

Abstract
Simulation studies are useful in various disciplines for a number of reasons including the development and evaluation of new computational and statistical methods. This is particularly true in human genetics and genetic epidemiology where new analytical methods are needed for the detection and characterization of disease susceptibility genes whose effects are complex, nonlinear, and partially or solely dependent on the effects of other genes (i.e. epistasis or gene-gene interaction). Despite this need, the development of complex genetic models that can be used to simulate data is not always intuitive. In fact, only a few such models have been published. We have previously developed a genetic algorithm approach to discovering complex genetic models in which two single nucleotide polymorphisms (SNPs) influence disease risk solely through nonlinear interactions. In this paper, we extend this approach for the discovery of high-order epistasis models involving three to five SNPs. We demonstrate that the genetic algorithm is capable of routinely discovering interesting high-order epistasis models in which each SNP influences risk of disease only through interactions with the other SNPs in the model. This study opens the door for routine simulation of complex gene-gene interactions among SNPs for the development and evaluation of new statistical and computational approaches for identifying common, complex multifactorial disease susceptibility genes.
Keywords: Gene-Gene Interactions, Simulation, Penetrance, Genetic Epidemiology
Introduction
One goal of human genetics is to identify genes that confer increased susceptibility to certain diseases. The identification of disease susceptibility genes has the potential to improve human health through the development of new prevention, diagnosis, and treatment strategies. Although achieving this goal is an important public health endeavor, it is not easily accomplished for common diseases, such as essential hypertension, due to the complex multifactorial etiology of the disease [8,13]. That is, risk of disease is due to a complex interplay between multiple genes and multiple environmental factors. Such gene-gene interactions (i.e. epistasis) and gene-environment interactions (i.e. plastic reaction norms) are expected to be ubiquitous in determining susceptibility to common human diseases [11], are examples of attribute interactions in data mining [4], and represent a significant statistical challenge in human genetics [11,13,16]. The statistical challenge is partly due to the curse of dimensionality [1]. That is, when high-order interactions are modeled, there are many genotype combinations (i.e. contingency table cells) for which there are no observed data. As a result, parametric statistical methods such as logistic regression have limited power and perhaps an increased false-positive or type I error rate. Multifactor dimensionality reduction (MDR) is a nonparametric and genetic-model free classification method that was developed to address this problem [7,13-15]. Although promising, the power of MDR to identify high-order gene-gene interactions has not been fully evaluated due to a lack of epistasis models in the literature that can be used to simulate data.
The lack of appropriate models that can be used for simulation is partly due to the combinatorial and computational complexities associated with the model discovery process [2]. The goal of the present study is to develop a genetic algorithm approach to model discovery that overcomes some of the combinatorial and computational limitations. We demonstrate here that the GA strategy is able to routinely discover models of gene-gene interaction effects among three to five total SNPs. The availability of high-order gene-gene interaction models will facilitate the simulation of data of varying complexity for the evaluation of new statistical and computational methods for identifying gene-gene interactions.
Related Research
Penetrance functions represent one approach to modeling the relationship between single nucleotide polymorphisms or SNPs (i.e. interindividual variation in a single nucleotide at a particular location in the DNA sequence of a gene) and risk of disease. A penetrance function is simply the probability (P) that an individual will have the disease (D) given a particular genotype or combination of genotypes (G) from multiple genes (i.e. P[D|G]). A single genotype is determined by one allele (i.e. a specific DNA sequence state) inherited from the mother and one allele inherited from the father. For most genetic variations (i.e. SNPs), only two alleles (A or a) exist in the biological population. Therefore, because the order of the alleles is unimportant, a genotype can have one of three values: AA, Aa or aa. Penetrance functions define the probability (P) of disease (D) for genotypes (G) for one or more genetic variations (e.g. P[D|G]=0.5). Once the penetrance functions are specified, genetic data can easily be simulated for people with the disease and for people without the disease. For example, consider the penetrance function for an autosomal recessive Mendelian disease (i.e. a disease that requires two copies of the same allele) such as cystic fibrosis in which only one of the three genotypes at a particular locus leads to disease. Here, individuals who inherit the AA or Aa genotypes have zero probability of disease while individuals who inherit the aa genotype are certain to have the disease. From this simple recessive Mendelian model, data can simply be simulated by giving affected individuals aa genotypes and unaffected individuals AA or Aa genotypes, in proportion to their defined population frequencies.
