

Abstract
Recessively inherited loss-of-function mutations in the parkin, DJ-1, or PINK1 gene are linked to familial cases of early-onset Parkinson's diseases (PD), and heterozygous mutations are associated with increased incidence of late-onset PD. We previously reported that single knockout mice lacking Parkin, DJ-1, or PINK1 exhibited no nigral degeneration, even though evoked dopamine release from nigrostriatal terminals was reduced and striatal synaptic plasticity was impaired. In this study, we tested whether inactivation of all three recessive PD genes, each of which was required for nigral neuron survival in the aging human brain, resulted in nigral degeneration during the lifespan of mice. Surprisingly, we found that triple knockout mice lacking Parkin, DJ-1, and PINK1 have normal morphology and numbers of dopaminergic and noradrenergic neurons in the substantia nigra and locus coeruleus, respectively, at the ages of 3, 16, and 24 months. Interestingly, levels of striatal dopamine in triple knockout mice were normal at 16 months of age but increased at 24 months. These results demonstrate that inactivation of all three recessive PD genes is insufficient to cause significant nigral degeneration within the lifespan of mice, suggesting that these genes may be protective rather than essential for the survival of dopaminergic neurons during the aging process. These findings also support the notion that mammalian Parkin and PINK1 may function in the same genetic pathway as in Drosophila.
Keywords: dopamine, dopaminergic, mouse, Parkinson's disease
Parkinson's disease (PD) is the most common movement disorder caused by the gradual loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc). While most cases are sporadic, 5–10% of PD patients carry mutations with a Mendelian inheritance. Mutations in the parkin, DJ-1, and PINK1 genes have been linked to autosomal recessive forms of PD (Kitada et al. 1998; Bonifati et al. 2003; Valente et al. 2004). However, it is still unclear how loss-of-function mutations in each of these genes cause degeneration of dopaminergic neurons, which results in reduced availability of dopamine (DA) in the striatum.
Clinical studies for the three recessive PD genes revealed that in addition to typical parkinsonian features (i.e., resting tremor, akinesia, muscle rigidity, and postural instability), patients carrying mutations in any of these genes sometimes show specific clinical features, such as prominent sleep benefit/diurnal fluctuation, morning foot dystonia, a tendency to develop dyskinesias in response to levodopa, and psychiatric problems (Yamamura et al. 1973, 2000; Bonifati et al. 2002; Dekker et al. 2003; Albanese et al. 2005). Thus, these clinical findings suggest that loss-of-function mutations in these recessive PD genes may cause similar dopaminergic dysfunction. Our generation and multidisciplinary analysis of single knockout (KO) mice lacking one of the three recessive PD genes indeed showed similar functional defects in the dopaminergic system, including reduced DA overflow from nigrostriatal terminals and impaired corticostriatal synaptic plasticity (Goldberg et al. 2003, 2005; Kitada et al. 2007, 2009). However, the morphology and numbers of DA neurons in the SNpc are normal in the absence of Parkin, DJ-1, or PINK1 up to 24 months of age (Goldberg et al. 2003, 2005; Kitada et al. 2007; Yamaguchi and Shen 2007). Thus, recapitulation of the genetic alterations in mice is insufficient to reproduce the final neuropathological feature of PD during the short lifespan of mice.
Interestingly, recent studies suggest functional interactions among three gene products. Genetic studies in Drosophila have demonstrated that PINK1 acts upstream of Parkin in the same genetic pathway (Greene et al. 2003; Pesah et al. 2004; Clark et al. 2006; Park et al. 2006; Yang et al. 2006). However, it is unclear whether the genetic interaction between Parkin and PINK1 is conserved in mice, especially because the drastic mitochondrial structural changes seen in mutant flies lacking Parkin or PINK1 are not present in single KO mice lacking Parkin or PINK1 (Greene et al. 2003; Pesah et al. 2004; Clark et al. 2006; Park et al. 2006; Yang et al. 2006), although both parkin and PINK1 KO mice exhibit mitochondrial functional defects (Palacino et al. 2004; Gautier et al. 2008). In addition, DJ-1 has been shown to protect cultured cells and fruit flies from the damages of oxidative stress (Yokota et al. 2003; Taira et al. 2004; Kim et al. 2005; Menzies et al. 2005; Meulener et al. 2005; Moore et al. 2005; Yang et al. 2005; Zhang et al. 2005; Andres-Mateos et al. 2007; Junn et al. 2009). Thus, Parkin, DJ-1, and PINK1 appear to function in concert to protect mitochondria against oxidative stress.
