

Abstract
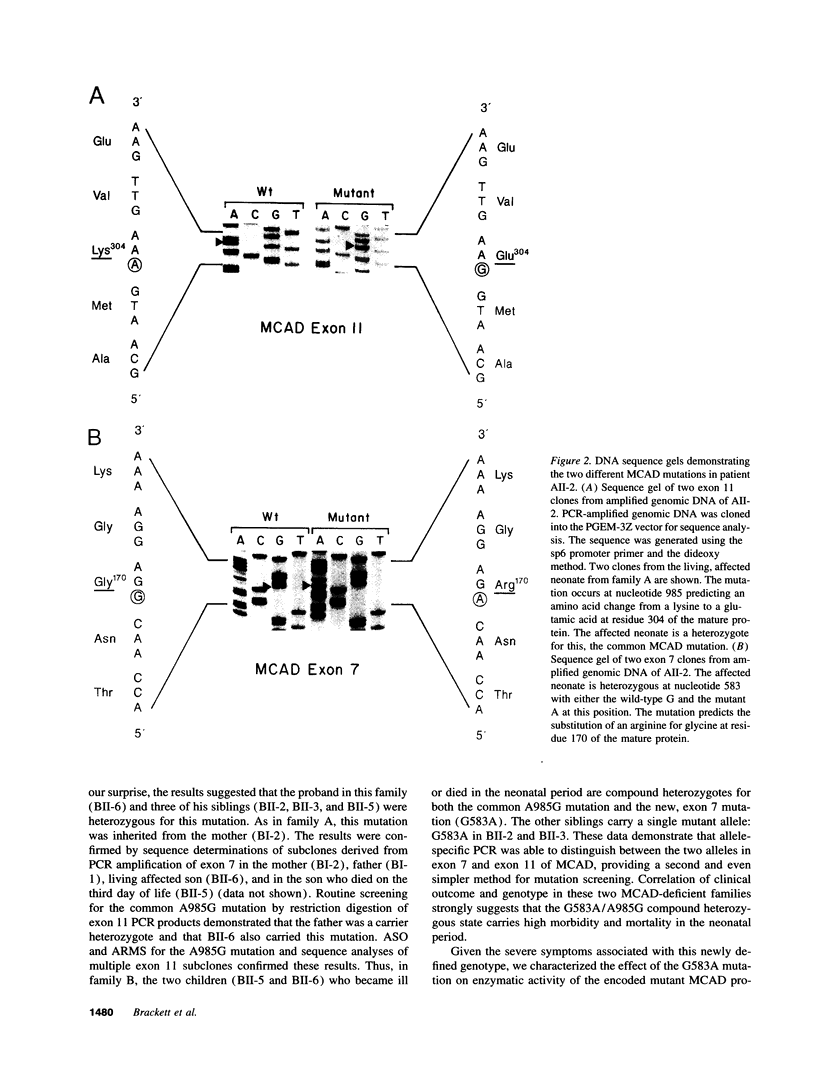
Medium chain acyl-CoA dehydrogenase (MCAD) deficiency is the most common known genetic disorder of fatty acid oxidation. Most (approximately 80%) cases are homozygous for a single mutation: A to G replacement at nucleotide 985 (A985G). MCAD deficiency typically presents in the second year of life as hypoketotic hypoglycemia associated with fasting and may progress to liver failure, coma, and death. Prompt diagnosis and management may prevent long-term sequelae. MCAD deficiency was verified by analysis of urinary acylglycine and serum acylcarnitine species from two neonates referred for diagnosis. Full-length cDNA and MCAD exon 7 and 11 genomic clones were prepared for sequence analysis. Normal and mutant cDNAs were expressed in bacteria, and enzymatic activity was assayed by the ferricenium hexaflurophosphate method. Four compound heterozygote individuals from two unrelated families with A985G on one allele and a novel G to A mutation at nucleotide 583 (G583A) as the second mutant allele presented with MCAD deficiency in the first week of life. The expressed G583A mutant protein lacks enzymatic activity. This novel mutation, G583A, is associated with severe MCAD deficiency causing hypoglycemia or sudden, unexpected neonatal death. This previously unrecognized phenotype of MCAD deficiency may contribute significantly to preventable infant deaths.
