oncol. 2009;27:2030–2037. doi: 10.1200/JCO.2008.19.4134. [DOI] [PubMed] [Google Scholar]
- 3.Rosenfeld N, Aharonov R, Meiri E, Rosenwald S, Spector Y, Zepeniuk M, Benjamin H, Shabes N, Tabak S, Levy A, et al. MicroRNAs accurately identify cancer tissue origin. Nat. Biotechnol. 2008;26:462–469. doi: 10.1038/nbt1392. [DOI] [PubMed] [Google Scholar]
- 4.Azuma-Mukai A, Oguri H, Mituyama T, Qian ZR, Asai K, Siomi H, Siomi MC. Characterization of endogenous human Argonautes and their miRNA partners in RNA silencing. Proc. Natl Acad. Sci. USA. 2008;105:7964–7969. doi: 10.1073/pnas.0800334105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bar M, Wyman SK, Fritz BR, Qi J, Garg KS, Parkin RK, Kroh EM, Bendoraite A, Mitchell PS, Nelson AM, et al. MicroRNA discovery and profiling in human embryonic stem cells by deep sequencing of small RNA libraries. Stem Cells. 2008;26:2496–2505. doi: 10.1634/stemcells.2008-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friedlander MR, Chen W, Adamidi C, Maaskola J, Einspanier R, Knespel S, Rajewsky N. Discovering microRNAs from deep sequencing data using miRDeep. Nat. Biotechnol. 2008;26:407–415. doi: 10.1038/nbt1394. [DOI] [PubMed] [Google Scholar]
- 7.Morin RD, O'Connor MD, Griffith M, Kuchenbauer F, Delaney A, Prabhu AL, Zhao Y, McDonald H, Zeng T, Hirst M, et al. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res. 2008;18:610–621. doi: 10.1101/gr.7179508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu JY, Pfuhl T, Motsch N, Barth S, Nicholls J, Grasser F, Meister G. Identification of novel Epstein-Barr virus microRNA genes from nasopharyngeal carcinomas. J. Virol. 2009;83:3333–3341. doi: 10.1128/JVI.01689-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001;294:858–862. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- 10.Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32:D493–D496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–D158. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hofacker IL. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31:3429–3431. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bentwich I, Avniel A, Karov Y, Aharonov R, Gilad S, Barad O, Barzilai A, Einat P, Einav U, Meiri E, et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat. Genet. 2005;37:766–770. doi: 10.1038/ng1590. [DOI] [PubMed] [Google Scholar]
- 15.Gilad S, Meiri E, Yogev Y, Benjamin S, Lebanony D, Yerushalmi N, Benjamin H, Kushnir M, Cholakh H, Melamed N, et al. Serum microRNAs are promising novel biomarkers. PLoS ONE. 2008;3:e3148. doi: 10.1371/journal.pone.0003148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 17.Marton S, Garcia MR, Robello C, Persson H, Trajtenberg F, Pritsch O, Rovira C, Naya H, Dighiero G, Cayota A. Small RNAs analysis in CLL reveals a deregulation of miRNA expression and novel miRNA candidates of putative relevance in CLL pathogenesis. Leukemia. 2008;22:330–338. doi: 10.1038/sj.leu.2405022. [DOI] [PubMed] [Google Scholar]
- 18.Cummins JM, He Y, Leary RJ, Pagliarini R, Diaz LA, Jr, Sjoblom T, Barad O, Bentwich Z, Szafranska AE, Labourier E, et al. The colorectal microRNAome. Proc. Natl Acad. Sci. USA. 2006;103:3687–3692. doi: 10.1073/pnas.0511155103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lui WO, Pourmand N, Patterson BK, Fire A. Patterns of known and novel small RNAs in human cervical cancer. Cancer Res. 2007;67:6031–6043. doi: 10.1158/0008-5472.CAN-06-0561. [DOI] [PubMed] [Google Scholar]