More complex genetic models can be developed by assigning disease risk to more than one genotype from one or more SNPs. Table 1 illustrates a penetrance function that relates two genetic variations, each with two alleles and three genotypes, to risk of disease. In this example, the alleles each have a biological population frequency of p = q = 0.5 with genotype frequencies of p2 for AA and BB, 2pq for Aa and Bb, and q2 for aa and bb, consistent with Hardy-Weinberg equilibrium. Thus, assuming the frequency of the AA genotype is 0.25, the frequency of Aa is 0.5, and the frequency of aa is 0.25, then the marginal penetrance of BB (i.e. the effect of just the BB genotype on disease risk) can be calculated as (0.25 * 0) + (0.5 * 0) + (0.25 * 1) = 0.25. This means that the probability of disease given the BB genotype is 0.25, regardless of the genotype at the other genetic variation. Similarly, the marginal penetrance of Bb can be calculated as (0.25 * 0) + (0.5 * 0.5) + (0.25 * 0) = 0.25. Note that for this model, all of the marginal penetrance values (i.e. the probability of disease given a single genotype, independent of the others) are equal, which indicates the absence of main effects (i.e. the genetic variations do not independently affect disease risk). This is true despite the table penetrance values not being equal. Here, risk of disease is greatly increased by inheriting exactly two high-risk alleles (e.g. a and b are defined as high risk). Thus, aa/BB, Aa/Bb, and AA/bb are the high-risk genotype combinations. This model was first described by Frankel and Schork [3] and was rediscovered by our two-SNP genetic algorithm [12]. What makes this model complex is the absence of a main effect for either of the SNPs. Thus, each SNP only has an effect on disease risk in the context of the other SNP. Such gene-gene interactions are believed to play an important role in determining an individual's risk for developing common diseases [11,13,16].
Table 1. Penetrance values for combinations of genotypes from two SNPs exhibiting interactions in the absence of independent main effects.
| Table penetrance | Margin penetrance | |||
|---|---|---|---|---|
| AA (.25) | Aa (.50) | aa (.25) | ||
| BB (.25) | 0 | 0 | 1 | 0.25 |
| Bb (.50) | 0 | 0.50 | 0 | 0.25 |
| bb (.25) | 1 | 0 | 0 | 0.25 |
| Margin penetrance | 0.25 | 0.25 | 0.25 | |
The gene-gene interaction model described in Table 1 was developed by trial and error. That is, a human derived this model by substituting various genotype frequencies and penetrance functions until a model was found that had gene-gene interaction effects without independent main effects. This is one of only a few complex genetic models that have been described in the literature. The scarcity of complex genetic models in the literature is primarily due to the extraordinary combinatorial complexity of the problem [2]. Effectively, there are an infinite number of possible penetrance functions that could be developed for just two genetic variations. Only some of these models would exhibit a complex relationship with disease risk. The size of the search space precludes the human-based trial and error approach as well as exhaustive computational searches without specific restrictions and assumptions about the genotype frequency and penetrance function values. For example, Li and Reich [10] enumerated every possible penetrance function using probability values restricted to zero and one (i.e. fully penetrant). This yielded a manageable 29 total models. Only one of these models exhibits interaction effects in the absence of main effects (see Table 2). Culverhouse et al. [2] have also enumerated a restricted set of models. The difficulties outlined here provided the motivation for the development of our genetic algorithm strategy for the routine discovery of two-locus epistasis models [12]. Using this GA strategy, we discovered more than 99,900 new models that were previously not described in the literature. The goal of the present study was to improve gene-gene interaction model discovery by developing a GA that can identify high-order models of interactions among three to five SNPs. To our knowledge, this is the first machine learning approach developed specifically for the discovery of high-order gene-gene interactions model. The next section describes the GA approach we used.
