


Abstract
The ribosome represents a major target for antibacterial drugs. Being a complex molecular machine, it offers many potential sites for functional interference. The high-resolution structures of ribosome in complex with various antibiotics provide a unique data set for understanding the universal features of drug-binding pockets on the ribosome. In this work, we have analyzed the structural and evolutionary properties of 65 antibiotic binding sites (ABSs) in the ribosome. We compared these sites to similar-size computed pockets extracted from the small and large ribosomal subunits. Based on this analysis, we defined properties of the known drug-binding sites, which constitute the signature of a ‘druggable’ site. The most noticeable properties of the ABSs are prevalence of non-paired bases, a strong bias in favor of unusual syn conformation of the RNA bases and an unusual sugar pucker. We propose that despite the different geometric and chemical properties of diverse antibiotics, their binding sites tend to have common attributes that possibly reflect the potency of the pocket for binding small molecules. Finally, we utilized the ensemble of properties to derive a druggability index, which can be used in conjunction with site functionality information to identify new drug-binding sites on the ribosome.
INTRODUCTION
The ribosome is responsible for protein synthesis in all living cells. The bacterial 70S ribosome is formed by the association of the large (50S) subunit and the small (30S) subunit. The small subunit interprets the genetic information encoded in the messenger RNA (mRNA), and the large subunit catalyzes polymerization of amino acids into a polypeptide. The ribosome is an RNA-based machine; the RNA accounts for about two-thirds of the molecular weight of each subunit. The 23S rRNA, ∼2900 nt long, and the 5S rRNA, comprised of ∼120 nt, associate with some 30 proteins to form the large ribosomal subunit. The small subunit contains an ∼1500-nt-long 16S rRNA plus 20 proteins (1). The high-resolution crystallographic and cryo-EM structures of the ribosome and its subunits support the results of many years of genetic and biochemical studies confirming that the main function of the ribosome is promoted by rRNA (2–4).
The ribosome has been an important target for antibiotics, both natural and synthetic (5). Structural and genetic studies show that most of the drugs inhibiting ribosome functions interact directly with rRNA. Despite the ribosome’s enormous size and the diversity of natural and synthetic inhibitors of translation, the majority of clinically effective antibiotics bind to only a few functional sites in the ribosome. For example, most antibiotics that act on the large ribosomal subunit, including diverse antibiotic classes such as phenicols, lincosamides, streptogramins, macrolides, oxazolidinones and others, bind at or near the peptidyl transferase center (PTC), which is located in a cavity on the interface side of the large subunit. The binding of these compounds interferes with the catalysis of peptide bond formation and/or the progression of the nascent peptide toward the exit tunnel. Similarly, several families of aminoglycoside drugs, including important and diverse antibiotics, such as neamine, ribostamycin, neomycin B, gentamycin, tobramycin, paromomycin, etc., interact with the decoding center in the bacterial 16S rRNA. Notably, some of the antibiotics (such as edeine or pactamycin), which broadly affect protein synthesis in all kingdoms of life, are not clinically relevant due to their limited selectivity (6). In addition, it is important to mention that some antibiotics, e.g. tetracyclines, were demonstrated to bind to both a primary (clinically relevant) site as well as to secondary sites which may not be clinically relevant (7).
High evolutionary conservation of many rRNA regions on the ribosome suggests their functional importance (8). Key ribosomal activities have been assigned to several major functional centers. However, the functionality of many other sites in the ribosome remains an enigma. Although it is clear that complex processes, such as translocation, signal transduction, interaction with multiple cellular ligands and many other ribosome-associated activities that are central to protein synthesis, must involve specific sites on the ribosome. In theory, some of these cryptic functional sites could be good candidates for antibiotic action. Indeed, in recent years, the binding of antibiotics to previously unrecognized sites on the ribosome has been reported (9–14). Furthermore, genetic and biochemical screening methods have been employed to reveal ribosomal sites whose structure is essential for protein synthesis (15–17). It remains unknown, however, which of these sites could be targeted by antibiotics.
A number of high-resolution structures of ribosome–antibiotic complexes have been obtained recently using X-ray crystallography. These structures provide fertile ground for attempts to identify common features of antibiotic binding sites (ABSs) and to deduce what distinguishes a ‘true’ ABS from other ‘pockets’ in the ribosome. Moreover, the observed variability in antibiotic binding justifies the search for new sites and improved compounds. Pursuing the general goal of identifying new antibiotic sites in the ribosome, here, we comprehensively analyzed 65 experimentally confirmed ABSs extracted from high-resolution structures of ribosome–drug complexes and identified common characteristics of the drug-binding pockets.
MATERIALS AND METHODS
Pocket extraction and parameter calibration
Extracting known ABS
Structures of Thermus thermophilus and Escheruchia coli 30S, as well as Haloarcula marismortui and Deinococcus radiodurans 50S ribosomal subunits in complex with various ligands (antibiotics) were obtained from the Protein Data Bank (PDB). The complete list of 65 crystal structures of ribosome–antibiotics complexes is presented in Table 1. A binding site was defined to include all atoms within a radius of 6 Å from any atom of the ligand. PDB ID 2HHH with identifier KSG 1524 was excluded from the table due to the low number of nucleotides at its binding site. Furthermore, the atoms of the binding sites were defined in the apo structures of the small ribosomal subunit of T. thermophilus (PDB 1J5E) (18) and E. coli (PDB 2AVY) (19), and the large ribosomal subunit of H. marismortui (PDB 1JJ2) (20) and D. radiodurans (PDB 2ZJR) (21). These structures were then used to calculate the structural properties for centrality measures calculations and for extracting putative pockets in the ribosome, as described below.
Table 1.
