

Abstract
Maternal infection during pregnancy with a wide range of RNA and DNA viruses is associated with increased risk for schizophrenia and autism in their offspring. A common feature in these exposures is that virus replication induces innate immunity through interaction with Toll-like receptors (TLRs). We employed a mouse model wherein pregnant mice were exposed to polyinosinic-polycytidylic acid [poly(I ⋅ C)], a synthetic, double-stranded RNA molecular mimic of replicating virus. Poly(I ⋅ C) inhibited embryonic neuronal stem cell replication and population of the superficial layers of the neocortex by neurons. Poly(I ⋅ C) also led to impaired neonatal locomotor development and abnormal sensorimotor gating responses in adult offspring. Using Toll-like receptor 3 (TLR3)-deficient mice, we established that these effects were dependent on TLR3. Inhibition of stem cell proliferation was also abrogated by pretreatment with the nonsteroidal anti-inflammatory drug (NSAID) carprofen, a cyclooxygenase (COX) inhibitor. Our findings provide insights into mechanisms by which maternal infection can induce subtle neuropathology and behavioral dysfunction, and they may suggest strategies for reducing the risk of neuropsychiatric disorders subsequent to prenatal exposures to pathogens and other triggers of innate immunity.
IMPORTANCE
Maternal infection during gestation increases the risk of neuropsychiatric disorders in their offspring. Furthermore, work in animal models indicates that pre- or neonatal infections with a wide range of viruses results in similar neurodevelopmental outcomes. These observations are consistent with a mechanism whereby damage is mediated through common pathways. Exposure of pregnant mice to polyinosinic-polycytidylic acid [poly(I ⋅ C)], a synthetic, double-stranded RNA (dsRNA) molecular mimic of replicating virus, inhibited embryonic neuronal stem cell replication and led to behavioral abnormalities in their offspring. These effects were mediated through TLR3 and abrogated by pretreatment with the nonsteroidal anti-inflammatory drug (NSAID) carprofen. Our findings provide insights into mechanisms by which maternal infection can induce subtle neuropathology and may suggest strategies for reducing the risk of neuropsychiatric diseases following exposures to infectious agents and other triggers of innate immunity during gestation.
INTRODUCTION
Infection during pregnancy is associated with increased risk of schizophrenia and autism in offspring (1). Several lines of evidence indicate that the maternal immune response, rather than direct infection of the fetus, may be responsible for the increased incidence of schizophrenia and autism in the offspring of mothers who suffer infection during pregnancy (2, 3). In a rodent model of maternal immune activation, changes in the behavior and neuroanatomy of the offspring are elicited by injection into the mother of synthetic double-stranded RNA (dsRNA) [poly(I ⋅ C)], a compound that evokes an antiviral-like innate immune response (4, 5). Behavioral abnormalities observed in this model are attributed to the disrupted balance of proinflammatory and anti-inflammatory cytokines produced in the mother (2). Using a similar mouse model, Meyer et al. showed in vivo evidence for a modulation of fetal dopaminergic system development by maternal immune activation during pregnancy (6). Furthermore, injection of poly(I ⋅ C) into pregnant rodents induces strong innate immune responses in the absence of an infection, leading to abnormal gene regulation and defective corticogenesis (5, 7). The breadth of microbes linked to neurodevelopmental abnormalities, including rubella virus, cytomegalovirus, influenza virus, and herpes simplex viruses, as well as Toxoplasma gondii and Corynebacterium diphtheriae (2, 8–14), is consistent with a mechanism whereby infection triggers innate immunity through common signaling pathways. Leading candidates for mediating these effects of pathogens include the Toll-like receptors (TLRs). TLRs bind a wide range of molecules associated with microbes, including flagellar structures, lipids and lipopeptides, zymosan, dsRNA molecules, U-rich single-stranded RNA, and unmethylated CpG dinucleotides (15, 16). Binding of these pathogen-associated molecular patterns (PAMPs) to their cognate TLRs triggers signal transduction events culminating in transcription of genes encoding interferon and cytokines and other genes that contribute to both innate and adaptive immunity (15). The dsRNA viral mimic poly(I ⋅ C) induces innate immunity via Toll-like receptor 3 (TLR3) (17). TLRs in mammals, like Toll receptors in Drosophila, also play pivotal roles in development. TLR2, -3, -4, and -8 are expressed by glial cells and neurons (17–21). Expression of TLR2 and -4 in adult neural stem/progenitor cells (NPC) may influence their proliferation and differentiation. TLR2 deficiency in mice impairs adult hippocampal neurogenesis, whereas the absence of TLR4 results in enhanced proliferation and neuronal differentiation (22).
A fundamental feature of cerebral corticogenesis in mammals is that positioning of neuronal constituents into horizontal and vertical arrays defines their function and connectivity. As cortical neurogenesis proceeds, newly generated neurons migrate radially out of the proliferative neuroepithelial cell layers lining the lateral ventricles, the ventricular zone (VZ) and subventricular zone (SVZ) according to a precise schedule and along well-defined pathways (23, 24). Newly generated neurons migrate past cells generated earlier to settle in progressively more superficial layers, ultimately forming a six-layered cortex wherein a neuron’s laminar position is determined by its birth date (25, 26). Reports both in schizophrenia of decreased numbers and/or incomplete clustering of neurons in layer II of entorhinal, frontal, and cingulate cortex (10) and in autism of more numerous, but smaller and less compact minicolumn architecture (27) indicate dysregulation in later stages of corticogenesis.