The lack of dopaminergic degeneration in single KO mice prompt us to test whether inactivation of all three recessive PD genes, presumably factors promoting nigral neuron survival in humans during aging would accelerate the nigral degeneration process and permit development of dopaminergic degeneration during the short lifespan of mice. We found that triple knockout (TKO) mice were grossly normal and had normal morphology and numbers of dopaminergic and noradrenergic neurons in the SNpc and locus coeruleus (LC) at the ages of 3, 16, and 24 months. Tyrosine hydroxylase (TH) expression level and DA synthesis in the striatum of TKO mice are also unchanged. However, TKO mice at the age of 24 months showed significantly higher DA levels in the striatum than wild-type controls. These results indicate that loss of all three recessive PD gene products is insufficient to cause nigral degeneration within the lifespan of mice but causes disruption of DA homeostasis in the striatum.
Materials and methods
Histology and neuron count
Dissected mouse brains were processed for paraffin embedding after 2 h formalin fixation. Each paraffin block contained four TKO and four wild-type brains and was sectioned at 16 μm thickness. Immunohistochemistry for TH (1 : 1000, rabbit anti-TH, Millipore, Billerica, MA, USA) was carried out using Vector Elite ABC kit (Vector Laboratories, Burlingame, CA, USA) with 3,3′-diaminobenzidine on every tenth midbrain sections and every fourth LC sections throughout the entire substantia nigra (SN) and LC. The number of DA neurons in the SN was determined by counting TH-immunoreactive neurons in coronal sections of TKO and wild-type brains using the fractionator and optical dissector methods of unbiased stereology under a microscope (Olympus, Center Valley, PA, USA) equipped with a CCD camera connected to a computer running Bioquant image analysis software (Bioquant, Nashville, TN, USA). The diameter (the longest axis) and the size (area) of TH-positive neurons were determined at 40× on the same light microscopy. The diameter of 15 neurons selected randomly was measured in images of four to seven different fields for each section. Each area of these neurons was measured by ImageJ version 1.37 software (NIH, Bethesda, MD, USA). The number of TH-immunoreactive neurons in the LC was assessed by direct counting through a light microscope at 2× magnification. Cell counts for LC were taken every fourth section spanning the mid rostral-caudal extent of the left side nucleus and the total numbers were evaluated directly followed by procedures described in (German et al. 2000). Cell counts were carried out genotype blind. Values were expressed as means ± SEM.
Western blotting
Striatal proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electroblotted onto the nitrocellulose membrane (Thermo Scientific, Rockford, IL, USA), and the membranes were blocked with 0.5% dry milk in 135 mM NaCl, 2.5 mM KCl, 50 mM Tris, and 0.1% Tween-20 and incubated with rabbit anti-goat polyclonal antibodies specific for TH (1 : 500). After washing in 135 mM NaCl, 2.5 mM KCl, 50 mM Tris, and 0.1% Tween-20, the membranes were incubated with peroxidase-conjugated anti-rabbit secondary antibodies (Bio-Rad Laboratories, Hercules, CA, USA). Then, they were washed, treated with chemiluminescence reagent (PerkinElmer, Waltham, MA, USA), and exposed to film. Each value of relative protein expressions was normalized to that of α-tubulin.
In vivo TH activity assay
To examine whether lack of the three recessive PD genes altered the activity of TH, which was the rate-limiting enzyme in DA synthesis, we measured TH activity in vivo by monitoring 3,4-dihydroxy-L-phenylalanine (DOPA) formation (Arrue et al. 2003). Shortly, mice at the age of 2–5 months were injected intraperitoneally with 100 mg/kg of NSD-1015 (Sigma-Aldrich, St Louis, MO, USA), a selective aromatic L-amino acid decarboxylase inhibitor. Thirty minutes after NSD-1015 injection, the mice were killed, and the striata were dissected out and put into 300 μL of 0.1 N perchloric acid and sonicated. An aliquot (50 μL) was used to determine protein concentration, and the remainder was centrifuged at 20 000 g for 10 min at 4°C. After filtration of supernatant, the levels of DOPA were determined by HPLC.
Measurements of striatal DA and its metabolites by HPLC
Dorsal striata were dissected, weighed, and stored at −80°C until use. Frozen striata were sonicated in an ice-cold solution (0.1 M perchloric acid, 0.2 mM sodium bisulfite) and centrifuged for 10 min at 14 000 g at 4°C. The supernatant was filtered and applied to C18 reverse phase HPLC columns connected to an Alexys LC-100 system (Antec-Leyden, Hanover, MD, USA) with electrochemical detection (DECADE II) and a VT-03 electrochemical flow cell. DA measurements were made using a detection potential of 320 mV and isocratic elution (50 mM phosphoric acid, 500 mg/L octane sulfonic acid, 0.1 mM EDTA, 8 mM KCl, pH 6.0, and 16.5% methanol) through a 50 × 2.1 mm HPLC column at the flow rate of 0.2 mL/min. DA metabolites [homovanillic acid (HVA) and 3,4-dihydroxyphenylacetic acid (DOPAC)] eluted isocratically on a 150 × 2.1 mm HPLC column at lower pH (50 mM phosphoric acid, 50 mM citric acid, 400 mg/L octane sulphonic acid, 0.1 mM EDTA, 8 mM KCl, pH 3.75, and 3% methanol) at the flow rate 0.2 mL/min and detected at 590 mV. To measure DOPA accumulation in the striatum, DOPA in the samples was eluted isocratically on a 150 × 2.1 mm HPLC column using a mobile phase (50 mM phosphoric acid, 50 mM citric acid, 500 mg/L octane sulphonic acid, 0.1 mM EDTA, 8 mM KCl, pH 3.25, and 5% methanol) at the flow rate 0.2 mL/min. Concentrations of DA, its metabolites, and DOPA in the striatal samples were obtained by comparing with a series of catecholamine standards of known concentration which was run under the same conditions. The levels of DA, its metabolites, and DOPA were normalized to tissue weights.