Crystallographic structures of T. thermophilus (T.t) and E.coli (E.c) small subunit, and H. marismortui (H.m) and D. radiodurans (D.r) large subunit complexed to antibiotics
| Index | Subunit | Antibiotics | MW (Da) | Organism | PDB | Identifier | Resolution | Refs |
|---|---|---|---|---|---|---|---|---|
| 1 | 30S | Hygromycin B | 527.52 | T.t | 1HNZ | HYG | 3.3 Å | (53) |
| 2 | 30S | Pactamycin | 558.62 | T.t | 1HNX | PCY | 3.4 Å | (53) |
| 3 | 30S | Tetracycline | 444.44 | T.t | 1HNW | TAC 1001 | 3.4 Å | (53) |
| 4 | 30S | Tetracycline | 444.44 | T.t | 1HNW | TAC 1002 | 3.4 Å | (53) |
| 5 | 30S | Tetracycline | 444.44 | T.t | 1I97 | TAC 2001 | 4.5 Å | (7) |
| 6 | 30S | Tetracycline | 444.44 | T.t | 1I97 | TAC 2003 | 4.5 Å | (7) |
| 7 | 30S | Tetracycline | 444.44 | T.t | 1I97 | TAC 2004 | 4.5 Å | (7) |
| 8 | 30S | Tetracycline | 444.44 | T.t | 1I97 | TAC 2005 | 4.5 Å | (7) |
| 9 | 30S | Tetracycline | 444.44 | T.t | 1I97 | TAC 2006 | 4.5 Å | (7) |
| 10 | 30S | Edeine | 781.9 | T.t | 1I95 | EDE | 4.5 Å | (7) |
| 11 | 30S | Spectinomycin | 332.35 | T.t | 1FJG | SCM | 3 Å | (54) |
| 12 | 30S | Streptomycin | 581.57 | T.t | 1FJG | SRY | 3 Å | (54) |
| 13 | 30S | Paromomycinaromomycin | 615.63 | T.t | 1FJG | PAR | 3 Å | (54) |
| 14 | 30S | Kasugamycin | 379.36 | T.t | 2HHH | KSG 1523 | 3.35 Å | (57) |
| 15 | 30S | Hygromycin B | 527.52 | E.c | 3DF1 | HYG | 3.5 Å | (58) |
| 16 | 30S | Spectinomycin | 332.35 | E.c | 2QOU | SCM | 3.93 Å | (59) |
| 17 | 30S | Kasugamycin | 379.36 | E.c | 1VS5 | KSG | 3.5 Å | (60) |
| 18 | 30S | Gentamicin | 449.54 | E.c | 2QB9 | LLL 2356 | 3.54 Å | (10) |
| 19 | 30S | Gentamicin | 449.54 | E.c | 2QB9 | LLL 2357 | 3.54 Å | (10) |
| 20 | 30S | Gentamicin | 449.54 | E.c | 2QB9 | LLL 2357 | 3.54 Å | (10) |
| 21 | 30S | Neomycin | 614.64 | E.c | 2QAL | NMY | 3.21 Å | (10) |
| 22 | 50S | Erythromycin | 733.93 | H.m | 1YI2 | ERY | 2.65 Å | (56) |
| 23 | 50S | Azithromycin | 748.99 | H.m | 1YHQ | ZIT | 2.4 Å | (56) |
| 24 | 50S | Telithromycin | 812.01 | H.m | 1YIJ | TEL | 3.4 Å | (56) |
| 25 | 50S | Quinupristin | 1022.22 | H.m | 1YJW | SYB | 2.9 Å | (56) |
| 26 | 50S | Virginiamycin S | 823.89 | H.m | 1YIT | VRS | 2.8 Å | (56) |
| 27 | 50S | Virginiamycin M | 525.59 | H.m | 1YIT | VIR | 2.8 Å | (56) |
| 28 | 50S | Clindamycin | 424.98 | H.m | 1YJN | CLY | 3 Å | (56) |
| 29 | 50S | Carbomycin | 841.98 | H.m | 1K8A | CAI | 3 Å | (35) |
| 30 | 50S | Spiramycin | 843.05 | H.m | 1KD1 | SPR | 3 Å | (35) |
| 31 | 50S | Tylosin | 916.1 | H.m | 1K9M | TYK | 3 Å | (35) |
| 32 | 50S | Sparsomycin | 361.44 | H.m | 1M90 | SPS | 2.8 Å | (55) |
| 33 | 50S | Chloramphenicol | 323.13 | H.m | 1NJI | CLM | 2.55 Å | (3) |
| 34 | 50S | Anisomycin | 265.31 | H.m | 1K73 | ANM | 3.01 Å | (3) |
| 35 | 50S | Blasticidin S | 422.44 | H.m | 1KC8 | BLS 9001 | 3.01 Å | (3) |
| 36 | 50S | Blasticidin S | 422.44 | H.m | 1KC8 | BLS 9002 | 3.01 Å | (3) |
| 37 | 50S | Homoharringtonine | 545.62 | H.m | 3G6E | HMT | 2.7 Å | (61) |
| 38 | 50S | Bruceantin | 548.58 | H.m | 3G71 | WIN | 3.85 Å | (61) |
| 39 | 50S | Tiamulin | 493.74 | H.m | 3G4S | MUL | 3.2 Å | (61) |
| 40 | 50S | Oxazolidinone | 513.52 | H.m | 3CXC | SLD | 3 Å | (62) |
| 41 | 50S | Girodazole | 190.63 | H.m | 2OTL | GIR | 2.7 Å | (12) |
| 42 | 50S | 13-deoxytedanolide | 594.73 | H.m | 2OTJ | 13T | 2.9 Å | (12) |
| 43 | 50S | Erythromycin | 733.93 | D.r | 1JZY | ERY | 3.5 Å | (63) |
| 44 | 50S | Clindamycin | 424.98 | D.r | 1JZX | CLY | 3.1 Å | (63) |
| 45 | 50S | Clarithromycin | 747.95 | D.r | 1J5A | CTY | 3.5 Å | (63) |
| 46 | 50S | Roxithromycin | 837.05 | D.r | 1JZZ | ROX | 3.8 Å | (63) |
| 47 | 50S | Chloramphenicol | 323.13 | D.r | 1K01 | CLM | 3.5 Å | (63) |
| 48 | 50S | Azithromycin | 748.99 | D.r | 1NWY | ZIT 1 | 3.3 Å | (64) |
| 49 | 50S | Azithromycin | 748.99 | D.r | 1NWY | ZIT 2 | 3.3 Å | (64) |
| 50 | 50S | Cethromycin | 765.93 | D.r | 1NWX | 773 | 3.5 Å | (64) |
| 51 | 50S | Telithromycin | 812.01 | D.r | 1P9X | TEL | 3.4 Å | (65) |
| 52 | 50S | Quinupristin | 1022.22 | D.r | 1SM1 | SYB | 3.42 Å | (66) |
| 53 | 50S | Dalfopristin | 690.85 | D.r | 1SM1 | DOL | 3.42 Å | (66) |
| 54 | 50S | Sparsomycin | 361.44 | D.r | 1NJN | SPS | 3.7 Å | (67) |
| 55 | 50S | Troleandomycin | 813.97 | D.r | 1OND | TAO | 3.4 Å | (68) |
| 56 | 50S | Tiamulin | 493.74 | D.r | 1XBP | MUL | 3.5 Å | (69) |
| 57 | 50S | Thiostrepton | 1664.89 | D.r | 3CF5 | TXX | 3.3 Å | (21) |
| 58 | 50S | Oxazolidinone | 337.35 | D.r | 3DLL | ZLD | 3.5 Å | (70) |
| 59 | 50S | SB-571519 | 498.57 | D.r | 2OGM | G19 | 3.5 Å | (45) |
| 60 | 50S | SB-280080 | 477.70 | D.r | 2OGN | G80 | 3.56 Å | (45) |
| 61 | 50S | SB-275833 | 517.76 | D.r | 2OGO | G34 | 3.66 Å | (45) |
| 62 | 50S | Rapamycin | 914.17 | D.r | 1Z58 | RAP | 3.8 Å | (13) |
| 63 | 50S | Josamycin | 828.00 | D.r | 2O44 | JOS | 3.3 Å | (71) |
| 64 | 50S | RU-69874 | 972.22 | D.r | 2O45 | RU6 | 3.6 Å | (71) |
| 65 | 50S | Erythromycylamine | 734.96 | D.r | 2O43 | ERN | 3.6 Å | (71) |
Extracting putative pockets in the ribosome
The program ‘solvent’ from the 3V package (http://geometry.molmovdb.org/3v/) was applied to identify putative pockets in the ribosome (22). Briefly, the program gets as an input a PDB file and the radius of a small and a large probe sphere, and outputs the void in the ribosome structures that can accommodate a small probe size but not a large probe size. The void itself, which corresponds to a possible binding pocket, is represented by the oxygen atoms of water molecules that define the borders of the void (i.e. the surface of the pocket). The program was applied to the PDB IDs 1J5E, 2AVY, 1JJ2 and 2ZJR files, excluding ions and including all ATOM coordinates (rRNA and ribosomal proteins). Grid spacing was set to 1 Å.
Small probe and large probe radii calibration
The calibration procedure was applied to define the parameters generating computed pockets that best fit the average dimension of the known binding pockets (Table 1). The calibration procedure was performed individually for each of the 11 selected known binding pockets that have ∼30% atoms in common (Table 1, indices; 2, 3, 4, 11, 12, 13, 23, 25, 31, 34 and 35).