During middle and late embryonic development, neocortical neurons are generated from two distinct types of progenitor cells, radial glia (RG) and intermediate progenitor cells (IPC). RG are self-renewing stem cells that produce neurons and IPC, divide at the ventricular surface, and express PAX6, a homeodomain transcription factor. IPC produce only neurons and divide away from the ventricular surface. Transition from RG to IPC is associated with upregulation of TBR2, a T-domain transcription factor, and downregulation of PAX6 (28–30). IPC contribute neurons to most or all cortical layers (31); in late neurogenesis, however, only subventricular IPC give rise to upper-layer neurons.
Here we investigate the effects of TLR3 activation on neurogenesis and fetal neocortical development and on neonatal and adult offspring behavior in a mouse model of gestational virus infection. Gestational exposure to poly(I ⋅ C) induced cell cycle arrest in RG, resulting in decreased neuronal output. Wild-type (WT) offspring exposed to poly(I ⋅ C) exhibited impaired early locomotor capacity; poly(I ⋅ C)-exposed WT adult offspring also had deficits in sensorimotor gating. Inhibition of embryonic neuronal progenitor proliferation was not observed in TLR3-deficient mice or in WT mice pretreated with the cyclooxygenase (COX) inhibitor, carprofen.
RESULTS
Poly(I ⋅ C) treatment during gestation impairs early locomotor development in neonates and PPI in adult mice.
We observed robust behavioral changes in the offspring of WT mice receiving a single injection of 5 mg/ml of poly(I ⋅ C) on gestational day 16 (GD16); hence, all behavioral testing was performed with animals receiving a single dose of poly(I ⋅ C) on GD16. WT pregnant C57BL/6 mice were treated on GD16 with either poly(I ⋅ C) (5 mg/kg of body weight) or phosphate-buffered saline (PBS). Open-field locomotor activity was tested in the offspring from the treated pregnant mice on postnatal day 8. The offspring from WT pregnant mice exposed to poly(I ⋅ C) on GD16 had reduced locomotor activity on postnatal day 8 compared to WT PBS-injected mice (n = 37 to 40) (P = 0.038 for the number of floor plane moves by the Mann-Whitney U test) (Fig. 1a). The offspring from poly(I ⋅ C)-treated WT mice exhibited a deficit in sensorimotor gating, as measured by the prepulse inhibition (PPI) of the startle response at 4 dB and 8 dB, but not at 2 dB or 16 dB (n = 16 to 22) (PPI values at +2 dB, P = 0.183; PPI values at +4 dB, P = 0.008; PPI values at +8 dB, P = 0.050; PPI values at +16 dB, P = 0.128) (Fig. 1b). The mean percent PPI across all prepulse intensities was disrupted by prenatal poly(I ⋅ C) treatment in offspring from WT dams (dose group main effect, P = 0.018; data not shown). Sex did not influence this dose group effect on the mean percent PPI (data not shown).
FIG 1 .
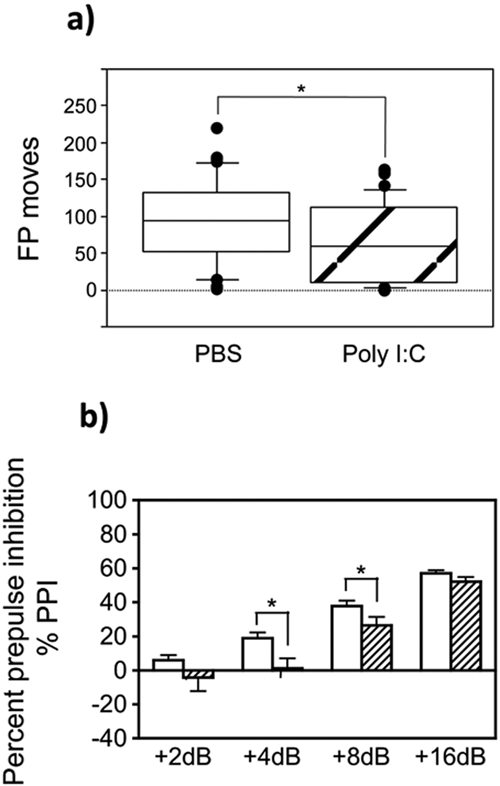
Poly(I ⋅ C) treatment during gestation impairs early locomotor development in neonates and prepulse inhibition (PPI) in adult mice. (a) WT C57BL/6 pregnant mice were treated with either PBS or poly(I ⋅ C) (5 mg/kg) on gestational day 16 (GD16). Protoambulatory behavior of the offspring from pregnant mice treated with PBS or poly(I ⋅ C) was measured in an open-field paradigm. Data are presented as the number of floor plane (FP) moves exhibited by the mice on postnatal day 8. The values for the poly(I ⋅ C)-treated group are significantly different (P < 0.05 by the Mann-Whitney U test) compared to the values for the PBS-treated control group as indicated by the bracket and asterisk. The height of the box plot shows the interquartile range, and the horizontal line indicates the median. The range is indicated by the error bars, and the circles represent outlier values. (b) Sensorimotor gating of the acoustic startle reflex was measured as a percentage of prepulse inhibition in the adult offspring from WT C57BL/6 dams injected on GD16 with either PBS (white bars) or poly(I ⋅ C) (5 mg/kg) (hatched bars). Data are presented as the percent PPI exhibited at each of the four prepulse intensity trials (2, 4, 8, and 16 dB above background noise) by the mice during postnatal weeks 15 to 17. The values for the poly(I ⋅ C)-treated group are significantly different (P < 0.05 by Fisher’s protected least-significant difference [PLSD] test) compared to the values for the PBS-treated control group as indicated by the bracket and asterisk.