Results
Generation of TKO mice
The generation and characterization of Parkin, DJ-1, and PINK1 single KO mice have been described previously (Goldberg et al. 2003, 2005; Kitada et al. 2007). TKO mice were generated by first crossing parkin−/− mice with DJ-1−/− mice to obtain parkin+/−; DJ-1+/− mice, which were interbred to obtain parkin−/−; DJ-1−/− mice. Second, we bred PINK1−/− mice with parkin−/−; DJ-1−/− mice to obtain PINK1+/−, parkin+/−, and DJ-1+/− mice, which were crossed with parkin−/−; DJ-1−/− mice to obtain PINK1+/−, parkin−/−, and DJ-1+/− mice. Third, PINK1+/−, parkin−/−, and DJ-1+/− mice were interbred to obtain parkin−/−, DJ-1−/−, and PINK1−/− mice. As the DJ-1 and PINK1 genes were both localized on the same chromosome (chromosome 4 for mice; Fig. 1a), with a 12.5 cM (centimorgan) distance between the two loci, we expected to obtain the right genotype at a reduced ratio (1/4 × 1/8 = 1/32). Indeed, out of 69 offspring from this breeding, we obtained two parkin−/−; DJ-1−/−, PINK1−/− mice (~1/35). The experimental triple KO mice and DJ-1−/−; PINK1−/− double KO were obtained from intercross of parkin+/−, DJ-1−/−; PINK1−/− mice, whereas wild-type and parkin−/− mice were obtained from intercross of parkin+/− mice. All mice were kept in the C57BL/6 and 129/Sv hybrid background with similar breeding schemes. Loss of Parkin and DJ-1 in TKO mice was confirmed by western blot analysis, and loss of PINK1 mRNAs was confirmed by northern blot analysis, because of the lack of specific PINK1 antibodies (Fig. 1b–d). TKO mice were viable and fertile without obvious abnormalities. Nissl staining revealed normal brain morphology in TKO mice (data not shown).
Fig. 1.
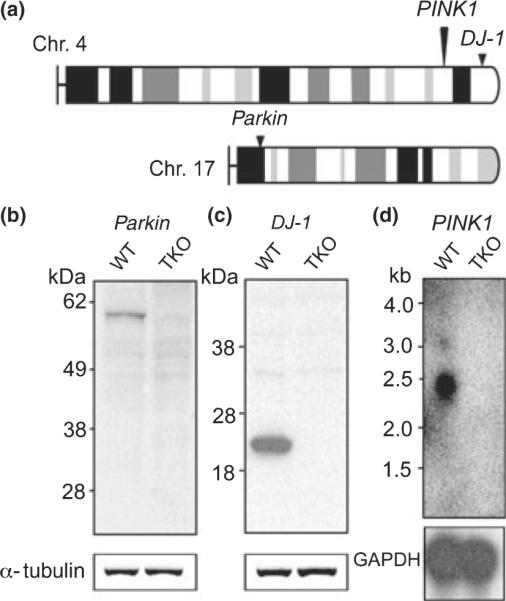
(a) The mouse parkin gene is located on chromosome 17, whereas the mouse DJ-1 and PINK1 genes are both on chromosome 4, with an allelic distance of 12.5 cM. (b, c) Western blot analysis confirms the absence of Parkin (b) and DJ-1 (c) in TKO brains. The same blots were incubated with an anti-α-tubulin antibody as loading controls. (d) Northern analysis using a probe specific for exon 8 shows the absence of PINK1 transcripts in TKO brains. The same blot was hybridized with a GAPDH cDNA probe as control. WT, wild-type; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Normal morphology and numbers of dopaminergic neurons in TKO mice
To investigate whether loss of all three recessive PD genes affected the survival of SNpc neurons, we performed quantitative immnohistological analysis using an antibody specific for TH on TKO mice and wild-type controls at the ages of 3, 16, and 24 months. This analysis indicated normal TH staining of SNpc and normal morphology of DA neurons in TKO mice (Fig. 2a–d). We furthermore counted TH-immunoreactive neurons in the SNpc using unbiased stereological methods. There was no significant difference in the number of dopaminergic neurons between TKO mice and wild-type controls at the ages of 3 months (+/+: 12720 ± 992, n = 4; −/−: 11080 ± 740, n = 4; p > 0.05), 16 months (+/+: 11460 ± 1735, n = 4; −/−: 11880 ± 605, n = 4; p > 0.05), and 24 months (+/+: 11008 ± 736, n = 5; −/−: 10440 ± 396, n = 4; p > 0.05; Fig. 2e). Consistent with this finding, we found similar numbers of TH-immunoreactive neurons in the SNpc between PINK1−/− and wild-type mice at the age of 24 months (+/+: 12853 ± 1880, n = 3; −/−: 11824 ± 1154, n = 5; p > 0.05). We further quantified the size of dopaminergic neurons by measuring both the diameter (+/+: 29.4 ± 1.4 μm; −/−: 29.0 ± 0.9 μm; n = 4 per genotype; p > 0.05) and the area (+/+: 318.6 ± 11.6 μm2; −/−: 314.1 ± 14.2 μm2; n = 4 per genotype; p > 0.05) of TH+ neurons in TKO and wild-type mice at 24 months of age, and found no genotypic differences.