Previous studies have shown that the large ribosomal subunit (PDB 1JJ2) contains no cavities big enough to enclose a sphere having a radius greater than 9.5 Å. Thus, in the first round of calibration, the large probe radius was fixed to 9.5 Å, while the small probe radius was gradually reduced (1 Å decline) in the 1.5–8.5 Å range. After each individual run of the ‘solvent’ algorithm with a defined set of parameters, the surface of the pockets was delineated by the coordinates of computed oxygen atoms of water molecules. The relativity between a computed pocket and a known binding site was described by the minimal distance between the computed oxygen atoms and the center of mass of all atoms included in the known binding sites. This procedure was repeated for each known ABS, testing the full range of small probe radii in order to define the optimal parameters for which the shortest distance between known and computed pockets was reached. Finally, a small probe radius of 1.5 Å was chosen as the optimal radius that gave the best results for all known binding pockets tested (Supplementary Table S1). The size of the small probe indicates accessibility of the computed pockets to solvent water molecules.
To decrease the number of computed binding pockets and eliminate pockets that do not match the dimension of known binding sites, a second round of calibration was performed. In the second round, only the size of the large probe radius was decreased, while the small probe radius was fixed to 1.5 Å. Finally, after gradually decreasing the large probe, a radius of 5.5 Å was chosen, keeping the distance from the computed pocket to the known binding sites at a minimum (Supplementary Table S2).
Size of the computed pockets
To define the computed pocket boundaries, we examined various radii cutoffs around the computed oxygen atoms (Supplementary Table S3). For each radius, we defined the pocket size as the number of nts it comprises, i.e. a nt was considered in the computed pocket if it had at least one atom within a defined radius. A radius of 9 Å around the computed oxygen atoms was chosen, as it resulted in approximately the same average number of nts as in the known binding sites (Supplementary Table S3). Finally, we retained only pockets that comprise 12 nt and differ in at least 1 nt, resulting in 17 610 (calculated for PDB 1J5E and 1JJ2) pockets that we refer to as the background. The number of pockets calculated for all structures used in this study is listed in Supplementary Table S4.
Specificity and sensitivity of binding site identification
Specificity and sensitivity were calculated based on the degree of overlap between the computed pockets and the known binding sites (Supplementary Table S5). ‘Overlapping specificity’ equals the fraction of common nts in the putative and the known pockets divided by the number of nts in the known binding site. ‘Overlapping sensitivity’ equals the fraction of common nts divided by the number of nts included in the putative (computed) pocket.
Physicochemical properties
The nitrogen and oxygen atoms in the base were classified as donors or acceptors. Backbone oxygen atoms were labeled as sugar or phosphate.
Structural properties
For the RNA structural properties classification, including base-pairing (23,24) and base-base stacking interactions (25), the MC-Annotate (version 1.6.2) program (26) was used. Base-pairing was defined according to three edges of the corresponding RNA bases available for H-bonding interactions, labeled the Watson–Crick (W) edge, the Hoogsteen edge (H) and the Sugar edge (S). For more details see refs. 25 and 28. Base–base stacking interactions were categorized as stacking interactions between adjacent and non-adjacent nts. For more details see ref. 27.
The features were extracted as described in ref. 28 using an in-house Perl script converting the MC-Annotate output files into binary format, i.e. each nt was given a score of ‘1’ when a specific property was present and a score of ‘0’ when it was absent. To calculate the relative abundance of a specific property, the fraction of nts in the pocket having this property was calculated. In order to generate the signature of the binding sites (Table 1), only properties covering at least 1% of all rRNA nts were included.
Functional sites
Interface residues of the 16S rRNA and 23S rRNA with ribosomal proteins mRNA and tRNA were calculated using the Intervor web server (http://cgal.inria.fr/abs/Intervor) (29) with default parameters. The interface with ribosomal proteins was calculated on the PDB IDs 1JJ2 and 1J5E. The interface with the tRNA and mRNA was calculated on the PDB IDs 2J00 and 2J01 (30). The A-minor motif annotation (PDB ID’s 1VQO, 2J00) was downloaded from http://www.biomath.nyu.edu/motifs/rrna.html.
Centrality measures
We modeled the 16S rRNA (PDB IDs 1J5E, 2AVY) and the 23S rRNA (PDB IDs 1JJ2, 2ZJR) as a structure-based network of interacting nts (31). Briefly, each nt was considered as a node in the network, and chemical interactions between nts defined the edges. The degree of a node i is defined as the number of edges connected to i. The betweenness of a node i is defined by the number of shortest paths that cross node i. The closeness of a node i is defined as the inverse average length of the shortest paths to all other nodes in the graph (32). The closeness of node i is given in Equation (1)
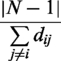 |
(1) |
where N is the total number of nodes in the network, and dij is the shortest path length to node i. For each known binding site and computed pocket, we calculated the average closeness, betweenness and degree.
Evolutionary conservation
Aligned sequences and a guide tree for both the 16S/18S and the 23S/28S were downloaded from the ARB-SILVA database (release 96) (http://www.arb-silva.de). The 16S/18S alignment comprises high-quality sequences, with a minimum length of 1200 bases for bacteria and eukarya and 900 bases for archaea. The 23S/28S alignment comprises high-quality rRNA sequences with a minimum length of 1900 bases. Positional variability in bacteria was calculated with the ARB package using parsimony function (37). The score of the evolutionary rate was multiplied by −1 to create values of evolutionary conservation.
Statistical analysis
The relative abundance of each property in a known site was evaluated relative to a background of putative pockets. Since we could not assume normality, an individual score (ui) of each property was standardized as given in Equation (2):
| (2) |
MAD denotes the median absolute deviation, given by Equation (3):
| (3) |
The statistical significance of the structural properties and 16 types of base pairing presentation in ABSs was evaluated based on the hyper geometric distribution using the Fisher’s exact test.
RESULTS AND DISCUSSION
Identifying putative ligand-binding pockets in the ribosome
To study the distinguishing characteristics of ABSs in the ribosome, we sought to compare the properties of known ABSs (Table 1) to a collection of computed structural pockets having similar dimensions. We defined a ‘known ABS’ as all rRNA atoms within 6 Å from any atom of the ligand (antibiotic) observed in the crystal structure of an antibiotic-ribosome complex. We then selected the coordinates of the corresponding rRNA atoms in the apo-structure, which represented the unbound drug-binding pocket (see ‘Materials and Methods’ section). To generate the collection of structural pockets in both ribosomal subunits, we first calibrated the parameters of the pocket-extraction protocol to correctly identify the known ABSs, and then used these parameters to identify all other similar pockets. Since the majority of antibiotics that bind the ribosome interact mostly with the ribosomal RNA (rRNA) (33), we chose to concentrate on pockets that consist exclusively of rRNA. The location and composition of the pockets were computed using the ‘small and the large probe spheres’ approach, previously applied for identifying ligand-binding sites in proteins (34). Here, we defined a pocket as a volume in the ribosome into which a small probe can enter but a large probe cannot. For automatic extraction of the pockets, we adopted the program ‘solvent’ from the 3V package (http://geometry.molmovdb.org/3v/) on a 1 Å grid space (22).
Testing the pocket extraction procedure
A plausible way of evaluating the pocket extraction procedure would be to examine the degree of inclusion of the known ABSs in the collection of all computed pockets (Supplementary Tables S1–S4). To achieve a maximum overlap between the extracted pockets and the known binding sites, we applied a calibration procedure that tested different small and large probe radii (see ‘Materials and Methods’ section). After selecting the optimal probe radii (Supplementary Tables S1–S2), we set the size of the computed pocket to be equal to the average number of nts in the known ABS (Supplementary Table S3). Finally, we defined the optimal parameters for the pockets (1.5 Å for the small probe radius, 5.5 Å for the large probe radius, and a pocket size of 12 nts). These parameters yielded a minimal number of computed pockets that provides a maximal overlap with the known ABSs. Overall, the overlapping values between the known and computed pockets were in the range between 50% and 100 % (Supplementary Table S5). The high accuracy of pocket extraction is exemplified in Figure 1, which illustrates the overlap between a computed pocket and the known ABS of tetracycline in the 16S rRNA (Figure 1A), and the erythromycin binding site in the 23S rRNA (Figure 1B). The high overlap between computed and known sites indicates that the computational procedure used in this study is suitable for identifying putative pockets with similar dimensions to the known ABSs.