Poly(I ⋅ C) treatment during gestation induces defective proliferation in fetal cerebral cortical cells.
Pregnant mice on GD16 were treated with either poly(I ⋅ C) or PBS and injected with the DNA replication marker, bromodeoxyuridine (BrdU), to label dividing cells in the proliferative zone, destined for superficial cortical layers 2 and 3 (23). Cell suspensions of dissociated cortex obtained from embryos on GD18 were analyzed by flow cytometry. Viable cerebral cortical cells were gated according to forward scatter (FSC) and side scatter (SSC) profile (Fig. 2a). The progeny of PBS-treated mice displayed two subpopulations of BrdU-positive cells that differed in the amount of BrdU incorporated, BrdUlow cells and BrdUhigh cells. Poly(I ⋅ C) treatment resulted in a significant decrease in the percentage of BrdUhigh cells (n = 7; P = 0.002 by the Mann-Whitney U test) (Fig. 2b and d). No significant change in BrdUlow labeling was observed with poly(I ⋅ C) treatment (Fig. 2b). The mean fluorescent intensity of BrdU labeling was also decreased in cells isolated from pups of poly(I ⋅ C)-treated pregnant mice (n = 4; P = 0.034 by the Mann-Whitney U test) (Fig. 2c).
FIG 2 .

Poly(I ⋅ C) treatment during gestation induces defective proliferation in fetal cerebral cortical cells. Pregnant mice were treated with either PBS or poly(I ⋅ C) on 3 consecutive days (GD15 to GD17) and injected with bromodeoxyuridine (BrdU) on GD16. Cerebral cortical cells were isolated from embryos on GD18, and quantitation of BrdU-labeled cells was performed by flow cytometric analysis. (a) Viable cerebral cortical cells were gated according to forward scatter (FSC) and side scatter (SSC) profiles. The percentage of live cells (85.5%) is shown. 50K, 50,000. (b) Representative histogram showing intracellular BrdU incorporation in cerebral cortical cells. The numbers above the brackets indicate the percentages of brightly labeled BrdU cells (BrdUhigh) and a subpopulation of cells that incorporated low levels of BrdU (BrdUlow). Gating for the BrdU-positive cells was based on the anti-BrdU antibody staining observed with cells obtained from mice that were not injected with BrdU (negative control). Anti-BrdU antibody conjugated to allophycocyanin (APC) (BrdU-APC) was used. (c) Representative histogram showing the mean fluorescence intensity (MFI) of BrdU-labeled cells represented as a percentage of maximum BrdU incorporation. Values for the different groups are indicated as follows: PBS-treated mice (red line), poly(I ⋅ C)-treated mice (blue line), and negative-control mice (gray line). The experiments were performed with cerebral cortical cells isolated from embryos (n = 7); data from one representative cortex are shown. (d) Percentage of BrdUhigh cells in cerebral cortical cells from mice treated with PBS or poly(I ⋅ C). Results are expressed as percentages of total gated cells. The values for the poly(I ⋅ C)-treated group are significantly different (P < 0.05 by the Mann-Whitney U test) compared to the values for the PBS-treated control group as indicated by the bracket and asterisk. The height of the box plot shows the interquartile range, and the horizontal line indicates the median. The range is indicated by the error bars, and the circles represent outlier values.
Relative importance of apoptosis and cell cycle arrest to fetal cerebral cortical cell development after poly(I ⋅ C) treatment during gestation.
We postulated that poly(I ⋅ C)-induced reduction in cortical cell proliferation could reflect apoptosis or cell cycle arrest. To assess the contribution of apoptosis, we measured the expression of activated caspase 3 in embryonic cerebral cortical cells. Pregnant mice on GD16 were injected with BrdU after treatment with either PBS or poly(I ⋅ C). Cell suspensions of dissociated cortex obtained from embryos on GD18 were analyzed using flow cytometry. Poly(I ⋅ C) treatment resulted in a significant increase in the percentage of cells expressing activated caspase 3 (n = 4) (P = 0.021 by the Mann-Whitney U test) (Fig. 3a and b). Expression of caspase 3 alone is not pathognomic of cell death. Indeed, we observed no differences between poly(I ⋅ C) and control groups when cells were gated according to their FSC and side scatter profiles (data not shown) or 7-aminoactinomycin D (7-AAD) staining (see Fig. S1 in the supplemental material).
FIG 3 .