Fig. 2.

No neurodegeneration in the SNpc and LC of TKO mice. (a–d) Normal immunoreactivity and morphology of SN neurons in TKO mice at the age of 24 months. Scale bars: 100 μm. (e) Similar numbers of TH-immunoreactive neurons are present in the SNpc of TKO mice and wild-type controls at the ages of 3 months (+/+: 12720 ± 992, n = 4; −/−: 11080 ± 740, n = 4; p > 0.05), 16 months (+/+: 11460 ± 1735, n = 4; −/−: 11880 ± 605, n = 4; p > 0.05), and 24 months (+/+: 11008 ± 736, n = 5; −/−: 10440 ± 396, n = 4; p > 0.05). All data are expressed as mean ± SEM. (f–i) Normal immunoreactivity and morphology of TH+ neurons in the LC of TKO mice at the age of 24 months. Scale bars: 100 μm. (j) Similar numbers of TH-immunoreactive neurons are present in the LC of TKO mice and wild-type controls at the ages of 3 months (+/+: 308 ± 16, n = 3; −/−: 298 ± 13, n = 4; p > 0.05), 16 months (+/+: 284 ± 38, n = 3; −/−: 236 ± 17, n = 4; p > 0.05), and 24 months (+/+: 231 ± 21, n = 4; −/−: 265 ± 16, n = 4; p > 0.05). All data are expressed as mean ± SEM.
Normal morphology and numbers of noradrenergic neurons in the LC of TKO mice
In addition to dopaminergic neurons in the SNpc, noradrenergic neurons in the LC degenerated and died in recessive and sporadic PD (Hughes et al. 1992; Yamamura et al. 1993, 2000; Takahashi et al. 1994). Our immunostaining analysis showed unchanged immunoreactivity and normal morphology of TH+ LC neurons (Fig. 2f–i) in TKO mice at the ages of 3, 16, and 24 months. We directly counted TH-immunoreactive neurons in the LC through a light microscope and found similar numbers of LC neurons in TKO mice at the ages of 3 months (+/+: 308 ± 16, n = 3; −/−: 298 ± 13, n = 4; p > 0.05), 16 months (+/+: 284 ± 38, n = 3; −/−: 236 ± 17, n = 4; p > 0.05), and 24 months (+/+: 231 ± 21, n = 4; −/−: 265 ± 16, n = 4; p > 0.05; Fig. 2j).
Unchanged TH expression and DA synthesis in the striatum of TKO mice
To quantify expression levels of TH, we performed western blotting using an anti-TH antibody. Western analysis of striatal samples from TKO and wild-type mice showed similar levels of TH at the ages of 16 months (+/+: 1.00 ± 0.11, n = 4; −/−: 1.07 ± 0.15, n = 4; p > 0.05) and 24 months (+/+: 1.00 ± 0.19, n = 5; −/−: 1.08 ± 0.12, n = 5; p > 0.05; Fig. 3a and b). To determine whether DA synthesis was affected in the absence of Parkin, DJ-1, and PINK1, we measured TH activity in vivo through HPLC quantification of DOPA accumulated in the presence of NSD-1015, an inhibitor of the aromatic L-amino acid DOPA decarboxylase, which could pass the blood–brain barrier and blocked the conversion of DOPA to DA. Levels of DOPA in the dissected striatum were unchanged in TKO mice at the age of 2–5 months (0.33 ± 0.04 ng/mg, n = 4), compared with wild-type controls (0.30 ± 0.07 ng/mg, n = 4; p > 0.05; Fig. 3c), suggesting normal TH activity in the absence of Parkin, DJ-1, and PINK1.