Figure 1.

Overlapping between the computed pockets and the observed binding sites. (A) Tetracycline bound to its primary binding site (53). The rRNA molecular surface (PDB ID 1HNW) is presented in grey; tetracycline is presented as blue sticks. The computed pocket showing the highest overlapping sensitivity (67%) and specificity (89%) is colored yellow. The pocket was calculated on the crystal structure of T. thermophilus (PDB ID 1J5E) and was superimposed to the bound structure (PDB ID 1HNW). (B) Erythromycin binding pocket (56). The rRNA molecular surface (PDB ID 1YI2) is presented in gray; erythromycin is presented as blue sticks. The computed pocket showing the highest overlapping sensitivity (75%) and specificity (82%) is colored yellow. The pocket was calculated on the crystal structure of H. marismortui, (PDB ID 1JJ2), and was superimposed to the bound structure (PDB ID 1YI2).
Having applied the procedure to the apo-structures of the T. thermophilus small ribosomal subunit (PDB ID 1J5E) and the H. marismortui large subunit (PDB ID 1JJ2), we extracted a total of 17 610 overlapping pockets that differ in at least 1 nt (Supplementary Table S4). The large number of pockets is not surprising given the enormous size of the ribosome, and is consistent with the notion that the RNA component of the ribosome presents a large network of interconnected channels that are readily accessible to small ligands (22). Furthermore, the discrepancy between the large number of putative pockets and the number of experimentally defined ABSs implies that the dimension parameter alone is not a sufficient criterion to explain the specific binding of antibiotics to the rRNA.
Properties of the drug-binding pockets
Despite the high number of potential binding pockets for small molecules in the ribosome, known ribosomal inhibitors act on a limited number of sites. In order to identify the properties distinguishing a real ABS from other random pockets on the ribosome (Table 1), we computed a set of physical, chemical and structural features (listed in Supplementary Table S6), as well as the extent of evolutionary conservation for both the experimentally validated ABSs and for all computed pockets. Clearly, apart from evolutionary conservation, a very important feature that is expected from clinically affective antibiotic binding sites is their specificity to the drug (i.e. the binding site in pathogenic bacteria should differ from the corresponding site in the host ribosome). However, since bacterial specificity usually boils down to a single nucleotide [as in the macrolide binding pockets (35)] which could not be identified in our statistic-based method and is not expected from all ABSs that we have included in our analysis (i.e. the archaeal ABSs), we did not incorporate specificity among the computed parameters. The selected features (Supplementary Table S6) were further compared between known binding sites and all computed pockets. To further test whether the properties that over represented in ABSs in of both the T. thermophilus, and H. marismortui are unique to druggable sites, we examined their abundance in both ribosomal protein-binding interfaces and in sites that consist of ribosome–ligands interactions. Specifically, we included nucleotides that interact with the mRNA and the tRNA, i.e. the A, P and E sites (see ‘Materials and Methods’ section). The properties of the 65 known binding sites, which exhibit statistically significant bias compared to all computed pockets, are presented in Figure 2 and are discussed in more detail in the subsequent paragraphs.
Figure 2.

The ‘RNA signature’ of antibiotic binding sites. Binding site indices are presented in Table 1. The color scheme refers to the standardized score against a background of 17 610 computed pockets [‘Materials and Methods’ section, Equation (3)]. Scores were scaled to range from −1 to 1. Significant biases relative to the background median are colored red and blue, respectively. (A) Physicochemical properties, (B) Nucleotides types, (C) Structural classification according to the program MC-annotate (26), (D) Centrality measures, (E) Evolutionary conservation. For convenience, the Watson–Crick edge was abbreviated to W, the sugar edge to S and the Hoogsteen edge to H.
RNA nucleotides and bases
When analyzing the structural composition of the known ABSs in comparison to the computed pockets, we noticed an increased representation of atoms belonging to the nitrogen bases rather than atoms of the sugar-phosphate backbone. This phenomenon was prominent specifically in binding sites of the large ribosomal subunit (Figure 2A, indices 22–63). The overabundance of atoms of the nitrogen bases in the ABSs may reflect the known sequence specific interactions of many antibiotics with the ribosome that form the basis of selectivity of clinically important medicines. Nevertheless, we did not identify a statistically significant preference for a specific type of nitrogen base or an explicit donor/acceptor group in the known ABSs (Figure 2A). Upon examining the occurrence of the 16 possible base pairs (26) in ABSs (see ‘Materials and Methods’ section), we noticed a significant overrepresentation of the non canonical UC base pair and an underrepresentation of the canonical UA pair in the 23S rRNA binding sites. Furthermore, the GC was underrepresented in ABSs in both 16S and 23S rRNA. Notably, such bias was specific to ABSs and was not observed in the ribosomal protein-binding interface, while the GC and UA underrepresentations were found in the natural ligand-binding sites in the large subunit (Supplementary Table S7).
Structural properties
The most apparent bias in the characteristics of the known ABSs was the overrepresentation of non-paired bases (Figure 2C). This phenomenon was more prominent in the large ribosomal subunit (Supplementary Table S7 and S8). Non-paired bases obviously offer a greater variation in the chemical groups available for structure-specific interaction with the drug (or natural ligand) compared to the nitrogen bases engaged in the inter-nucleotide interactions. Therefore, rRNA pockets with the higher representation of non-paired bases could be evolutionary selected as preferred ABSs. We further analyzed various types of interactions between the nts in the pockets. Interactions were annotated according to three edges of H-bonding interactions [Watson–Crick (W)/Hoogsteen (H)/Sugar (S)] (24,27). Further, base–base interactions were categorized in terms of stacking interactions between adjacent and non-adjacent nts (26). We observed a bias to the non-canonical W-H pairing in the known ABSs. Additionally, we noted an overrepresentation of non-adjacent base–base stacking interactions in the large ribosomal subunit (Figure 2C).
The A-minor motif, which involves inserting the edges of adenine bases into the minor groove of RNA helices, was previously found to be the most abundant structural motif in the large ribosomal subunit (36–39). Consistent with previous observations showing the importance of the A-minor motif in functional sites (51), we found that a considerable number (Supplementary Table S9) of ABSs, specifically in the 16S rRNA, comprise diverse elements of the A-minor motif. Overall, the results of structural analysis of RNA bases in the ABSs are consistent with the notion that antibiotic binding, both in the large and small subunits, involves RNA nts belonging to single-stranded RNA regions or distortions in the RNA helices such as in bulges, loops, etc. (40).
Interestingly, we also observed a preference for syn over anti conformation of the RNA bases in the majority of known ABSs (Supplementary Tables S7 and S8). Syn and anti conformations are defined by the glycosidic torsion angle χ. It was previously shown that the syn conformation has higher energies than the anti conformation in pyrimidine nucleosides (41). The sugar conformation is known to have a great effect on the distribution of glycosidic torsion angles. Specifically, the C2 endo sugar pucker provides less steric hindrance than the C3 endo, allowing a wider range of χ values and less energy cost for a syn conformation (19,30). Consistent with the latter, we found that the ribose sugar pucker in C3 endo conformation was underrepresented in the vast majority of ABSs. The relative overabundance of the unusual sugar pucker conformation (both C2 endo and C4 exo) and syn conformation of RNA bases in ligand-binding sites could be associated with the higher flexibility of rRNA in these sites (42,43). The flexibility of the glycosidic bonds of the bases in the antibiotic binding sites of the large ribosomal subunit (specifically at the PTC and the tunnel region) was recently suggested to explain antibiotic selectivity and action (44). Possibly during evolution, antibiotics were selected to target flexible RNA pockets that are capable of the induced fit-binding mode (45). Consistent with the result in the 23S rRNA, a preference for non-paired bases and the syn conformation was also observed in the 16S rRNA (Figure 2).