Relative importance of apoptosis and cell cycle arrest to fetal cerebral cortical cell development after poly(I ⋅ C) treatment during gestation. (a) Pregnant mice were treated with either PBS or poly(I ⋅ C) and injected with BrdU on GD16. Cerebral cortical cells were isolated from the embryos on GD18, and quantification of activated caspase 3-positive (caspase 3+) cells was performed by flow cytometric analysis. Representative histograms show intracellular activated caspase 3+ cells in cerebral cortical cells. The numbers above the brackets indicate the percentages of activated caspase 3+ cells. Gating for the caspase 3+ cells was based on the staining observed with the isotype control. Experiments were performed with cerebral cortical cells isolated from embryos (n = 4); data from one representative cortex are shown. (b) Percentage of activated caspase 3+ cells in cerebral cortex from mice treated with PBS or poly(I ⋅ C). The values for the poly(I ⋅ C)-treated group are significantly different (P < 0.05 by the Mann-Whitney U test) compared to the values for the PBS-treated control group as indicated by the bracket and asterisk. (c) Pregnant mice were treated with either PBS or poly(I ⋅ C) and simultaneously injected with 5-iodo-2′-deoxyuridine (IdU) on GD16. After 10 h of IdU exposure, mice received a single injection of 5-chloro-2′-deoxyuridine (CldU). IdU and CldU incorporation was quantified by flow cytometry. Dot plots of IdU+ versus CldU+ cerebral cortical cells from embryos of PBS- or poly(I ⋅ C)-injected dams are shown. (d) Isotype control. The numbers in the four quadrants indicate the percentages of each subpopulation. Gating for the IdU- or CldU-positive cells was based on the staining observed with the isotype control. Experiments were performed with cells isolated from embryos (n = 3 to 4); data from one representative cortex are shown. (e) Percentage of IdU+ and IdU+ CldU+ in cerebral cortical cells from mice treated with PBS or poly(I ⋅ C). Results are expressed as percentages of total viable cells. The values for PBS-treated mice (white bars) and poly(I ⋅ C)-treated mice (hatched bars) are shown. The values for the poly(I ⋅ C)-treated group are significantly different (P < 0.05 by the Mann-Whitney U test) compared to the values for the PBS-treated control group as indicated by the bracket and asterisk. The height of the box plot shows the interquartile range, and the horizontal line indicates the median. The range is indicated by the error bars, and the circles represent outlier values.
Caspase 3 expression may also vary as a function of cell cycle dynamics. To assess the effects of poly(I ⋅ C) on the cell cycle, we treated pregnant mice with two different halogenated nucleotide analogs at different time points. Pregnant mice were treated with either PBS or poly(I ⋅ C) and simultaneously injected with 5-iodo-2′-deoxyuridine (IdU) on GD16. After 10 h of IdU exposure, mice received a single injection of 5-chloro-2′-deoxyuridine (CldU). Cell suspensions of dissociated cortex obtained from pups on postnatal day 0 (PND0) were analyzed using flow cytometry for the incorporation of IdU and CldU.
The interval between the injections allows the majority of cells to exit the S phase after the first nucleotide injection but is shorter than the whole cell cycle, ensuring that cells reentering the cell cycle incorporate the second nucleotide (32). Poly(I ⋅ C) resulted in a reduction of the percentage of cells incorporating both nucleotide analogs (n = 3 to 4) (P = 0.034 by the Mann-Whitney U test) (Fig. 3c, top right quadrant, and Fig. 3e). The quadrants were defined on the basis of staining observed with the antibody isotype controls (Fig. 3d). No change in the percentage of cells incorporating IdU was observed with poly(I ⋅ C) (Fig. 3c, top two quadrants, and Fig. 3e). This is consistent with the BrdU data obtained with GD18 embryos wherein no change in total BrdU incorporation was observed (Fig. 2b). Taken together, these data suggest that prenatal poly(I ⋅ C) induces cell cycle arrest in cortical cells.
Poly(I ⋅ C) treatment during gestation negatively regulates the transition of PAX6+ RG cells to TBR2+ PAX6+ IPC in a TLR3-dependent manner.
To evaluate the effect of poly(I ⋅ C) on neural stem/progenitor cells (NPC) proliferation, we quantitated the intracellular expression of PAX6 and TBR2 in subtypes of NPC, radial glia (RG) cells and intermediate progenitor cells (IPC). Embryonic cerebral cortical cells were isolated on GD18 from pregnant dams injected with poly(I ⋅ C) and BrdU and analyzed by flow cytometry for expression of TBR2 and PAX6. Poly(I ⋅ C) treatment resulted in a decrease in the percentage of TBR2-positive (TBR2+) PAX6-positive (PAX6+) IPC (n = 3) (P = 0.049 by the Mann-Whitney U test) (Fig. 4a and e). The mean fluorescent intensity of TBR2+ PAX6+ IPC was also decreased in cells isolated from pups of poly(I ⋅ C)-treated pregnant dams (n = 3) (P = 0.049 by the Mann-Whitney U test) (Fig. 4c). Poly(I ⋅ C) treatment resulted in a concomitant increase in the percentage of PAX6+ RG cells (n = 3) (P = 0.049 by the Mann-Whitney U test) (Fig. 4b and e). The mean fluorescent intensity of PAX6+ RG also increased in embryos of poly(I ⋅ C)-treated dams (n = 3) (P = 0.049 by the Mann-Whitney U test) (Fig. 4d). These results are consistent with a mechanism wherein poly(I ⋅ C) treatment inhibits the transition of RG cells to IPC, thereby decreasing cortical NPC expansion.
FIG 4 .