Fig. 3.

Increased DA in the striatum of TKO mice at 24 months of age. (a and b) Western analysis showed similar levels of TH in TKO mice at the ages of 16 months (+/+: 1.00 ± 0.11, n = 4; −/−: 1.07 ± 0.15, n = 4; p > 0.05) and 24 months (+/+: 1.00 ± 0.19, n = 5; −/−: 1.08 ± 0.12, n = 5; p > 0.05). Each value of the TH protein level is normalized to that of α-tubulin. (c) TH activity assay. DOPA content in the TKO striatum following NSD-1015 treatment at the age of 2–5 months (0.33 ± 0.04 ng/mg, n = 4) was unchanged compared with wild-type controls (0.30 ± 0.07 ng/mg, n = 4; p > 0.05). Each data point represents the mean ± SEM. (d) Levels of striatal DA content are similar in TKO mice and wild-type controls at the age of 16 months (+/+: 18.4 ± 2.0 ng/mg, n = 5; −/−: 18.5 ± 2.3 ng/mg, n = 5; p > 0.05) and increased in TKO mice at 24 months (+/+: 16.3 ± 1.8 ng/mg, n = 5; −/−: 24.6 ± 3.4 ng/mg, n = 4; *p < 0.05). (e) Levels of striatal HVA in TKO mice and wild-type controls at the ages of 16 months (+/+: 1.76 ± 0.28 ng/mg, n = 5; −/−: 1.98 ± 0.18 ng/mg, n = 5; p > 0.05) and 24 months (+/+: 1.65 ± 0.23 ng/mg, n = 5; −/−: 2.67 ± 0.65 ng/mg, n = 4; p > 0.05). (f) Levels of striatal DOPAC in TKO mice and wild-type controls at the ages of 16 months (+/+: 1.53 ± 0.20 ng/mg, n = 5; −/−: 1.42 ± 0.18 ng/mg, n = 5; p > 0.05) and 24 months (+/+: 1.42 ± 0.13 ng/mg, n = 5; −/−: 2.09 ± 0.46 ng/mg, n = 4; p > 0.05). All data are expressed as mean ± SEM.
Normal levels of striatal DA at 16 months but increased levels of striatal DA at 24 months in TKO mice
Through measurement of striatal levels of DA and its metabolites DOPAC and HVA using HPLC, we detected normal levels of DA in TKO mice at 16 months of age (+/+: 18.4 ± 2.0 ng/mg, n = 5; −/−: 18.5 ± 2.3 ng/mg, n = 5; p > 0.05), and significantly increased DA levels at 24 months (+/+: 16.3 ± 1.8 ng/mg, n = 5; −/−: 24.6 ± 3.4 ng/mg, n = 4; p < 0.05; Fig. 3d). Levels of HVA (+/+: 1.76 ± 0.28 ng/mg, n = 5; −/−: 1.98 ± 0.18 ng/mg, n = 5; p > 0.05) and DOPAC (+/+: 1.53 ± 0.20 ng/mg, n = 5; −/−: 1.42 ± 0.18 ng/mg, n = 5; p > 0.05) were normal in the striatum of TKO mice at 16 months of age. At 24 months, levels of HVA (+/+: 1.65 ± 0.23 ng/mg, n = 5; −/−: 2.67 ± 0.65 ng/mg, n = 4; p > 0.05) and DOPAC (+/+: 1.42 ± 0.13 ng/mg, n = 5; −/−: 2.09 ± 0.46 ng/mg, n = 4; p > 0.05) appeared to show increases in the TKO striatum, although the differences were not statistically significant (Fig. 3e and f).
Discussion
Recent studies have shown that Parkin, DJ-1, and PINK1 play important roles in mitochondrial function and protection against oxidative stress (Palacino et al. 2004; Menzies et al. 2005; Meulener et al. 2005; Clark et al. 2006; Park et al. 2006; Yang et al. 2006; Andres-Mateos et al. 2007; Gautier et al. 2008), disruption of which is thought to be involved in the pathogenesis of PD (Beal 2003). Thus, inactivation of all three recessive PD genes may have synergistic effects and cause more severe PD pathology than single gene inactivation. In fact, patients with digenic parkin and PINK1 mutations had a lower age of onset than those carrying the same single parkin mutation (Funayama et al. 2008). In another clinical report, individuals with digenic DJ-1 and PINK1 mutations suffered resting tremor of the left leg in their twenties, before it spread to all limbs (Tang et al. 2006). Thus, digenic mutations in recessive PD genes appeared to modify the clinical course and hasten the disease process, suggesting a synergistic effect among these genes.