- 20.Seitz H, Ghildiyal M, Zamore PD. Argonaute loading improves the 5′ precision of both MicroRNAs and their miRNA strands in flies. Curr. Biol. 2008;18:147–151. doi: 10.1016/j.cub.2007.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okamura K, Phillips MD, Tyler DM, Duan H, Chou YT, Lai EC. The regulatory activity of microRNA* species has substantial influence on microRNA and 3′ UTR evolution. Nat. Struct. Mol. Biol. 2008;15:354–363. doi: 10.1038/nsmb.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi W, Hendrix D, Levine M, Haley B. A distinct class of small RNAs arises from pre-miRNA-proximal regions in a simple chordate. Nat. Struct. Mol. Biol. 2009;16:183–189. doi: 10.1038/nsmb.1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Langenberger D, Bermudez-Santana C, Hertel J, Hoffmann S, Khaitovich P, Stadler PF. Evidence for human microRNA-offset RNAs in small RNA sequencing data. Bioinformatics. 2009;25:2298–2301. doi: 10.1093/bioinformatics/btp419. [DOI] [PubMed] [Google Scholar]
- 25.Denman RB. Using RNAFOLD to predict the activity of small catalytic RNAs. Biotechniques. 1993;15:1090–1095. [PubMed] [Google Scholar]
- 26.Ender C, Krek A, Friedlander MR, Beitzinger M, Weinmann L, Chen W, Pfeffer S, Rajewsky N, Meister G. A human snoRNA with microRNA-like functions. Mol. Cell. 2008;32:519–528. doi: 10.1016/j.molcel.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 27.Scott MS, Avolio F, Ono M, Lamond AI, Barton GJ. Human miRNA precursors with box H/ACA snoRNA features. PLoS Comput Biol. 2009;5:e1000507. doi: 10.1371/journal.pcbi.1000507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taft RJ, Glazov EA, Lassmann T, Hayashizaki Y, Carninci P, Mattick JS. Small RNAs derived from snoRNAs. RNA. 2009;15:1233–1240. doi: 10.1261/rna.1528909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Piriyapongsa J, Jordan IK. A family of human microRNA genes from miniature inverted-repeat transposable elements. PLoS One. 2007;2:e203. doi: 10.1371/journal.pone.0000203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smalheiser NR, Torvik VI. Mammalian microRNAs derived from genomic repeats. Trends Genet. 2005;21:322–326. doi: 10.1016/j.tig.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 31.Krude T, Christov CP, Hyrien O, Marheineke K. Y RNA functions at the initiation step of mammalian chromosomal DNA replication. J. Cell Sci. 2009;122:2836–2845. doi: 10.1242/jcs.047563. [DOI] [PubMed] [Google Scholar]
- 32.Sim S, Weinberg DE, Fuchs G, Choi K, Chung J, Wolin SL. The subcellular distribution of an RNA quality control protein, the Ro autoantigen, is regulated by noncoding Y RNA binding. Mol. Biol. Cell. 2009;20:1555–1564. doi: 10.1091/mbc.E08-11-1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rutjes SA, van der Heijden A, Utz PJ, van Venrooij WJ, Pruijn GJ. Rapid nucleolytic degradation of the small cytoplasmic Y RNAs during apoptosis. J. Biol. Chem. 1999;274:24799–24807. doi: 10.1074/jbc.274.35.24799. [DOI] [PubMed] [Google Scholar]
- 34.Watanabe T, Totoki Y, Toyoda A, Kaneda M, Kuramochi-Miyagawa S, Obata Y, Chiba H, Kohara Y, Kono T, Nakano T, et al. Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature. 2008;453:539–543. doi: 10.1038/nature06908. [DOI] [PubMed] [Google Scholar]
- 35.Bandres E, Cubedo E, Agirre X, Malumbres R, Zarate R, Ramirez N, Abajo A, Navarro A, Moreno I, Monzo M, et al. Identification by Real-time PCR of 13 mature microRNAs differentially expressed in colorectal cancer and non-tumoral tissues. Mol. Cancer. 2006;5:29. doi: 10.1186/1476-4598-5-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schetter AJ, Leung SY, Sohn JJ, Zanetti KA, Bowman ED, Yanaihara N, Yuen ST, Chan TL, Kwong DL, Au GK, et al. MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. JAMA. 2008;299:425–436. doi: 10.1001/jama.299.4.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl Acad. Sci. USA. 2006;103:2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baffa R, Fassan M, Volinia S, O'Hara B, Liu CG, Palazzo JP, Gardiman M, Rugge M, Gomella LG, Croce CM, et al. MicroRNA expression profiling of human metastatic cancers identifies cancer gene targets. J. Pathol. 2009;219:214–221. doi: 10.1002/path.2586. [DOI] [PubMed] [Google Scholar]