Treatment with poly(I ⋅ C) during gestation negatively regulates the transition of PAX6+ radial glia (RG) cells to TBR2+ PAX6+ intermediate progenitor cells (IPC) in a TLR3-dependent manner. WT C57BL/6 or TLR3−/− C57BL/6 pregnant mice were treated with either PBS or poly(I ⋅ C) for three consecutive days (GD15 to GD17) and injected with BrdU on GD16. Cerebral cortical cells were isolated from embryos on GD18. Total viable cells were analyzed for the expression of TBR2 and PAX6 by flow cytometry. (a) Representative histograms showing TBR2+ PAX6+ positive IPC in WT and TLR3−/− cerebral cortical cells isolated from embryos (n = 3) obtained from mice treated with PBS or poly(I ⋅ C). The numbers above the brackets indicate the percentages of TBR2+ PAX6+ IPC. (b) Representative histograms showing PAX6+ RG in WT and TLR3−/− cerebral cortical cells isolated from embryos obtained from PBS- and poly(I ⋅ C)-treated mice. The numbers above the brackets indicate the percentages of PAX6+ RG cells. Gating for the PAX6- or TBR2-positive cells was based on the staining observed with the isotype control. (c) Representative histograms showing the mean fluorescence intensity of TBR2 and PAX6 expression. The mean fluorescence intensity (MFI) is shown as a percentage of the maximum expression. MFI values for the different groups are indicated as follows: PBS-treated group (red line), poly(I ⋅ C)-treated group (blue line), and isotype control group (gray line). The MFI for PBS-treated mice was 1,158.7 ± 57.4, and the MFI for poly(I ⋅ C)-treated mice was 809.3 ± 28.4. (d) Representative histograms showing the MFI of PAX6 expression. Experiments were performed with cerebral cortical cells isolated from embryos (n = 3); data from one representative cortex are shown. MFI values for the different groups are indicated as follows: PBS-treated group (red line), poly(I ⋅ C)-treated group (blue line), and isotype control group (gray line). The MFI for PBS-treated mice was 13,784.0 ± 1,527.3, and the MFI for poly(I ⋅ C)-treated mice was 16,842.3 ± 799.1. (e) Percentages of TBR2+ PAX6+ IPC and PAX6+ RG in WT and TLR3−/− cerebral cortical cells isolated from embryos obtained from PBS- and poly(I ⋅ C)-treated mice. Results are expressed as percentages of total viable cells. The values for PBS-treated mice (white bars) and poly(I ⋅ C)-treated mice (hatched bars) are shown. Values that are significantly different (P < 0.05 by the Mann-Whitney U test) compared to the values for the PBS-treated control group are indicated by the bracket and asterisk. Values that are significantly different (P < 0.05 by the Mann-Whitney U test) compared to the values for the PBS-treated control group are indicated by the bracket and two asterisks. The height of the box plot shows the interquartile range, and the horizontal line indicates the median. The range is indicated by the error bars, and the circles represent outlier values.
TLR3-deficient (TLR3−/−) mice were used to assess the role of TLR3 in the poly(I ⋅ C)-mediated inhibition of the transition of RG to IPC. Embryonic cerebral cortical cells were isolated from WT and TLR3−/− dams injected with poly(I ⋅ C) and BrdU. Whereas poly(I ⋅ C) treatment of WT dams resulted in decreased TBR2+PAX6+ IPC, decreased mean fluorescent intensity of TBR2+PAX6 IPC, and increased PAX6+ RG cells, no differences were observed in the labeling patterns of embryonic cells isolated from poly(I ⋅ C)-treated dams compared to those from PBS-treated, TLR3−/− dams (Fig. 4a to e). The percentages of PAX6+ (RG) cells and of TBR2+ PAX6+ cells (IPC) were significantly reduced in poly(I ⋅ C)-treated WT mice versus poly(I ⋅ C)-treated TLR−/− mice (n = 3) (P = 0.049 by the Mann-Whitney U test) (Fig. 4e). Taken together, these results implicate TLR3 signaling in the impaired NPC proliferation induced by poly(I ⋅ C) during late corticogenesis.
Poly(I ⋅ C) treatment during gestation induces deficits in cerebral cortical neurogenesis.
To investigate whether poly(I ⋅ C) induced long-term deficits in cortical neurogenesis, we examined BrdU-labeled cells from the postnatal cerebral cortices of pups from WT C57BL/6 dams that had previously been injected with poly(I ⋅ C) or PBS. Immunofluorescence analysis of coronal cortical slices (see Text S1 in the supplemental material) showed that on PND0 and PND8, BrdU+ cells migrated towards the pial surface and reached positions in cortical layers II and III (see Fig. S5a in the supplemental material). On PND0, BrdU-positive cells were found in the cortex of the progeny in the ventricular, subventricular, and intermediary layers. On PND8, BrdU-labeled cells were restricted to the cortical gray matter (see Fig. S5a in the supplemental material). BrdU-labeled, CUX1-labeled pyramidal neurons were reduced in the cerebral cortices of PND8 offspring of poly(I ⋅ C)-treated mice (see Fig. S5b and S5c in the supplemental material).
Treatment with the nonselective cyclooxygenase (COX) inhibitor carprofen prevents poly(I ⋅ C)-induced decrease in cerebral cortical neurogenesis.