Since the identification of Parkin, DJ-1, and PINK1 as causative genes for recessive PD, a large number of studies have been carried out to determine whether inactivation of each of these genes in mice or fruit flies results in progressive and selective loss of dopaminergic neurons (Goldberg et al. 2003, 2005; Greene et al. 2003; Itier et al. 2003; Pesah et al. 2004; Kim et al. 2005; Menzies et al. 2005; Meulener et al. 2005; Perez and Palmiter 2005; Yang et al. 2005, 2006; Clark et al. 2006; Park et al. 2006; Andres-Mateos et al. 2007; Gorner et al. 2007; Kitada et al. 2007; Wood-Kaczmar et al. 2008). Virtually all of the studies, including our series of genetic studies on parkin, DJ-1, and PINK1 single KO mice, reported no loss of dopaminergic neurons in the SNpc in mice lacking any one of the three genes (Goldberg et al. 2003, 2005; Itier et al. 2003; Kim et al. 2005; Perez and Palmiter 2005; Andres-Mateos et al. 2007; Gorner et al. 2007; Kitada et al. 2007; Wood-Kaczmar et al. 2008). Recently, double mutant mice carrying a parkin deletion and a transgene for Pael receptor (a putative Parkin substrate) were reported to exhibit progressive loss of neurons in the SN and LC (Wang et al. 2008). Generation of an animal model recapitulating selective, progressive dopaminergic degeneration, and neuronal death in the SNpc is crucial and will aid the investigation and elucidation of PD pathogenic mechanisms. We therefore generated TKO mice to create an exaggerated genetic alteration in an effort to produce a mouse model that would develop progressive dopaminergic neurodegeneration.
To our great surprise, inactivation of all three recessive PD genes which encoded proteins presumed to be essential for the survival of aging nigral neurons failed to reproduce the nigral cell loss in patients carrying mutations in Parkin, DJ-1, or PINK1. Our immunohistological analysis followed by stereological neuron count revealed similar numbers of dopaminergic neurons in the SNpc between triple KO and control mice up to 24 months of age. However, the total level of DA in the striatum was increased in an age-dependent manner in triple KO mice, with normal levels of striatal DA at 16 months and increased levels of striatal DA at 24 months of age. This was unlikely to be caused by increased DA synthesis as TH expression and activity were normal in triple KO mice. It is unclear how loss of all three recessive PD genes results in age-dependent disruption of DA homeostasis in the striatum.
In summary, our findings from triple KO studies demonstrated that inactivation of Parkin, DJ-1, and PINK1 was still insufficient to cause dopaminergic degeneration in mice during their lifespan. These results further support the notion that Parkin, DJ-1, and PINK1 are not absolutely required for the survival of nigral neurons. Rather, they are likely to provide protection to nigral neurons against insults, such as oxidative stress, during the decades-long aging process in human patients. Furthermore, genetic studies in Drosophila have shown that mutant flies lacking Parkin and/or PINK1 exhibit similar phenotypes, suggesting that they act in the same genetic pathway (Clark et al. 2006; Park et al. 2006; Yang et al. 2006). It is therefore possible that mammalian Parkin and PINK1 function in the same pathway, even though mutant mice lacking Parkin or PINK1 do not exhibit striking mitochondrial structural defects seen in mutant flies. If this were the case, triple KO mice essentially would be the same as double KO mice lacking DJ-1 and Parkin or PINK1. Thus, the lack of striking phenotypes in triple KO mice is consistent with the notion that mammalian Parkin and PINK1 may indeed function in the same pathway. Future investigation awaits us to determine whether the dopaminergic and mitochondrial dysfunctions caused by the loss of either Parkin or PINK1 worsen in the absence of both Parkin and PINK1, and whether these functional defects exacerbate in aged mutant mice.
Acknowledgements
We thank Huailong Zhao for assistance. This work was supported by a grant from the NINDS (R01 NS041779). The authors declare that they have no competing interests.