Table 2. Penetrance values for combinations of genotypes from two SNPs exhibiting interactions in the absence of independent main effects.
| Table penetrance | Margin penetrance | |||
|---|---|---|---|---|
| AA (.25) | Aa (.50) | aa (.25) | ||
| BB (.25) | 0 | 1 | 0 | 0.5 |
| Bb (.50) | 1 | 0 | 1 | 0.5 |
| bb (.25) | 0 | 1 | 0 | 0.5 |
| Margin penetrance | 0.5 | 0.5 | 0.5 | |
The Genetic Algorithm Strategy
Overview of Genetic Algorithms
Genetic algorithms, neural networks, case-based learning, rule induction, and analytic learning are some of the more popular paradigms in machine learning [9]. Genetic algorithms perform a beam or parallel search of the solution space that is analogous to the problem solving abilities of biological populations undergoing evolution by natural selection [5,6]. With this procedure, a randomly generated ‘population’ of solutions to a particular problem are generated and then evaluated for their ‘fitness’ or ability to solve the problem. Here, solutions are encoded as binary arrays or chromosomes. The highest fit chromosomes in the population are selected and then undergo exchanges of random model pieces, a process that is also referred to as recombination. Recombination generates variability among the solutions and is the key to the success of the beam search, just as it is a key part of evolution by natural selection. Following recombination, the models are reevaluated and the cycle of selection, recombination, and evaluation continues until an optimal solution is identified.
Solution Representation and Fitness Determination
In the two-SNP case [12], a solution or model consists of a set of nine penetrance values or probabilities on the interval from zero to one in increments of 0.001. Thus, the entire search space consisted of 10019 possible models. Each penetrance value represents the probability of disease given a particular combination of two genotypes. Each of the nine real-valued probabilities was encoded as 32 bits for a total GA chromosome length of 288 bits.
Using the GA to generate high-order models of three to five SNPs requires larger GA chromosomes. In the three-SNP case, there are a total of 27 penetrance values or probabilities (i.e. one for each multilocus genotype combination) on the interval from zero to one in increments of 0.1. Thus, with each real-valued probability encoded as 32 bits, a total GA chromosome length of 864 bits is required (e.g. see Figure 1). Similarly, in the four-SNP case, there are a total of 81 penetrance values requiring a total GA chromosome length of 2592 bits. In the five-SNP case, there are a total of 243 penetrance values requiring a total GA chromosome length of 7776 bits. The size of the search space is 1127 for three SNPs, 1181 for four SNPs, and 11243 for five SNPs. Thus, the identification of high-order gene-gene interaction models requires searching an effectively-infinite solution space.
Figure 1.
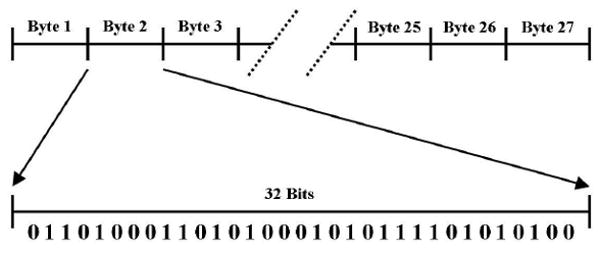
Illustration of a GA chromosome encoding 27 penetrance values using 32-bit bytes.
Fitness was determined by maximizing the variance of the table penetrance values (Vt) and minimizing the variance of the marginal penetrance values (Vm). Maximizing Vt ensures that we identify interesting patterns of genotypes while minimizing Vm ensures the size of the main effect of each genotype is small. We stopped the GA when a model satisfied both Vt ≥ 0.1 and Vm ≤ 0.0001 for three-locus models and Vt ≥ 0.2 and Vm ≤ 0.0001 for four- and five-locus models. These values were selected to ensure gene-gene interaction effects with minimal main effects for each SNP.
Genetic Algorithm Parameter Settings
A total population size of 200 chromosomes or solutions was used. The fitness function used was Vt - Vm. A recombination frequency of 0.6 and a mutation frequency of 0.01 were used. Stochastic uniform selection was used in addition to a uniform crossover operator. Additional details are given by Moore et al. [12].
Hardware and Software
Our GA implementation used GAlib, a C++ class library for UNIX, Windows and Mac operating systems (http://lancet.mit.edu/ga/). Coarse-grained parallelism, utilizing 10 processors to perform 10 sets of 100 runs, for a total of 1,000 runs, used the MPICH parallel programming library on a 110-node Beowulf-style parallel computing cluster running Linux.