To investigate whether blocking inflammation could reduce deficits in cortical neurogenesis associated with poly(I ⋅ C), we examined BrdU-labeled cells isolated from the postnatal cerebral cortices of pups from WT pregnant C57BL/6 mice that had previously been injected with poly(I ⋅ C) or PBS, with or without the nonsteroidal anti-inflammatory drug (NSAID), carprofen (gestational days 15, 16, and 17). Cortical cells obtained from the pups of these dams on postnatal day 8, an endpoint of cortical neuron differentiation, were analyzed for BrdU incorporation by flow cytometry. Poly(I ⋅ C) treatment alone resulted in a decreased percentage of BrdU+ cells (4 pups obtained from 4 different dams) (P = 0.021 by the Mann-Whitney U test) (Fig. 5a and c). The mean fluorescent intensity of BrdU labeling was also reduced in pups from dams treated with poly(I ⋅ C) alone (4 pups obtained from 4 different dams) (P = 0.021 by the Mann-Whitney U test) (Fig. 5b). To examine the effect of poly(I ⋅ C) on the BrdU-labeled neurons destined to occupy the upper layers of the cerebral cortex, we measured BrdU incorporation into neurons expressing CUX1, a marker of cortical layers II/III (33, 34). The percentage of CUX1+ neurons labeled with BrdU was reduced in offspring obtained from dams treated with poly(I ⋅ C) alone (4 pups obtained from 4 different dams) (P = 0.021 by the Mann-Whitney U test) (Fig. 5d and f). The mean fluorescent intensity of BrdU labeling was also reduced in CUX1+ neurons of these pups (4 pups obtained from 4 different dams) (P = 0.021 by the Mann-Whitney U test) (Fig. 5e).
FIG 5 .

Treatment with the nonselective cyclooxygenase (COX) inhibitor carprofen prevents poly(I ⋅ C)-induced decreases in cerebral cortical neurogenesis. WT C57BL/6 pregnant mice were treated with a subcutaneous dose of either carprofen or PBS, followed by intraperitoneal injection of either PBS or poly(I ⋅ C) for three consecutive days (GD15 to GD17). On GD16, the mice were injected with BrdU. Cerebral cortical cells were isolated from pups on postnatal day 8 (PND8), and quantification of BrdU-labeled cells was performed by flow cytometric analysis. Gating for the BrdU-positive cells was based on the anti-BrdU antibody staining observed with cells obtained from mice that were not injected with BrdU (negative control). (a) Representative histograms showing intracellular BrdU incorporation in cerebral cortical cells isolated from pups obtained from the different groups of pregnant mice. The mice were first injected with carprofen or PBS. Two hours later, the mice were injected with PBS or poly(I ⋅ C). The four treatment groups were as follows (the first injection is shown before the slash, and the second injection is shown after the slash): (i) PBS/PBS, (ii) PBS/poly(I ⋅ C), (iii) carprofen/PBS, and (iv) carprofen/poly(I ⋅ C). The numbers above the brackets indicate the percentages of BrdU-labeled cells. (b) Histograms showing the mean fluorescence intensity (MFI) of BrdU-labeled cells represented as a percentage of maximum BrdU incorporation. The treatment groups are as explained above for panel a. The values for the different groups are indicated as follows: PBS/PBS group (red line), carprofen/PBS group (purple line), PBS/poly(I ⋅ C) group (blue line), carprofen/poly(I ⋅ C) group (green line), and negative-control group (gray line). The MFI values for the different groups were as follows: 4,171.0 ± 184.4 for the PBS/PBS group, 3,348.0 ± 204.3 for the carprofen/PBS group, 2,302.3 ± 47.4 for the PBS/poly(I ⋅ C) group, and 3,012.8 ± 102.5 for the carprofen/poly(I ⋅ C) group. Experiments were performed with cerebral cortical cells isolated from pups (n = 4); data from one representative cortex are shown. (c) Percentage of BrdU+ in cerebral cortical cells from pups obtained from pregnant mice pretreated with carprofen or not pretreated with carprofen, followed by treatment with PBS or poly(I ⋅ C). Results are expressed as percentages of total gated cells. The values that are significantly different (P < 0.05 by the Mann-Whitney U test) compared to the values for the PBS-treated control group are indicated by the bracket and asterisk. (d) Representative histograms show intracellular BrdU incorporation in CUX1+ cortical cells isolated from pups obtained from the same treatment groups of pregnant mice as explained above for panel a. The numbers above the brackets indicate the percentages of BrdU+ cells. (e) Representative histogram shows the MFI of BrdU labeling in CUX1+ cerebral cortical cells. Experiments were performed with cerebral cortical cells isolated from pups (n = 4); data from one representative cortex are shown. The values for the different groups are indicated as follows: PBS/PBS group (red line), carprofen/PBS group (purple line), PBS/poly(I ⋅ C) group (blue line), carprofen/poly(I ⋅ C) group (green line), and negative-control group (gray line). The MFI values for the different groups were as follows: 9,180.3 ± 600.5 for the PBS/PBS group, 5,218.5 ± 624.9 for the carprofen/PBS group, 4,453.8 ± 253.6 for the PBS/poly(I ⋅ C) group, and 4,958.5 ± 179.8 for the carprofen/poly(I ⋅ C) group. (f) Percentage of BrdU+ CUX1+ cerebral cortical cells from pups obtained from pregnant mice treated with carprofen or not treated with carprofen, followed by treatment with PBS or poly(I ⋅ C). Results are expressed as percentages of total CUX1+ cells. Values for PBS-treated mice (white bars) and poly(I ⋅ C)-treated mice (hatched bars) are shown. The values for the poly(I ⋅ C)-treated group are significantly different (P < 0.05 by the Mann-Whitney U test) compared to the values for the PBS-treated control group as indicated by the bracket and asterisk. The height of the box plot shows the interquartile range, and the horizontal line indicates the median. The range is indicated by the error bars, and the circles represent outlier values.