Abbreviations used
- DA
dopamine
- DOPA
3,4-dihydroxy-L-phenylalanine
- DOPAC
3,4-dihydroxyphenylacetic acid
- HPLC
high performance liquid chromatography
- HVA
homovanillic acid
- LC
locus coeruleus
- PD
Parkinson's disease
- SN
substantia nigra
- SNpc
substantia nigra pars compacta
- TH
tyrosine hydroxylase
- TKO
triple knockout
References
- Albanese A, Valente EM, Romito LM, Bellacchio E, Elia AE, Dallapiccola B. The PINK1 phenotype can be indistinguishable from idiopathic Parkinson disease. Neurology. 2005;64:1958–1960. doi: 10.1212/01.WNL.0000163999.72864.FD. [DOI] [PubMed] [Google Scholar]
- Andres-Mateos E, Perier C, Zhang L, et al. DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proc. Natl. Acad. Sci. USA. 2007;104:14807–14812. doi: 10.1073/pnas.0703219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrue A, Ruiz-Ortega JA, Ugedo L, Giralt MT. Short-term effects of 3,4-methylenedioximethamphetamine on noradrenergic activity in locus coeruleus and hippocampus of the rat. Neurosci. Lett. 2003;337:123–126. doi: 10.1016/s0304-3940(02)01252-1. [DOI] [PubMed] [Google Scholar]
- Beal MF. Mitochondria, oxidative damage, and inflammation in Parkinson's disease. Ann. N Y Acad. Sci. 2003;991:120–131. doi: 10.1111/j.1749-6632.2003.tb07470.x. [DOI] [PubMed] [Google Scholar]
- Bonifati V, Breedveld GJ, Squitieri F, et al. Localization of autosomal recessive early-onset parkinsonism to chromosome 1p36 (PARK7) in an independent dataset. Ann. Neurol. 2002;51:253–256. doi: 10.1002/ana.10106. [DOI] [PubMed] [Google Scholar]
- Bonifati V, Rizzu P, van Baren MJ, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- Dekker M, Bonifati V, van Swieten J, Leenders N, Galjaard RJ, Snijders P, Horstink M, Heutink P, Oostra B, van Duijn C. Clinical features and neuroimaging of PARK7-linked parkinsonism. Mov. Disord. 2003;18:751–757. doi: 10.1002/mds.10422. [DOI] [PubMed] [Google Scholar]
- Funayama M, Li Y, Tsoi TH, et al. Familial Parkinsonism with digenic parkin and PINK1 mutations. Mov. Disord. 2008;23:1461–1465. doi: 10.1002/mds.22143. [DOI] [PubMed] [Google Scholar]
- Gautier CA, Kitada T, Shen J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. USA. 2008;105:11364–11369. doi: 10.1073/pnas.0802076105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German DC, Liang CL, Manaye KF, Lane K, Sonsalla PK. Pharmacological inactivation of the vesicular monoamine transporter can enhance 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurodegeneration of midbrain dopaminergic neurons, but not locus coeruleus noradrenergic neurons. Neuroscience. 2000;101:1063–1069. doi: 10.1016/s0306-4522(00)00385-7. [DOI] [PubMed] [Google Scholar]
- Goldberg MS, Fleming SM, Palacino JJ, et al. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J. Biol. Chem. 2003;278:43628–43635. doi: 10.1074/jbc.M308947200. [DOI] [PubMed] [Google Scholar]
- Goldberg MS, Pisani A, Haburcak M, et al. Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron. 2005;45:489–496. doi: 10.1016/j.neuron.2005.01.041. [DOI] [PubMed] [Google Scholar]
- Gorner K, Holtorf E, Waak J, Pham TT, Vogt-Weisenhorn DM, Wurst W, Haass C, Kahle PJ. Structural determinants of the C-terminal helix-kink-helix motif essential for protein stability and survival promoting activity of DJ-1. J. Biol. Chem. 2007;282:13680–13691. doi: 10.1074/jbc.M609821200. [DOI] [PubMed] [Google Scholar]
- Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. USA. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinicopathological study of 100 cases. J. Neurol. Neurosurg. Psychiatry. 1992;55:181–184. doi: 10.1136/jnnp.55.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itier JM, Ibanez P, Mena MA, et al. Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum. Mol. Genet. 2003;12:2277–2291. doi: 10.1093/hmg/ddg239. [DOI] [PubMed] [Google Scholar]
- Junn E, Jang WH, Zhao X, Jeong BS, Mouradian MM. Mitochondrial localization of DJ-1 leads to enhanced neuroprotection. J. Neurosci. Res. 2009;87:123–129. doi: 10.1002/jnr.21831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim RH, Smith PD, Aleyasin H, et al. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc. Natl. Acad. Sci. USA. 2005;102:5215–5220. doi: 10.1073/pnas.0501282102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Kitada T, Pisani A, Porter DR, Yamaguchi H, Tscherter A, Martella G, Bonsi P, Zhang C, Pothos EN, Shen J. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc. Natl. Acad. Sci. USA. 2007;104:11441–11446. doi: 10.1073/pnas.0702717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Pisani A, Karouani M, Haburcak M, Martella G, Tscherter A, Platania P, Wu B, Pothos EN, Shen J. Impaired dopamine release and synaptic plasticity in the striatum of Parkin−/− mice. J. Neurochem. 2009;110:613–621. doi: 10.1111/j.1471-4159.2009.06152.x. [DOI] [PubMed] [Google Scholar]