Experimental Design
We ran the GA a total of 1,000 times with each run consisting of a maximum of 1,000 generations for three-locus models and 5,000 generations for four- and five-locus models. The GA was executed for the maximum number of generations unless a single best model was identified that met the fitness criteria of Vt ≥ 0.1 and Vm ≤ 0.0001 for three-SNP models and Vt ≥ 0.2 and Vm ≤ 0.0001 for four- and five-SNP models.
Results
For each of the 1,000 runs of the GA, a best model was identified that met the criteria of Vt ≥ 0.1 and Vm ≤ 0.0001 for three-SNP models and Vt ≥ 0.2 and Vm ≤ 0.0001 for four- and five-SNP models. Thus, the GA routinely identified epistasis models with no or little independent main effects. Further, of the 1,000 models identified for each number of SNPs, there were no duplicates. Thus, the GA discovered 1,000 unique three-, four-, and five-SNP gene-gene interaction models exhibiting minimal independent main effects.
Interaction Models Including Three SNPs
Table 3 illustrates one of 1,000 three-SNP gene-gene interaction penetrance functions discovered by the GA (Vt = 0.1023, Vm = 0.000037). Note the nonlinear distribution of probability values for each combination of three SNP genotypes. This is indicative of gene-gene interaction in the sense that the effect of any one genotype on disease risk is dependent on genotypes from the other SNPs. For example, the independent main effect of the AA genotype (i.e. P[D|AA]) is 0.475. However, the probability of disease given the genotype combination AA, Bb and CC is 0.1 while the probability of disease given the genotype combination AA, BB and cc is 1.0. Thus, the effect of the AA genotype is dependent on the context of the other genotypes. The independent main effects of the other genotypes are as follows: P(D|Aa) = 0.487, P(D|aa) = 0.475, P(D|BB) = 0.475, P(D|Bb) = 0.481, P(D|bb) = 0.487, P(D|CC) = 0.475, P(D|Cc) = 0.487, and P(D|cc) = 0.475.
Table 3. Penetrance values for combinations of genotypes from three SNPs exhibiting interactions in the absence of independent main effects.
| CC | Cc | cc | |||||||
|---|---|---|---|---|---|---|---|---|---|
| AA | Aa | aa | AA | Aa | aa | AA | Aa | aa | |
| BB | 0.4 | 0.9 | 0.7 | 0.2 | 0.2 | 0.6 | 1.0 | 0.4 | 0.5 |
| Bb | 0.9 | 0.0 | 0.9 | 0.6 | 0.9 | 0.0 | 0.3 | 0.1 | 0.6 |
| bb | 0.1 | 0.2 | 0.6 | 0.3 | 0.6 | 0.3 | 0.3 | 0.9 | 1.0 |
Interaction Models Including Four SNPs
Table 4 illustrates one of 1,000 four-SNP gene-gene interaction penetrance functions discovered by the GA (Vt = 0.2004, Vm = 0.000032). Again, note the nonlinear distribution of probability values for each combination of four SNP genotypes. For example, probability of disease given the genotype combination Aa, BB, CC and DD is 1.0 while the probability of disease given the genotype combination Aa, Bb, Cc and dd is 0. The independent main effects or marginal penetrance of Aa is 0.5. Thus, the effect of the Aa genotype is dependent on the context of the other genotypes. The independent main effects of the other genotypes are also 0.5.
Table 4. Penetrance values for combinations of genotypes from four SNPs exhibiting interactions in the absence of independent main effects.