Full text
PDF






Images in this article



Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Catzeflis C., Bachmann C., Hale D. E., Coates P. M., Wiesmann U., Colombo J. P., Joris F., Délèze G. Early diagnosis and treatment of neonatal medium-chain acyl-CoA dehydrogenase deficiency: report of two siblings. Eur J Pediatr. 1990 May;149(8):577–581. doi: 10.1007/BF01957697. [DOI] [PubMed] [Google Scholar]
- Chirgwin J. M., Przybyla A. E., MacDonald R. J., Rutter W. J. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry. 1979 Nov 27;18(24):5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- Coates P. M., Hale D. E., Stanley C. A., Corkey B. E., Cortner J. A. Genetic deficiency of medium-chain acyl coenzyme A dehydrogenase: studies in cultured skin fibroblasts and peripheral mononuclear leukocytes. Pediatr Res. 1985 Jul;19(7):671–676. doi: 10.1203/00006450-198507000-00007. [DOI] [PubMed] [Google Scholar]
- Ding J. H., Roe C. R., Iafolla A. K., Chen Y. T. Medium-chain acyl-coenzyme A dehydrogenase deficiency and sudden infant death. N Engl J Med. 1991 Jul 4;325(1):61–62. doi: 10.1056/NEJM199107043250113. [DOI] [PubMed] [Google Scholar]
- Duran M., Wanders R. J., de Jager J. P., Dorland L., Bruinvis L., Ketting D., Ijlst L., van Sprang F. J. 3-Hydroxydicarboxylic aciduria due to long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency associated with sudden neonatal death: protective effect of medium-chain triglyceride treatment. Eur J Pediatr. 1991 Jan;150(3):190–195. doi: 10.1007/BF01963564. [DOI] [PubMed] [Google Scholar]
- Goelz S. E., Hamilton S. R., Vogelstein B. Purification of DNA from formaldehyde fixed and paraffin embedded human tissue. Biochem Biophys Res Commun. 1985 Jul 16;130(1):118–126. doi: 10.1016/0006-291x(85)90390-0. [DOI] [PubMed] [Google Scholar]
- Howat A. J., Bennett M. J., Variend S., Shaw L., Engel P. C. Defects of metabolism of fatty acids in the sudden infant death syndrome. Br Med J (Clin Res Ed) 1985 Jun 15;290(6484):1771–1773. doi: 10.1136/bmj.290.6484.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly D. P., Gordon J. I., Alpers R., Strauss A. W. The tissue-specific expression and developmental regulation of two nuclear genes encoding rat mitochondrial proteins. Medium chain acyl-CoA dehydrogenase and mitochondrial malate dehydrogenase. J Biol Chem. 1989 Nov 15;264(32):18921–18925. [PubMed] [Google Scholar]
- Kelly D. P., Hale D. E., Rutledge S. L., Ogden M. L., Whelan A. J., Zhang Z., Strauss A. W. Molecular basis of inherited medium-chain acyl-CoA dehydrogenase deficiency causing sudden child death. J Inherit Metab Dis. 1992;15(2):171–180. doi: 10.1007/BF01799626. [DOI] [PubMed] [Google Scholar]
- Kelly D. P., Whelan A. J., Ogden M. L., Alpers R., Zhang Z. F., Bellus G., Gregersen N., Dorland L., Strauss A. W. Molecular characterization of inherited medium-chain acyl-CoA dehydrogenase deficiency. Proc Natl Acad Sci U S A. 1990 Dec;87(23):9236–9240. doi: 10.1073/pnas.87.23.9236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. J., Wang M., Paschke R. Crystal structures of medium-chain acyl-CoA dehydrogenase from pig liver mitochondria with and without substrate. Proc Natl Acad Sci U S A. 1993 Aug 15;90(16):7523–7527. doi: 10.1073/pnas.90.16.7523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. J., Wu J. Structure of the medium-chain acyl-CoA dehydrogenase from pig liver mitochondria at 3-A resolution. Proc Natl Acad Sci U S A. 1988 Sep;85(18):6677–6681. doi: 10.1073/pnas.85.18.6677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman T. C., Hale D. E., Bhala A., Thorpe C. An acyl-coenzyme A dehydrogenase assay utilizing the ferricenium ion. Anal Biochem. 