- Menzies FM, Yenisetti SC, Min KT. Roles of Drosophila DJ-1 in survival of dopaminergic neurons and oxidative stress. Curr. Biol. 2005;15:1578–1582. doi: 10.1016/j.cub.2005.07.036. [DOI] [PubMed] [Google Scholar]
- Meulener M, Whitworth AJ, Armstrong-Gold CE, Rizzu P, Heutink P, Wes PD, Pallanck LJ, Bonini NM. Drosophila DJ-1 mutants are selectively sensitive to environmental toxins associated with Parkinson's disease. Curr. Biol. 2005;15:1572–1577. doi: 10.1016/j.cub.2005.07.064. [DOI] [PubMed] [Google Scholar]
- Moore DJ, Zhang L, Troncoso J, Lee MK, Hattori N, Mizuno Y, Dawson TM, Dawson VL. Association of DJ-1 and parkin mediated by pathogenic DJ-1 mutations and oxidative stress. Hum. Mol. Genet. 2005;14:71–84. doi: 10.1093/hmg/ddi007. [DOI] [PubMed] [Google Scholar]
- Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J. Biol. Chem. 2004;279:18614–18622. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- Perez FA, Palmiter RD. Parkin-deficient mice are not a robust model of parkinsonism. Proc. Natl. Acad. Sci. USA. 2005;102:2174–2179. doi: 10.1073/pnas.0409598102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesah Y, Pham T, Burgess H, Middlebrooks B, Verstreken P, Zhou Y, Harding M, Bellen H, Mardon G. Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development. 2004;131:2183–2194. doi: 10.1242/dev.01095. [DOI] [PubMed] [Google Scholar]
- Taira T, Saito Y, Niki T, Iguchi-Ariga SM, Takahashi K, Ariga H. DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 2004;5:213–218. doi: 10.1038/sj.embor.7400074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Ohama E, Suzuki S, Horikawa Y, Ishikawa A, Morita T, Tsuji S, Ikuta F. Familial juvenile parkinsonism: clinical and pathologic study in a family. Neurology. 1994;44:437–441. doi: 10.1212/wnl.44.3_part_1.437. [DOI] [PubMed] [Google Scholar]
- Tang B, Xiong H, Sun P, et al. Association of PINK1 and DJ-1 confers digenic inheritance of early-onset Parkinson's disease. Hum. Mol. Genet. 2006;15:1816–1825. doi: 10.1093/hmg/ddl104. [DOI] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- Wang HQ, Imai Y, Inoue H, Kataoka A, Iita S, Nukina N, Takahashi R. Pael-R transgenic mice crossed with parkin deficient mice displayed progressive and selective catecholaminergic neuronal loss. J. Neurochem. 2008;107:171–185. doi: 10.1111/j.1471-4159.2008.05607.x. [DOI] [PubMed] [Google Scholar]
- Wood-Kaczmar A, Gandhi S, Yao Z, et al. PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS ONE. 2008;3:e2455. doi: 10.1371/journal.pone.0002455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Shen J. Absence of dopaminergic neuronal degeneration and oxidative damage in aged DJ-1-deficient mice. Mol. Neurodegener. 2007;2:10. doi: 10.1186/1750-1326-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamura Y, Sobue I, Ando K, Iida M, Yanagi T. Paralysis agitans of early onset with marked diurnal fluctuation of symptoms. Neurology. 1973;23:239–244. doi: 10.1212/wnl.23.3.239. [DOI] [PubMed] [Google Scholar]
- Yamamura Y, Arihiro K, Kohriyama T, Nakamura S. Early-onset parkinsonism with diurnal fluctuation: clinical and pathological studies. Rinsho Shinkeigaku. 1993;33:491–496. [PubMed] [Google Scholar]
- Yamamura Y, Hattori N, Matsumine H, Kuzuhara S, Mizuno Y. Autosomal recessive early-onset parkinsonism with diurnal fluctuation: clinicopathologic characteristics and molecular genetic identification. Brain Dev. 2000;22(Suppl. 1):S87–91. doi: 10.1016/s0387-7604(00)00130-3. [DOI] [PubMed] [Google Scholar]
- Yang Y, Gehrke S, Haque ME, et al. Inactivation of Drosophila DJ-1 leads to impairments of oxidative stress response and phosphatidylinositol 3-kinase/Akt signaling. Proc. Natl. Acad. Sci. USA. 2005;102:13670–13675. doi: 10.1073/pnas.0504610102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang Y, Wang JW, Yang L, Beal MF, Vogel H, Lu B. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc. Natl. Acad. Sci. USA. 2006;103:10793–10798. doi: 10.1073/pnas.0602493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota T, Sugawara K, Ito K, Takahashi R, Ariga H, Mizusawa H. Down regulation of DJ-1 enhances cell death by oxidative stress, ER stress, and proteasome inhibition. Biochem. Biophys. Res. Commun. 2003;312:1342–1348. doi: 10.1016/j.bbrc.2003.11.056. [DOI] [PubMed] [Google Scholar]
- Zhang L, Shimoji M, Thomas B, et al. Mitochondrial localization of the Parkinson's disease related protein DJ-1: implications for pathogenesis. Hum. Mol. Genet. 2005;14:2063–2073. doi: 10.1093/hmg/ddi211. [DOI] [PubMed] [Google Scholar]