| CC | Cc | cc | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| AA | Aa | aa | AA | Aa | aa | AA | Aa | aa | ||
| DD | BB | 0.9 | 1.0 | 0.0 | 0.9 | 0.1 | 0.1 | 1.0 | 0.9 | 1.0 |
| Bb | 0.9 | 0.0 | 0.8 | 1.0 | 0.0 | 1.0 | 0.1 | 0.0 | 0.0 | |
| bb | 0.1 | 1.0 | 0.9 | 1.0 | 0.1 | 0.8 | 0.9 | 0.9 | 1.0 | |
| Dd | BB | 0.0 | 0.0 | 0.8 | 0.1 | 1.0 | 0.1 | 0.0 | 1.0 | 0.1 |
| Bb | 1.0 | 0.1 | 1.0 | 0.0 | 0.8 | 0.7 | 0.0 | 1.0 | 0.0 | |
| bb | 0.1 | 1.0 | 0.0 | 1.0 | 0.3 | 0.1 | 0.0 | 0.0 | 0.2 | |
| dd | BB | 0.9 | 0.1 | 0.0 | 1.0 | 0.0 | 0.0 | 0.9 | 1.0 | 0.7 |
| Bb | 0.9 | 0.0 | 0.0 | 0.0 | 0.5 | 1.0 | 0.0 | 1.0 | 1.0 | |
| bb | 1.0 | 0.9 | 1.0 | 1.0 | 0.2 | 0.3 | 0.9 | 0.0 | 0.0 | |
Interaction Models Including Five SNPs
Table 5 below illustrates one of 1,000 five-SNP gene-gene interaction penetrance functions discovered by the GA (Vt = 0.2004, Vm = 0.000032). As with the three- and four-SNP models, there is a nonlinear distribution of probability values for each combination of five SNP genotypes.
Table 5. Penetrance values for combinations of genotypes from five SNPs exhibiting interactions in the absence of independent main effects.
| CC | Cc | cc | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| AA | Aa | aa | AA | Aa | aa | AA | Aa | aa | |||
| EE | DD | BB | 0.6 | 0.6 | 0.0 | 0.1 | 1.0 | 1.0 | 0.1 | 1.0 | 0.0 |
| Bb | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 1.0 | 0.3 | 0.9 | ||
| bb | 1.0 | 0.1 | 0.0 | 0.9 | 1.0 | 0.0 | 0.0 | 0.1 | 1.0 | ||
| Dd | BB | 1.0 | 1.0 | 0.0 | 0.0 | 1.0 | 1.0 | 0.8 | 0.1 | 1.0 | |
| Bb | 0.0 | 1.0 | 0.9 | 0.0 | 0.0 | 0.9 | 0.0 | 1.0 | 0.9 | ||
| bb | 1.0 | 0.1 | 0.0 | 0.4 | 0.9 | 1.0 | 1.0 | 0.9 | 0.1 | ||
| dd | BB | 0.9 | 0.8 | 0.4 | 1.0 | 0.0 | 1.0 | 0.1 | 0.0 | 0.0 | |
| Bb | 0.3 | 1.0 | 0.0 | 1.0 | 0.8 | 0.0 | 0.0 | 1.0 | 0.0 | ||
| bb | 1.0 | 0.9 | 0.0 | 0.1 | 0.0 | 0.1 | 0.0 | 0.1 | 0.0 | ||
| Ee | DD | BB | 1.0 | 0.0 | 1.0 | 0.8 | 0.9 | 0.0 | 0.0 | 0.0 | 0.0 |
| Bb | 0.5 | 1.0 | 0.0 | 0.7 | 1.0 | 0.8 | 1.0 | 0.1 | 0.1 | ||
| bb | 1.0 | 0.0 | 0.2 | 0.0 | 1.0 | 1.0 | 0.3 | 0.0 | 1.0 | ||
| Dd | BB | 0.0 | 1.0 | 0.0 | 0.1 | 0.0 | 0.9 | 0.0 | 0.9 | 1.0 | |
| Bb | 1.0 | 0.9 | 0.0 | 1.0 | 0.0 | 1.0 | 1.0 | 0.1 | 1.0 | ||
| bb | 0.0 | 0.7 | 1.0 | 0.9 | 0.0 | 0.3 | 0.1 | 0.0 | 0.9 | ||
| dd | BB | 1.0 | 0.9 | 0.4 | 0.8 | 0.1 | 0.0 | 1.0 | 0.1 | 0.0 | |
| Bb | 0.2 | 0.2 | 0.2 | 1.0 | 0.0 | 0.1 | 0.9 | 0.9 | 0.0 | ||
| bb | 0.0 | 1.0 | 0.5 | 0.0 | 1.0 | 0.4 | 0.9 | 1.0 | 0.0 | ||
| ee | DD | BB | 0.8 | 1.0 | 1.0 | 0.1 | 1.0 | 0.9 | 0.9 | 1.0 | 0.1 |
| Bb | 0.7 | 0.0 | 0.2 | 0.8 | 0.0 | 1.0 | 0.9 | 0.1 | 0.9 | ||
| bb | 0.1 | 0.0 | 0.1 | 1.0 | 0.2 | 0.7 | 0.0 | 0.4 | 1.0 | ||
| Dd | BB | 0.0 | 0.0 | 1.0 | 0.0 | 1.0 | 1.0 | 0.1 | 0.0 | 0.1 | |
| Bb | 0.9 | 0.2 | 0.0 | 0.0 | 1.0 | 0.0 | 0.1 | 0.0 | 0.9 | ||
| bb | 0.0 | 1.0 | 1.0 | 0.3 | 0.9 | 0.0 | 1.0 | 0.0 | 1.0 | ||
| dd | BB | 0.0 | 0.1 | 0.9 | 0.0 | 1.0 | 0.0 | 1.0 | 0.8 | 1.0 | |
| Bb | 0.6 | 0.1 | 0.0 | 0.0 | 1.0 | 0.0 | 1.0 | 1.0 | 0.8 | ||
| bb | 0.1 | 1.0 | 1.0 | 0.0 | 1.0 | 0.9 | 0.0 | 1.0 | 1.0 | ||
Discussion
We have introduced a genetic algorithm (GA) approach to identifying penetrance functions that model epistasis or gene-gene interactions among three to five SNPs in the absence of independent main effects. The development of this GA approach and our initial two-SNP version [12] was motivated by a lack of published gene-gene interaction models and a lack of methods for generating them. We find that the GA is capable of routinely discovering interesting epistasis penetrance functions with up to five SNPs in a search space that consists of as many as 11243 possible models. We anticipate this approach will be useful for generating complex genetic models in the form of penetrance functions that can be used to simulate data for the development and evaluation of new analytical methods for the