1990 May 1;186(2):280–284. doi: 10.1016/0003-2697(90)90080-s. [DOI] [PubMed] [Google Scholar]
- Leung K. C., Hammond J. W., Chabra S., Carpenter K. H., Potter M., Wilcken B. A fatal neonatal case of medium-chain acyl-coenzyme A dehydrogenase deficiency with homozygous A-->G985 transition. J Pediatr. 1992 Dec;121(6):965–968. doi: 10.1016/s0022-3476(05)80353-1. [DOI] [PubMed] [Google Scholar]
- Lundemose J. B., Gregersen N., Kølvraa S., Nørgaard Pedersen B., Gregersen M., Helweg-Larsen K., Simonsen J. The frequency of a disease-causing point mutation in the gene coding for medium-chain acyl-CoA dehydrogenase in sudden infant death syndrome. Acta Paediatr. 1993 Jun-Jul;82(6-7):544–546. doi: 10.1111/j.1651-2227.1993.tb12749.x. [DOI] [PubMed] [Google Scholar]
- Matsubara Y., Narisawa K., Miyabayashi S., Tada K., Coates P. M., Bachmann C., Elsas L. J., 2nd, Pollitt R. J., Rhead W. J., Roe C. R. Identification of a common mutation in patients with medium-chain acyl-CoA dehydrogenase deficiency. Biochem Biophys Res Commun. 1990 Aug 31;171(1):498–505. doi: 10.1016/0006-291x(90)91421-n. [DOI] [PubMed] [Google Scholar]
- Matsubara Y., Narisawa K., Tada K., Ikeda H., Yao Y. Q., Danks D. M., Green A., McCabe E. R. Prevalence of K329E mutation in medium-chain acyl-CoA dehydrogenase gene determined from Guthrie cards. Lancet. 1991 Aug 31;338(8766):552–553. doi: 10.1016/0140-6736(91)91110-g. [DOI] [PubMed] [Google Scholar]
- Newton C. R., Graham A., Heptinstall L. E., Powell S. J., Summers C., Kalsheker N., Smith J. C., Markham A. F. Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS). Nucleic Acids Res. 1989 Apr 11;17(7):2503–2516. doi: 10.1093/nar/17.7.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobukuni Y., Yokoo T., Ohtani Y., Endo F., Aoki S., Yoshinaga M., Matsumoto T., Yoshimoto M., Tsuji Y., Matsuda I. Neonatal onset of medium-chain acyl-CoA dehydrogenase deficiency in two siblings. Brain Dev. 1988;10(2):129–134. doi: 10.1016/s0387-7604(88)80084-6. [DOI] [PubMed] [Google Scholar]
- Rinaldo P., O'Shea J. J., Coates P. M., Hale D. E., Stanley C. A., Tanaka K. Medium-chain acyl-CoA dehydrogenase deficiency. Diagnosis by stable-isotope dilution measurement of urinary n-hexanoylglycine and 3-phenylpropionylglycine. N Engl J Med. 1988 Nov 17;319(20):1308–1313. doi: 10.1056/NEJM198811173192003. [DOI] [PubMed] [Google Scholar]
- Saijo T., Welch W. J., Tanaka K. Intramitochondrial folding and assembly of medium-chain acyl-CoA dehydrogenase (MCAD). Demonstration of impaired transfer of K304E-variant MCAD from its complex with hsp60 to the native tetramer. J Biol Chem. 1994 Feb 11;269(6):4401–4408. [PubMed] [Google Scholar]