To test whether modulation of the immune response might restore the impairment of NPC proliferation triggered by poly(I ⋅ C), we injected the COX inhibitor, carprofen, or PBS into pregnant mice prior to treatment with poly(I ⋅ C) or PBS on GD15 to GD17. BrdU was injected once on GD16. Flow cytometric analysis of cortical cells from PND8 pups of dams treated with carprofen and poly(I ⋅ C) or PBS revealed no difference in either the percentage of BrdU-labeled cells or the intensity of BrdU labeling (Fig. 5a to c). Similarly, the poly(I ⋅ C)-related decrease in the percentage of BrdU-labeled, CUX1+ neurons and in the intensity of BrdU labeling was blocked by carprofen (Fig. 5d to f). No significant differences in total BrdU labeling were observed in cells isolated from PBS-injected dams exposed to carprofen or not exposed to carprofen (4 pups obtained from 4 different dams) (P = 0.083 by the Mann-Whitney U test) (see Fig. S2 in the supplemental material). Fluorescence-activated cell sorting (FACS) data indicate that carprofen pretreatment abrogates the poly(I ⋅ C)-induced decrease in PAX6+ TBR2+ IPC and in CUX1+ neurons (data not shown).
DISCUSSION
Maternal gestational exposure to viruses is associated with increased risk for neurodevelopmental disorders, including autism (9, 27) and schizophrenia (35, 36). The data are most compelling for schizophrenia, wherein maternal report has been confirmed by serology, indicating increased risk in the offspring of women exposed to influenza virus during the second trimester of pregnancy (2, 37). The fact that different pathogens (e.g., influenza virus [36], rubella virus [38], herpesvirus [35]) are associated with similar outcomes is consistent with a model whereby infection per se triggers damage. The biological plausibility of this hypothesis is underscored by decades of observations in rodent models of neonatal infection using lymphocytic choriomeningitis virus (39, 40), Borna disease virus (41–43), and mumps virus (44), wherein behavioral disturbances have been linked to abnormal neuroanatomy. In mouse models of maternal infection with human influenza virus at mid-gestation, the adult offspring displayed behavioral, pharmacological, and histological abnormalities (5, 10). Viral RNA was not detected in fetal tissue; abnormalities in the offspring were reproduced by exposure to poly(I ⋅ C) (45). Building on this work and that of others who have also reported neurodevelopmental damage after gestational exposure to poly(I ⋅ C) (4, 45, 46), we selected poly(I ⋅ C) as a surrogate to represent a wide range of viral infections.
Impaired open-field locomotor activity was observed in the offspring of poly(I ⋅ C)-treated WT pregnant mice on postnatal day 8 (PND8). Sensorimotor gating, a brain function impaired in schizophrenia and autism, defines the ability to segregate components of a continuous stream of sensory and cognitive information (47). The primary mammalian acoustic startle circuit consists of three synapses, linking the auditory nerve with spinal motor function. The relatively simple startle reflex is also influenced by the forebrain circuitry and demonstrates other important forms of behavioral plasticity, including attention, habituation, and fear potentiation. We observed robust behavioral changes in WT mice that received a single injection of 5 mg/ml of poly(I ⋅ C). Hence, all behavior experiments were performed with animals that received a single dose of poly(I ⋅ C) on gestational day 16 (GD16).
Cerebral corticogenesis is accomplished by an orderly generation and radial migration of cortical neurons leading to the formation of six layers in an “inside-out” pattern (48). Deep layers (layers V and VI) are composed of early-born neurons, and upper layers (layers II and IV) are of late-born neurons that are derived from intermediate progenitor cells (IPC) (26, 48). We chose to administer poly(I ⋅ C) on embryonic day 15 (ED15), ED16, and ED17, a late time point in the rodent cortical neurodevelopment coinciding with the cortical development of layers II and III (24, 26). Although it is challenging to match the stages of pregnancy of rodents and humans, the neurogenesis of cortical layer II/III, peaking on GD16 in mice, correlates closely with the cortical development events of the second trimester of pregnancy in humans (postconception day, 100.4; http://translatingtime.net/) (49). On PND8, a time point when the terminal differentiation of upper cortical layers is achieved in mice, neurons occupying layers II /III express CUX1, a homeodomain transcription factor (33, 34). The decrease in the number of BrdU+ CUX1+ neurons in the progeny of mice treated with poly(I ⋅ C) suggests that prenatal immune activation can cause long-term defects in cortical development in their offspring (see Fig. S5a, S5b, and S6 in the supplemental material). In schizophrenia, a disorder wherein a subset of cases is associated with maternal infection during a period of development equivalent to the one we have tried to model here, incomplete clustering of neurons and disorganization in layer II of entorhinal, frontal, hippocampal, and anterior cingulate cortex are among the most frequently reported cytoarchitectonic abnormalities (10, 27).