detection and characterization of gene-gene interactions or epistasis. For example, we have recently developed the multifactor dimensionality reduction (MDR) method for detecting gene-gene interactions in epidemiological study designs [7,13-15]. The power of this approach has been evaluated using data simulated from the models illustrated in Tables 1 and 2 [14]. Although MDR has excellent power for identifying functional SNPs from these two-locus models, it is not known how MDR will perform on a variety of other perhaps more complex or higher-order models. The results of this study will facilitate a comprehensive evaluation of the power of MDR and other methods using simulated data.
Future Research and Conclusion
An important next step in the discovery of complex genetic models will be to determine the diversity and number of models that exhibit epistasis in the absence of main effects for any given number of SNPs. How many total models exist that have gene-gene interaction properties? How many exist for different allele and genotype frequencies? In this study, we identified 1,000 models that have different probability values for some or all of the genotype combinations. Even though the probabilities are different in the penetrance function matrix, are all of these really different models? Are there classes of models that have different probabilities but similar functional properties? Addressing this question will depend on developing a metric for summarizing the patterns of probabilities in a penetrance function for comparison with others. For example, two penetrance functions that are mirror images of one another should probably be grouped together in the same class since they will be functionally identical. Perhaps the most relevant metric for geneticists is heritability. Heritability is a measure on the scale from zero to one that reflects the proportion of variability in a trait that is due to genetic variation. A heritability of one indicates the trait is completely genetically determined. Culverhouse et al. [2] have outlined a mathematical approach to calculating heritability from penetrance tables such as those presented here. Determining the best way to classify models into functional groups will be the focus of future studies.
Although the models generated in this study exhibit gene-gene interactions in the absence of main effects of any single SNP, it is possible that intermediate sub-models exist that have significant effects. For example, in the five-locus models, it is possible that there are perhaps two of the five SNPs that exhibit an effect independent of the other three. Thus, a method like MDR [7,13-15] might identify the two-locus model in addition the higher-order five-locus model creating some confusion as to which is actually the functional set of SNPs. The fitness function we have designed in this study does not account for this possibility. We only require the individual loci not have an independent effect while the entire model does have an effect. In future studies, we will explore more complex fitness functions that provide more control over the effects of subgroups of loci in the model.
Finally, it will be of interest to begin relating some of the simulated models to biological models. How many of the models generated in this study are biologically plausible? Speculating on biological plausibility may require a collaborative effort among clinical, molecular, and statistical geneticists. Whether or not these models resemble biological models may not be important for the evaluation of new statistical and computational approaches. A diversity of models of varying complexity, regardless of biological plausibility, may be necessary to fully evaluate the performance of a particular method since the full spectrum of biological models is most likely not yet fully characterized.