Interestingly, we observed that stacking interactions (both adjacent and non-adjacent) are underrepresented in the ribosome–ligand-binding interface, but the non-adjacent interactions are overrepresented in both the ABSs and protein binding of the 23S rRNA (Supplementary Table S7). Remarkably, in the protein-binding interface, we could not identify bias to the non-paired bases, Watson–Hoogsteen pairing, syn conformation, or irregular sugar pucker. Comparison of structural properties of known antibiotic sites between the T. thermophilus and Escherichia coli ribosomes (34) showed that the preference for non-paired nts, non-adjacent base-base stacking interactions, and high occurrence of syn conformation appear to be conserved between ribosomes of different bacterial species (Figure S1). The conservation of unique properties in the binding pockets of these species emphasizes the importance of the unique structural properties to the function of these sites.
Centrality and evolutionary conservation
Recent studies have employed the network (graph) approach to analyze unique properties of macromolecule structures (31,46–48). In such structure-based networks, the amino-acids/bases are represented as nodes, and inter-residue interactions as edges. In a previous study, we showed that the centrality parameters (i.e. closeness, betweenness and degree) were highly correlated with functionality of the nt in the ribosome (31). As demonstrated in Figure 2D, we found that the nts in most known ABSs are characterized with high centrality values, and a specifically high closeness. When we further calculated the evolutionary conservation for the three domains of life (bacteria, archaea and eukarya) using the ARB-SILVA database (49), a high degree of conservation was observed in most ABSs. This observation is in accord with previously noted evolutionary conservation of rRNA in the functional centers of the ribosome (Figure 2E) (50).
Summarizing our findings discussed in the previous paragraphs, we note that the known sites of antibiotic binding in the ribosome are preferentially endowed with specific structural properties. Characteristics such as the prevalence of the A-minor motif (42,51) and high centrality values (31) have been previously shown to be associated with ribosome functional sites. Therefore, we tested whether the other structural properties distinguishing the ABSs could be attributable to the functional sites in general. Upon selecting a subset of top 25% highly conserved nts, we found that they exhibit a property bias similar to that found for the antibiotic sites, such as overabundance of non-paired bases, unusual sugar pucker and prevalence of syn conformation (Figure 3). These results are also consistent with the above analysis of mRNA and the A, P and E sites, in which we observed bias in favor of non-paired bases and the syn conformation, while an underrepresentation was observed in the C3 endo conformation and canonical Watson–Crick base pairing (GC) (Supplementary Table S7). Likely, the bias we observed in ABSs reflects the unique structural properties of functional sites on the ribosome that contribute to the high affinity binding of their natural ligands.
Figure 3.

Overrepresented and underrepresented properties in highly conserved sites (top 25%). The 16S rRNA is presented in grey bars. The 23S rRNA is presented in black bars. Bar height represents the statistical significance shown by the –log10 of the P-value. The red line marks the level of statistical significance (P = 0.05).
New antibiotic sites for future drug design
Knowledge of the properties of known ABSs can illuminate which other pockets in rRNA could serve as binding sites for new antibiotics. However, clearly further research on site functionality and specificity is required. In order to estimate whether a computed pocket can be a potential drug-binding site, we assigned a ‘druggability index’ (DI) score [Equation (4)] to each computed pocket. DI weights the properties that were found significantly biased in the known ABS in H. marismortui 50S subunit (Table 1) as follows:
| (4) |
where x is a property presented in Supplementary Table S7, Px is the P-value of property x, and Fx is the fraction of nts in the computed pocket with property x. It is important to note that the DI was derived from the preferences for specific structural properties we observed mainly in the large subunit (Figure 2). As a result, we applied the DI to identify new structural pockets exclusively in the large ribosomal subunit. Figure 4 plots the DI values of computed pockets in the large ribosomal subunit with DI >0 (comprising 63% of all computed pockets) versus their calculated average evolutionary conservation, usually employed to identify functional sites.
Figure 4.

The ‘druggability index’ (DI) versus evolutionary conservation. DI and evolutionary conservation values were rescaled from 0 to 1. All pockets with DI >0 (7482 pockets) calculated from the H. marismortui large ribosomal subunit (PDB ID 1JJ2) are shown; pockets overlapping known ABSs are colored red. Pockets including a deleterious mutation are colored blue. Pockets that include a deleterious mutation and are located outside the PTC region are colored cyan.
As shown, while many computed sites in the 23S rRNA possess a very high conservation (demonstrated by the high density of dots in the right-hand side of Figure 4), we found that the computed pockets that overlap with the known ABSs are both highly conserved and possess a high DI score, which mainly reflects their unique structural properties (Figure 4, red dots). Interestingly, among the known sites of antibiotic action, the computed site that overlapped the macrolides binding pocket in the nascent peptide exit tunnel had the highest DI value. The top four ranked computed pockets (with the highest DI scores) are presented in Figure 5. Three of them overlap experimentally validated ABSs. Pockets 1 and 2 (Figure 5A and B) overlap the binding sites of the macrolides and ketolides antibiotics. The third top-ranking pocket (Figure 5C) is located near the ribosomal E site and overlaps the binding site of girodazole (12). The site ranked in fourth place (Figure 5D) is located about 12 Å from the nascent peptide tunnel close to the loop of ribosomal protein L22. This computed site comprises a methylated residue m6A1618 (52) and is not targeted by any antibiotic currently in use. The overall distribution of the DI score over the 23S rRNA molecular surface is presented in Figure 6. As demonstrated, very high DI values were obtained in the vicinity of the PTC and the E-site region. A zoom to the E-site region showing a view into the exit tunnel is also presented. As shown, the red (high DI) patch decorating the tunnel walls is the region of macrolides-binding sites. Notably, other regions of the large ribosomal subunit demonstrate patches of high DI values, suggesting other potential binding sites.
Figure 5.

Binding pockets on the ribosome with the highest ‘druggability index’ score. The rRNA molecular surface is shown in gray. The computed pocket surface is shown in yellow. (A) The computed pocket with the highest DI score (DI = 1.00). Erythromycin is presented as blue sticks (PDB ID 1YI2). (B) The computed pocket ranked in second place (DI = 0.98). Telithromycin is presented as blue sticks (PDB ID 1YIJ). (C) The computed pocket ranked in third place (DI = 0.94). Girodazole is presented as blue sticks (PDB ID 2OTL). (D) The computed pocket ranked in fourth place (DI = 0.93).
Figure 6.

Druggability Index (DI) projected on the 23S rRNA molecular surface (PDB ID 1JJ2). A view into the exit tunnel is shown in the box. Low and high DIs are colored blue and red, respectively. Color bar is shown.
To test the validity of the DI score in clinically relevant sites, we examined the DI scores of the tetracycline computed pockets in the small subunit that comprise both the clinically relevant primary site and additional sites that may include non specific interactions with tetracycline (53). Interestingly, of all sites the highest DI was calculated for the primary binding site (1HNW, TAC1001, DI = 0.72; 1I97, TAC2001, DI = 0.65); this score was higher (Supplementary Table S10) than the secondary site (1HNW TAC1002, DI = 0.59) and the other observed sites (1I97) (7). To further evaluate the potential of DI to detect ABSs, we concentrated on sites possessing a DI value that fell in the range of the DI of known ABSs (0.44<DI<0.98). These sites accounted for 21% (i.e. 2487 pockets including the 606 known ABSs). Overall, we indentified 1181 overlapping pockets on the 23S rRNA as potential new druggable sites. Interestingly, several of them overlap our previously detected deleterious point mutations (16,17) (Figure 4, cyan and blue dots). As shown in Figure 4, the conservation and DI of these pockets resemble the values of known ABSs. Two 23S rRNA sites outside PTC, the sarcin–ricin loop (centered around nt 2664) and the internal loop in the A-site finger (around nt 864) are characterized by clusters of deleterious mutations that make them attractive candidates for drug development. Of these two sites, the sarcin–ricin loop pocket has a higher DI (DI = 0.68) than the A-site finger pocket (DI = 0.49). Yet, the DI values of both sites are high enough to place both of them in the list of potentially druggable ribosomal sites.