- Tanaka K., Yokota I., Coates P. M., Strauss A. W., Kelly D. P., Zhang Z., Gregersen N., Andresen B. S., Matsubara Y., Curtis D. Mutations in the medium chain acyl-CoA dehydrogenase (MCAD) gene. Hum Mutat. 1992;1(4):271–279. doi: 10.1002/humu.1380010402. [DOI] [PubMed] [Google Scholar]
- Touma E. H., Charpentier C. Medium chain acyl-CoA dehydrogenase deficiency. Arch Dis Child. 1992 Jan;67(1):142–145. doi: 10.1136/adc.67.1.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treem W. R., Rinaldo P., Hale D. E., Stanley C. A., Millington D. S., Hyams J. S., Jackson S., Turnbull D. M. Acute fatty liver of pregnancy and long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency. Hepatology. 1994 Feb;19(2):339–345. [PubMed] [Google Scholar]
- Treem W. R., Witzleben C. A., Piccoli D. A., Stanley C. A., Hale D. E., Coates P. M., Watkins J. B. Medium-chain and long-chain acyl CoA dehydrogenase deficiency: clinical, pathologic and ultrastructural differentiation from Reye's syndrome. Hepatology. 1986 Nov-Dec;6(6):1270–1278. doi: 10.1002/hep.1840060608. [DOI] [PubMed] [Google Scholar]
- Tsai M. Y., Schwichtenberg K., Tuchman M. Laboratory diagnosis of medium-chain acyl-coenzyme A dehydrogenase deficiency by the amplification refractory mutation system. Clin Chem. 1993 Feb;39(2):280–283. [PubMed] [Google Scholar]
- Van Hove J. L., Zhang W., Kahler S. G., Roe C. R., Chen Y. T., Terada N., Chace D. H., Iafolla A. K., Ding J. H., Millington D. S. Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency: diagnosis by acylcarnitine analysis in blood. Am J Hum Genet. 1993 May;52(5):958–966. [PMC free article] [PubMed] [Google Scholar]
- Walker V., Mills G. A., Radford M. Diagnosis of medium chain acyl-CoA dehydrogenase deficiency in the newborn. Lancet. 1990 May 26;335(8700):1288–1289. doi: 10.1016/0140-6736(90)91363-f. [DOI] [PubMed] [Google Scholar]
- Walker V., Mills G. A., Weavind G. P., Hall M. A., Johnston P. G. Diagnosis of medium chain acyl-CoA dehydrogenase (MCAD) deficiency in an asymptomatic neonate. Ann Clin Biochem. 1990 May;27(Pt 3):267–269. doi: 10.1177/000456329002700314. [DOI] [PubMed] [Google Scholar]
- Whelan A. J., Strauss A. W., Hale D. E., Mendelsohn N. J., Kelly D. P. Expression and characterization of human mutant (glutamic acid304) medium-chain acyl-coenzyme A dehydrogenase in mammalian cells. Pediatr Res. 1993 Nov;34(5):694–697. doi: 10.1203/00006450-199311000-00025. [DOI] [PubMed] [Google Scholar]
- Wilcken B., Carpenter K. H., Hammond J. Neonatal symptoms in medium chain acyl coenzyme A dehydrogenase deficiency. Arch Dis Child. 1993 Sep;69(3 Spec No):292–294. doi: 10.1136/adc.69.3_spec_no.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota I., Coates P. M., Hale D. E., Rinaldo P., Tanaka K. Molecular survey of a prevalent mutation, 985A-to-G transition, and identification of five infrequent mutations in the medium-chain Acyl-CoA dehydrogenase (MCAD) gene in 55 patients with MCAD deficiency. Am J Hum Genet. 1991 Dec;49(6):1280–1291. [PMC free article] [PubMed] [Google Scholar]
- Yokota I., Saijo T., Vockley J., Tanaka K. Impaired tetramer assembly of variant medium-chain acyl-coenzyme A dehydrogenase with a glutamate or aspartate substitution for lysine 304 causing instability of the protein. J Biol Chem. 1992 Dec 25;267(36):26004–26010. [PubMed] [Google Scholar]