In summary, we have developed a GA approach to generating penetrance functions that exhibit epistasis or gene-gene interactions effects among three to five SNPs in the absence of any independent main effects of each SNP. Each run of the GA identified a different model meeting the epistasis criteria suggesting model discovery by the GA is routine. This study provides a useful starting point for those hoping to use complex genetic models for simulating genetic data.
Acknowledgments
This work was supported by National Institutes of Health grants HL65234, HL65962, GM31304, AG19085, AG20135, and LM007450. We thank an anonymous referee for their thoughtful comments and suggestions.
Footnotes
Commercial Benefits: none
References
- 1.Bellman R. Adaptive Control Processes. Princeton University Press; 1961. [Google Scholar]
- 2.Culverhouse R, Suarez BK, Lin J, Reich T. A perspective on epistasis: limits of models displaying no main effect. American Journal of Human Genetics. 2002;70:461–71. doi: 10.1086/338759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frankel WN, Schork NJ. Who's afraid of epistasis? Nature Genetics. 1996;14:371–373. doi: 10.1038/ng1296-371. [DOI] [PubMed] [Google Scholar]
- 4.Freitas AA. Understanding the crucial role of attribute interaction in data mining. Artificial Intelligence Reviews. 2001;16:177–199. [Google Scholar]
- 5.Goldberg DE. Genetic Algorithms in Search, Optimization, and Machine Learning. Addison-Wesley; Reading: 1989. [Google Scholar]
- 6.Holland JH. Adaptation in Natural and Artificial Systems. University of Michigan Press; Ann Arbor: 1975. [Google Scholar]
- 7.Hahn LW, Ritchie MD, Moore JH. Multifactor dimensionality reduction software for detecting gene-gene and gene-environment interactions. Bioinformatics. 2003;19:376–82. doi: 10.1093/bioinformatics/btf869. [DOI] [PubMed] [Google Scholar]
- 8.Kardia SLR. Context-dependent genetic effects in hypertension. Current Hypertension Reports. 2000;2:32–38. doi: 10.1007/s11906-000-0055-6. [DOI] [PubMed] [Google Scholar]
- 9.Langley P. Elements of Machine Learning. Morgan Kaufmann Publishers; San Francisco: 1996. [Google Scholar]
- 10.Li W, Reich J. A complete enumeration and classification of two-locus disease models. Human Heredity. 2000;50:334–349. doi: 10.1159/000022939. [DOI] [PubMed] [Google Scholar]
- 11.Moore JH. The ubiquitous nature of epistasis in determining susceptibility to common human diseases. Human Heredity. 2003 doi: 10.1159/000073735. in press. [DOI] [PubMed] [Google Scholar]
- 12.Moore JH, Hahn LW, Ritchie MD, Thornton TA, White BC. Application of genetic algorithms to the discovery of complex genetic models for simulation studies in human genetics. In: Langdon WB, et al., editors. Proceedings of the Genetic and Evolutionary Computation Conference. Morgan Kaufmann Publishers; San Francisco: 2002. [PMC free article] [PubMed] [Google Scholar]
- 13.Moore JH, Williams SW. New strategies for identifying gene-gene interactions in hypertension. Annals of Medicine. 2002;34:88–95. doi: 10.1080/07853890252953473. [DOI] [PubMed] [Google Scholar]
- 14.Ritchie MD, Hahn LW, Moore JH. Power of multifactor dimensionality reduction for detecting gene-gene interactions in the presence of genotyping error, phenocopy, and genetic heterogeneity. Genetic Epidemiology. 2003;24:150–57. doi: 10.1002/gepi.10218. [DOI] [PubMed] [Google Scholar]
- 15.Ritchie MD, Hahn LW, Roodi N, Bailey LR, Dupont WD, Parl FF, Moore JH. Multifactor dimensionality reduction reveals high-order interactions among estrogen metabolism genes in sporadic breast cancer. American Journal of Human Genetics. 2001;69:138–147. doi: 10.1086/321276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Templeton AR. Epistasis and complex traits. In: Wolf J, Brodie B III, Wade M, editors. Epistasis and the Evolutionary Process. Oxford University Press; New York: 2000. [Google Scholar]