Figure 7 further demonstrates the potential to combine the DI with the existence of structural motifs preferentially found in known binding pockets, such as the A-minor motif. An example of such a putative site was found in the A-site finger. The pocket was computed from the H. marismortui structure (PDB 1JJ2) and has a DI value of 0.49. This pocket consists of 12 nt, six which form non-adjacent base–base stacking interactions. Additionally, the pocket has a UC mismatch base pair (Figure 6; positions 913, 914), and an unpaired adenine in position 910, which participates in an A-minor motif interaction. The nts that were identified as A-minor motif elements refer to positions 862 and 863, which form interactions with adenines in the 5S rRNA. As discussed above, these properties were commonly found in functional sites as well as in known ABSs. The functional role of this highly conserved region is also denoted by the presence of a strong deleterious mutation (position 864) (17). Although the function of this region is unclear due to its high DI value and overall functional characteristics, we suggest that this pocket could be a good candidate for a novel drug.
Figure 7.

Example of a computed pocket in H. marismortui with ‘druggable’ properties. The 12 nt of the computed pockets are labeled (E. coli numbering) and presented in balls and sticks. Six nucleotides in the pocket form non-adjacent stacking interactions. Positions 913 and 914 refer to UC base pairs. The nucleotides corresponding to the strong deleterious mutation in position 864 is colored red. The pocket includes nts that involve A-minor motifs (positions 862, 863 and 910).
CONCLUSIONS
In an attempt to better understand the atomic and structural features that could elucidate the druggability of ABSs on the ribosome, we studied the properties of 65 antibiotic binding pockets extracted from the available high-resolution structures of ribosome-antibiotic complexes (3,4,7,12,35,53–56). In addition, we applied an automatic method to compute structural pockets from the T. thermophilus and H. marismortui small and large ribosomal subunits. Overall, we extracted ∼17 000 overlapping computed pockets on the ribosome, all in the same range of dimensions as the known binding sites. The enormous number of computed binding sites clearly shows that the ribosome, which is comprised of a large network of interconnected channels, is much more accessible to small ligands than traditional protein drug targets. However, to date, only a small number of possible binding pockets in the ribosome has been verified to bind antibiotics.
A comprehensive analysis of the physicochemical and structural (local and global) features of RNA in the known antibiotic binding pockets revealed that while the sites differ in their dimension and atomic details, they share several unique properties, namely an ‘RNA signature’. The most noticeable structural properties found in the majority of known binding sites were the prevalence of non-paired bases, preference for the unusual syn base conformation and unusual ribose sugar pucker. We postulate that the unique base conformation and sugar pucker may reflect higher flexibility of the rRNA bases in the ABSs as in natural ligands. We believe that the properties we identified, specifically the suggested DI, will facilitate the identification of new functional sites suitable for drug design. Clearly, other considerations such as bacterial specificity of the site and experimental validation of site functionality will be required. Furthermore, we believe that a similar approach could be implemented for characterizing and identifying other ligand-binding sites in non-ribosomal RNA targets (such as non-coding RNAs) for which selective ligands are extremely scarce or do not exist.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The United States, Israel Binational Science Foundation (Grant Number 2007453 to Y.M.G. and A.S.M.); Israeli Science Foundation (Grant number 1297/09 to Y.M.G.); and by the J. and A. Taub Biological Research (H.D.E.). Funding for open access charge: The United States, Israel Binational Science Foundation (grant Number 2007453).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENT
We would like to thank Ada Yonath for her inspiring ideas and helpful suggestions throughout the course of this study. Many thanks to Anat Bashan and Chen Davidovich for their help in generating the ABSs list and for their many helpful suggestions.
REFERENCES
- 1.Ramakrishnan V. Ribosome structure and the mechanism of translation. Cell. 2002;108:557–572. doi: 10.1016/s0092-8674(02)00619-0. [DOI] [PubMed] [Google Scholar]
- 2.Green R, Noller HF. Ribosomes and translation. Annu. Rev. Biochem. 1997;66:679–716. doi: 10.1146/annurev.biochem.66.1.679. [DOI] [PubMed] [Google Scholar]
- 3.Hansen JL, Moore PB, Steitz TA. Structures of five antibiotics bound at the peptidyl transferase center of the large ribosomal subunit. J. Mol. Biol. 2003;330:1061–1075. doi: 10.1016/s0022-2836(03)00668-5. [DOI] [PubMed] [Google Scholar]
- 4.Ogle JM, Brodersen DE, Clemons WM, Jr, Tarry MJ, Carter AP, Ramakrishnan V. Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science. 2001;292:897–902. doi: 10.1126/science.1060612. [DOI] [PubMed] [Google Scholar]
- 5.Wilson DN. The A-Z of bacterial translation inhibitors. Crit Rev Biochem. Mol. Biol. 2009;44:393–433. doi: 10.3109/10409230903307311. [DOI] [PubMed] [Google Scholar]
- 6.Yonath A. Antibiotics targeting ribosomes: resistance, selectivity, synergism and cellular regulation. Annu. Rev. Biochem. 2005;74:649–679. doi: 10.1146/annurev.biochem.74.082803.133130. [DOI] [PubMed] [Google Scholar]
- 7.Pioletti M, Schlunzen F, Harms J, Zarivach R, Gluhmann M, Avila H, Bashan A, Bartels H, Auerbach T, Jacobi C, et al. Crystal structures of complexes of the small ribosomal subunit with tetracycline, edeine and IF3. EMBO J. 2001;20:1829–1839. doi: 10.1093/emboj/20.8.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cannone JJ, Subramanian S, Schnare MN, Collett JR, D'Souza LM, Du Y, Feng B, Lin N, Madabusi LV, Muller KM, et al. The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinform. 2002;3:2. doi: 10.1186/1471-2105-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belova L, Tenson T, Xiong L, McNicholas PM, Mankin AS. A novel site of antibiotic action in the ribosome: interaction of evernimicin with the large ribosomal subunit. Proc. Natl Acad. Sci. USA. 2001;98:3726–3731. doi: 10.1073/pnas.071527498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borovinskaya MA, Pai RD, Zhang W, Schuwirth BS, Holton JM, Hirokawa G, Kaji H, Kaji A, Cate JH. Structural basis for aminoglycoside inhibition of bacterial ribosome recycling. Nat. Struct. Mol. Biol. 2007;14:727–732. doi: 10.1038/nsmb1271. [DOI] [PubMed] [Google Scholar]
- 11.Johansen SK, Maus CE, Plikaytis BB, Douthwaite S. Capreomycin binds across the ribosomal subunit interface using tlyA-encoded 2'-O-methylations in 16S and 23S rRNAs. Mol. Cell. 2006;23:173–182. doi: 10.1016/j.molcel.2006.05.044. [DOI] [PubMed] [Google Scholar]