- 39.Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. A census of human cancer genes. Nat. Rev. Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Girard A, Sachidanandam R, Hannon GJ, Carmell MA. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature. 2006;442:199–202. doi: 10.1038/nature04917. [DOI] [PubMed] [Google Scholar]
- 42.Ruby JG, Jan CH, Bartel DP. Intronic microRNA precursors that bypass Drosha processing. Nature. 2007;448:83–86. doi: 10.1038/nature05983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tam OH, Aravin AA, Stein P, Girard A, Murchison EP, Cheloufi S, Hodges E, Anger M, Sachidanandam R, Schultz RM, et al. Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature. 2008;453:534–538. doi: 10.1038/nature06904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Christov CP, Trivier E, Krude T. Noncoding human Y RNAs are overexpressed in tumours and required for cell proliferation. Br. J. Cancer. 2008;98:981–988. doi: 10.1038/sj.bjc.6604254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li W, Duan R, Kooy F, Sherman SL, Zhou W, Jin P. Germline mutation of microRNA-125a is associated with breast cancer. J. Med. Genet. 2009;46:358–360. doi: 10.1136/jmg.2008.063123. [DOI] [PubMed] [Google Scholar]
- 46.Shen J, Ambrosone CB, Zhao H. Novel genetic variants in microRNA genes and familial breast cancer. Int. J. Cancer. 2009;124:1178–1182. doi: 10.1002/ijc.24008. [DOI] [PubMed] [Google Scholar]
- 47.Zhu Z, Gao W, Qian Z, Miao Y. Genetic variation of miRNA sequence in pancreatic cancer. Acta Biochim. Biophys. Sin. (Shanghai) 2009;41:407–413. doi: 10.1093/abbs/gmp023. [DOI] [PubMed] [Google Scholar]
- 48.Tian T, Shu Y, Chen J, Hu Z, Xu L, Jin G, Liang J, Liu P, Zhou X, Miao R, et al. A functional genetic variant in microRNA-196a2 is associated with increased susceptibility of lung cancer in Chinese. Cancer Epidemiol. Biomarkers Prev. 2009;18:1183–1187. doi: 10.1158/1055-9965.EPI-08-0814. [DOI] [PubMed] [Google Scholar]
- 49.Jazdzewski K, Liyanarachchi S, Swierniak M, Pachucki J, Ringel MD, Jarzab B, de la Chapelle A. Polymorphic mature microRNAs from passenger strand of pre-miR-146a contribute to thyroid cancer. Proc. Natl Acad. Sci. USA. 2009;106:1502–1505. doi: 10.1073/pnas.0812591106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nygaard S, Jacobsen A, Lindow M, Eriksen J, Balslev E, Flyger H, Tolstrup N, Moller S, Krogh A, Litman T. Identification and analysis of miRNAs in human breast cancer and teratoma samples using deep sequencing. BMC Med. Genomics. 2009;2:35. doi: 10.1186/1755-8794-2-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 52.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Akao Y, Nakagawa Y, Naoe T. MicroRNAs 143 and 145 are possible common onco-microRNAs in human cancers. Oncol. Rep. 2006;16:845–850. [PubMed] [Google Scholar]
- 55.Qian B, Katsaros D, Lu L, Preti M, Durando A, Arisio R, Mu L, Yu H. High miR-21 expression in breast cancer associated with poor disease-free survival in early stage disease and high TGF-beta1. Breast Cancer Res. Treat. 2009;117:131–140. doi: 10.1007/s10549-008-0219-7. [DOI] [PubMed] [Google Scholar]
- 56.Taylor DD, Gercel-Taylor C. MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol. Oncol. 2008;110:13–21. doi: 10.1016/j.ygyno.2008.04.033. [DOI] [PubMed] [Google Scholar]
- 57.Ng EK, Chong WW, Jin H, Lam EK, Shin VY, Yu J, Poon TC, Ng SS, Sung JJ. Differential expression of microRNAs in plasma of patients with colorectal cancer: a potential marker for colorectal cancer screening. Gut. 2009;58:1375–1381. doi: 10.1136/gut.2008.167817. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}