- 12.Schroeder SJ, Blaha G, Tirado-Rives J, Steitz TA, Moore PB. The structures of antibiotics bound to the E site region of the 50 S ribosomal subunit of Haloarcula marismortui: 13-deoxytedanolide and girodazole. J. Mol. Biol. 2007;367:1471–1479. doi: 10.1016/j.jmb.2007.01.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amit M, Berisio R, Baram D, Harms J, Bashan A, Yonath A. A crevice adjoining the ribosome tunnel: hints for cotranslational folding. FEBS Lett. 2005;579:3207–3213. doi: 10.1016/j.febslet.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 14.Brandi L, Fabbretti A, La Teana A, Abbondi M, Losi D, Donadio S, Gualerzi CO. Specific, efficient, and selective inhibition of prokaryotic translation initiation by a novel peptide antibiotic. Proc. Natl Acad. Sci. USA. 2006;103:39–44. doi: 10.1073/pnas.0507740102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laios E, Waddington M, Saraiya AA, Baker KA, O'Connor E, Pamarathy D, Cunningham PR. Combinatorial genetic technology for the development of new anti-infectives. Arch. Pathol. Lab Med. 2004;128:1351–1359. doi: 10.5858/2004-128-1351-CGTFTD. [DOI] [PubMed] [Google Scholar]
- 16.Yassin A, Fredrick K, Mankin AS. Deleterious mutations in small subunit ribosomal RNA identify functional sites and potential targets for antibiotics. Proc. Natl Acad. Sci. USA. 2005;102:16620–16625. doi: 10.1073/pnas.0508444102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yassin A, Mankin AS. Potential new antibiotic sites in the ribosome revealed by deleterious mutations in RNA of the large ribosomal subunit. J. Biol. Chem. 2007;282:24329–24342. doi: 10.1074/jbc.M703106200. [DOI] [PubMed] [Google Scholar]
- 18.Wimberly BT, Brodersen DE, Clemons WM, Jr, Morgan-Warren RJ, Carter AP, Vonrhein C, Hartsch T, Ramakrishnan V. Structure of the 30S ribosomal subunit. Nature. 2000;407:327–339. doi: 10.1038/35030006. [DOI] [PubMed] [Google Scholar]
- 19.Schuwirth BS, Borovinskaya MA, Hau CW, Zhang W, Vila-Sanjurjo A, Holton JM, Cate JH. Structures of the bacterial ribosome at 3.5 A resolution. Science. 2005;310:827–834. doi: 10.1126/science.1117230. [DOI] [PubMed] [Google Scholar]
- 20.Klein DJ, Schmeing TM, Moore PB, Steitz TA. The kink-turn: a new RNA secondary structure motif. EMBO J. 2001;20:4214–4221. doi: 10.1093/emboj/20.15.4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harms JM, Wilson DN, Schluenzen F, Connell SR, Stachelhaus T, Zaborowska Z, Spahn CM, Fucini P. Translational regulation via L11: molecular switches on the ribosome turned on and off by thiostrepton and micrococcin. Mol. Cell. 2008;30:26–38. doi: 10.1016/j.molcel.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 22.Voss NR, Gerstein M, Steitz TA, Moore PB. The geometry of the ribosomal polypeptide exit tunnel. J. Mol. Biol. 2006;360:893–906. doi: 10.1016/j.jmb.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 23.Lemieux S, Major RNA canonical and non-canonical base pairing types: a recognition method and complete repertoire. Nucleic Acids Res. 2002;30:4250–4263. doi: 10.1093/nar/gkf540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leontis NB, Westhof E. Geometric nomenclature and classification of RNA base pairs. RNA. 2001;7:499–512. doi: 10.1017/s1355838201002515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gabb HA, Sanghani SR, Robert CH, Prevost C. Finding and visualizing nucleic acid base stacking. J. Mol. Graph. 1996;14:611, 2314. doi: 10.1016/0263-7855(95)00086-0. [DOI] [PubMed] [Google Scholar]
- 26.Gendron P, Lemieux S, Major F. Quantitative analysis of nucleic acid three-dimensional structures. J. Mol. Biol. 2001;308:919–936. doi: 10.1006/jmbi.2001.4626. [DOI] [PubMed] [Google Scholar]
- 27.Leontis NB, Stombaugh J, Westhof E. The non-Watson-Crick base pairs and their associated isostericity matrices. Nucleic Acids Res. 2002;30:3497–3531. doi: 10.1093/nar/gkf481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Banatao DR, Altman RB, Klein TE. Microenvironment analysis and identification of magnesium binding sites in RNA. Nucleic Acids Res. 2003;31:4450–4460. doi: 10.1093/nar/gkg471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cazals F, Proust F, Bahadur RP, Janin J. Revisiting the Voronoi description of protein-protein interfaces. Protein Sci. 2006;15:2082–2092. doi: 10.1110/ps.062245906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Selmer M, Dunham CM, Murphy FVt, Weixlbaumer A, Petry S, Kelley AC, Weir JR, Ramakrishnan V. Structure of the 70S ribosome complexed with mRNA and tRNA. Science. 2006;313:1935–1942. doi: 10.1126/science.1131127. [DOI] [PubMed] [Google Scholar]
- 31.David-Eden H, Mandel-Gutfreund Y. Revealing unique properties of the ribosome using a network based analysis. Nucleic Acids Res. 2008;36:4641–4652. doi: 10.1093/nar/gkn433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Freeman LC. Centrality is social networks I: conceptual clarification. Soc. Networks. 1979;1:215–239. [Google Scholar]
- 33.Mankin A. Antibiotic blocks mRNA path on the ribosome. Nat. Struct. Mol. Biol. 2006;13:858–860. doi: 10.1038/nsmb1006-858. [DOI] [PubMed] [Google Scholar]
- 34.Kawabata T, Go N. Detection of pockets on protein surfaces using small and large probe spheres to find putative ligand binding sites. Proteins. 2007;68:516–529. doi: 10.1002/prot.21283. [DOI] [PubMed] [Google Scholar]
- 35.Hansen JL, Ippolito JA, Ban N, Nissen P, Moore PB, Steitz TA. The structures of four macrolide antibiotics bound to the large ribosomal subunit. Mol. Cell. 2002;10:117–128. doi: 10.1016/s1097-2765(02)00570-1. [DOI] [PubMed] [Google Scholar]
- 36.Batey RT, Rambo RP, Doudna JA. Tertiary motifs in RNA structure and folding. Angew. Chem. Int. Ed. Engl. 1999;38:2326–2343. doi: 10.1002/(sici)1521-3773(19990816)38:16<2326::aid-anie2326>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 37.Hermann T, Patel DJ. Stitching together RNA tertiary architectures. J. Mol. Biol. 1999;294:829–849. doi: 10.1006/jmbi.1999.3312. [DOI] [PubMed] [Google Scholar]
- 38.Moore PB. Structural motifs in RNA. Annu. Rev. Biochem. 1999;68:287–300. doi: 10.1146/annurev.biochem.68.1.287. [DOI] [PubMed] [Google Scholar]
- 39.Tinoco I, Jr, Bustamante C. How RNA folds. J. Mol. Biol. 1999;293:271–281. doi: 10.1006/jmbi.1999.3001. [DOI] [PubMed] [Google Scholar]
- 40.Saenger W. Priciples of Nucleic Acid Structure. New York: Springer; 1984. [Google Scholar]
- 41.Dalluge JJ, Hashizume T, Sopchik AE, McCloskey JA, Davis DR. Conformational flexibility in RNA: the role of dihydrouridine. Nucleic Acids Res. 1996;24:1073–1079. doi: 10.1093/nar/24.6.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nissen P, Ippolito JA, Ban N, Moore PB, Steitz TA. RNA tertiary interactions in the large ribosomal subunit: the A-minor motif. Proc. Natl Acad. Sci. USA. 2001;98:4899–4903. doi: 10.1073/pnas.081082398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xin Y, Laing C, Leontis NB, Schlick T. Annotation of tertiary interactions in RNA structures reveals variations and correlations. RNA. 2008;14:2465–2477. doi: 10.1261/rna.1249208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fulle S, Gohlke H. Statics of the ribosomal exit tunnel: implications for cotranslational peptide folding, elongation regulation, and antibiotics binding. J. Mol. Biol. 2009;387:502–517. doi: 10.1016/j.jmb.2009.01.037. [DOI] [PubMed] [Google Scholar]
- 45.Davidovich C, Bashan A, Auerbach-Nevo T, Yaggie RD, Gontarek RR, Yonath A. Induced-fit tightens pleuromutilins binding to ribosomes and remote interactions enable their selectivity. Proc. Natl Acad. Sci. USA. 2007;104:4291–4296. doi: 10.1073/pnas.0700041104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Amitai G, Shemesh A, Sitbon E, Shklar M, Netanely D, Venger I, Pietrokovski S. Network analysis of protein structures identifies functional residues. J. Mol. Biol. 2004;344:1135–1146. doi: 10.1016/j.jmb.2004.10.055. [DOI] [PubMed] [Google Scholar]
- 47.Chea E, Livesay DR. How accurate and statistically robust are catalytic site predictions based on closeness centrality? BMC Bioinform. 2007;8:153. doi: 10.1186/1471-2105-8-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.del Sol A, Fujihashi H, Amoros D, Nussinov R. Residue centrality, functionally important residues, and active site shape: analysis of enzyme and non-enzyme families. Protein Sci. 2006;15:2120–2128. doi: 10.1110/ps.062249106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glockner FO. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35:7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Noller HF. Ribosomal RNA and translation. Annu. Rev. Biochem. 1991;60:191–227. doi: 10.1146/annurev.bi.60.070191.001203. [DOI] [PubMed] [Google Scholar]
- 51.Lescoute A, Westhof E. The A-minor motifs in the decoding recognition process. Biochimie. 2006;88:993–999. doi: 10.1016/j.biochi.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 52.Sergiev PV, Serebryakova MV, Bogdanov AA, Dontsova OA. The ybiN gene of Escherichia coli encodes adenine-N6 methyltransferase specific for modification of A1618 of 23 S ribosomal RNA, a methylated residue located close to the ribosomal exit tunnel. J. Mol. Biol. 2008;375:291–300. doi: 10.1016/j.jmb.2007.10.051. [DOI] [PubMed] [Google Scholar]
- 53.Brodersen DE, Clemons WM, Jr, Carter AP, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. The structural basis for the action of the antibiotics tetracycline, pactamycin, and hygromycin B on the 30S ribosomal subunit. Cell. 2000;103:1143–1154. doi: 10.1016/s0092-8674(00)00216-6. [DOI] [PubMed] [Google Scholar]
- 54.Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature. 2000;407:340–348. doi: 10.1038/35030019. [DOI] [PubMed] [Google Scholar]
- 55.Hansen JL, Schmeing TM, Moore PB, Steitz TA. Structural insights into peptide bond formation. Proc. Natl Acad. Sci. USA. 2002;99:11670–11675. doi: 10.1073/pnas.172404099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tu D, Blaha G, Moore PB, Steitz TA. Structures of MLSBK antibiotics bound to mutated large ribosomal subunits provide a structural explanation for resistance. Cell. 2005;121:257–270. doi: 10.1016/j.cell.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 57.Schluenzen F, Takemoto C, Wilson DN, Kaminishi T, Harms JM, Hanawa-Suetsugu K, Szaflarski W, Kawazoe M, Shirouzu M, Nierhaus KH, et al. The antibiotic kasugamycin mimics mRNA nucleotides to destabilize tRNA binding and inhibit canonical translation initiation. Nat. Struct. Mol. Biol. 2006;13:871–878. doi: 10.1038/nsmb1145. [DOI] [PubMed] [Google Scholar]
- 58.Borovinskaya MA, Shoji S, Fredrick K, Cate JH. Structural basis for hygromycin B inhibition of protein biosynthesis. RNA. 2008;14:1590–1599. doi: 10.1261/rna.1076908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Borovinskaya MA, Shoji S, Holton JM, Fredrick K, Cate JH. A steric block in translation caused by the antibiotic spectinomycin. ACS Chem. Biol. 2007;2:545–552. doi: 10.1021/cb700100n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schuwirth BS, Day JM, Hau CW, Janssen GR, Dahlberg AE, Cate JH, Vila-Sanjurjo A. Structural analysis of kasugamycin inhibition of translation. Nat. Struct. Mol. Biol. 2006;13:879–886. doi: 10.1038/nsmb1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gurel G, Blaha G, Moore PB, Steitz TA. U2504 determines the species specificity of the A-site cleft antibiotics: the Structures of tiamulin, homoharringtonine, and bruceantin bound to the ribosome. J. Mol. Biol. 2009;389:146–156. doi: 10.1016/j.jmb.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou J, Bhattacharjee A, Chen S, Chen Y, Duffy E, Farmer J, Goldberg J, Hanselmann R, Ippolito JA, Lou R, et al. Design at the atomic level: design of biaryloxazolidinones as potent orally active antibiotics. Bioorg. Med. Chem. Lett. 2008;18:6175–6178. doi: 10.1016/j.bmcl.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 63.Schlunzen F, Zarivach R, Harms J, Bashan A, Tocilj A, Albrecht R, Yonath A, Franceschi F. Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature. 2001;413:814–821. doi: 10.1038/35101544. [DOI] [PubMed] [Google Scholar]
- 64.Schlunzen F, Harms JM, Franceschi F, Hansen HA, Bartels H, Zarivach R, Yonath A. Structural basis for the antibiotic activity of ketolides and azalides. Structure. 2003;11:329–338. doi: 10.1016/s0969-2126(03)00022-4. [DOI] [PubMed] [Google Scholar]
- 65.Berisio R, Harms J, Schluenzen F, Zarivach R, Hansen HA, Fucini P, Yonath A. Structural insight into the antibiotic action of telithromycin against resistant mutants. J. Bacteriol. 2003;185:4276–4279. doi: 10.1128/JB.185.14.4276-4279.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Harms JM, Schlunzen F, Fucini P, Bartels H, Yonath A. Alterations at the peptidyl transferase centre of the ribosome induced by the synergistic action of the streptogramins dalfopristin and quinupristin. BMC Biol. 2004;2:4. doi: 10.1186/1741-7007-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bashan A, Agmon I, Zarivach R, Schluenzen F, Harms J, Berisio R, Bartels H, Franceschi F, Auerbach T, Hansen HA, et al. Structural basis of the ribosomal machinery for peptide bond formation, translocation, and nascent chain progression. Mol. Cell. 2003;11:91–102. doi: 10.1016/s1097-2765(03)00009-1. [DOI] [PubMed] [Google Scholar]
- 68.Berisio R, Schluenzen F, Harms J, Bashan A, Auerbach T, Baram D, Yonath A. Structural insight into the role of the ribosomal tunnel in cellular regulation. Nat. Struct. Biol. 2003;10:366–370. doi: 10.1038/nsb915. [DOI] [PubMed] [Google Scholar]
- 69.Schlunzen F, Pyetan E, Fucini P, Yonath A, Harms JM. Inhibition of peptide bond formation by pleuromutilins: the structure of the 50S ribosomal subunit from Deinococcus radiodurans in complex with tiamulin. Mol. Microbiol. 2004;54:1287–1294. doi: 10.1111/j.1365-2958.2004.04346.x. [DOI] [PubMed] [Google Scholar]
- 70.Wilson DN, Schluenzen F, Harms JM, Starosta AL, Connell SR, Fucini P. The oxazolidinone antibiotics perturb the ribosomal peptidyl-transferase center and effect tRNA positioning. Proc. Natl. Acad. Sci. USA. 2008;105:13339–13344. doi: 10.1073/pnas.0804276105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pyetan E, Baram D, Auerbach-Nevo T, Yonath A. Chemical parameters influencing fine-tuning in the binding of macrolide antibiotics to the ribosomal tunnel. Pure Appl. Chem. 2007;79:955–968. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.