Embryonic progenitor cells generate diverse neuronal subtypes by undergoing repeated cell divisions. The proliferation of neural stem/progenitor cells (NPC) during late corticogenesis involves asymmetrical divisions of radial glia (RG) cells in the ventricular zone (VZ), yielding self-renewing RG cells and neurons. A second mechanism of neuron generation in the dorsal cortex involves production of IPC from RG cells (24, 48). IPC migrate to the subventricular zone (SVZ), where they produce neurons through symmetrical divisions. In the developing brain, sequential expression of multiple transcription factors is involved in control of progenitor proliferation and neuronal fate (24, 28, 29). In this cascade, the PAX6 transcription factor is initially expressed by RG. The transition from RG to IPC occurs with a sequential increase in the expression of TBR2, followed by a reduction in PAX6 expression. Our neuronal birth dating and double-labeling experiments indicate that poly(I ⋅ C) induces a cell cycle arrest in embryonic neuronal progenitor/stem cells. This is most definitively demonstrated by an increase in the expansion of PAX6+ RG stem cells and a concurrent decrease in the TBR2-expressing IPC (see Fig. S6 in the supplemental material). The majority of the BrdUhigh cells were TBR2+ PAX6+ IPC; a smaller proportion represented RG stem cells (see Fig. S3 in the supplemental material). These findings suggest that poly(I ⋅ C) treatment results in depletion of the BrdUhigh IPC subpopulation.
These data may have implications for understanding pathological cortical development in other gyrencephalic species. In primates as well as rodents, the SVZ is the source of upper-layer cortical neurons, and regional patterns of neurogenesis caused by IPC proliferation in the SVZ help determine patterns of gyrification (48, 50).
The Toll-like receptor (TLR) family plays a critical role in regulating the type I interferon (IFN) response as well as other inflammatory cytokine and chemokine responses to viral pathogens. Immune cells in vivo utilize distinctive mechanisms to detect viral RNA and to generate type I IFNs. In several viral infections, Toll-like receptor 3 (TLR3)-independent signaling mechanisms, such as mitochondrial antiviral signaling (MAVS) or TLR7/TLR9-mediated pathways, are responsible for inducing innate immunity against viral pathogens (51). Poly(I ⋅ C) administration leads to pronounced expression of proinflammatory cytokines through TLR3 activation (52). Cytokine levels are altered in maternal serum as well as in amniotic fluid, placenta, and fetal brain following maternal immune activation by influenza infection, as well as through exposure to lipopolysaccharide (LPS) or poly(I ⋅ C) (37, 45, 53). Smith and colleagues, using behavior as an outcome, identified interleukin 6 (IL-6) as a mediator of the effects of poly(I ⋅ C) on fetal murine brain development (54). In vivo administration of poly(I ⋅ C) has been reported to induce a transient, viral-infection-like sickness behavior, including changes in weight loss, body temperature, and anorexia (55). Elimination of TLR3 in our knockout mice largely abrogates the adverse effects of poly(I ⋅ C) on embryonic NPC proliferation. Despite these observations, it is unclear that poly(I ⋅ C) effects are mediated only by immune activation of the mother and/or the embryo. Direct effects of poly(I ⋅ C) on neural elements have been demonstrated in vitro, with reduced cell proliferation in WT embryonic cortical neurospheres, but not in NPC derived from TLR3−/− mice (56). These findings are consistent with our in vivo data wherein TLR3 expression is restricted to RG stem cells (see Fig. S4 in the supplemental material). Ongoing experiments involving experimental transfer of TLR3−/− embryos to WT surrogate dams and vice versa will more conclusively address the relative contributions of maternal and fetal immune responses to the phenotypes induced by poly(I ⋅ C).
Poly(I ⋅ C) signals through TLR3 via an NF-κB-dependent mechanism and induces proinflammatory cytokines, such as IL-1, IL-6, IL-8, tumor necrosis factor alpha (TNF-α) and RANTES, and type I and II interferons (17, 54). Cyclooxygenase (COX) inhibitors, such as carprofen, are used clinically to reduce pain, fever, and inflammation (57). In addition to inhibiting COX, some nonsteroidal anti-inflammatory drugs (NSAIDs) suppress the production of proinflammatory cytokines and IFN through NF-κB transactivation (57, 58). In our model, NSAID treatment may counter the TLR3-dependent poly(I ⋅ C) effect through a similar cytokine-mediated mechanism. Extrapolation from murine models to humans can be hazardous. Nonetheless, the impaired cerebral corticogenesis and inflammatory response during congenital viral infections suggest that an innate immune-related mechanism may underlie the postnatal development of behavioral malfunctions characteristic of many neuropsychiatric diseases. Additionally, the observation that a COX inhibitor eliminates poly(I ⋅ C) effects on embryonic neuronal progenitor/stem cell proliferation may provide a rationale for the use of anti-inflammatory drugs in prenatal infections, during periods of neural development when the fetus is particularly vulnerable to central nervous system (CNS) damage.
MATERIALS AND METHODS
Mice.
WT pregnant C57BL/6 mice on gestational day 14 (GD14) were obtained from Harlan Laboratories (Dublin, VA). All protocols were approved by the Institutional Animal Care and Use Committee at Columbia University. Congenic mice with a C57BL/6 background with Toll-like receptor 3 (TLR3) deleted were obtained by repeated breeding of heterozygous B6;129S1-Tlrtm1Flv/J TLR3 knockout mice (Jackson Laboratory, Bar Harbor, ME) with WT C57BL/6 mice for 10 generations. Heterozygous mice, at each generation, carrying the TLR3 deletion were identified by PCR analysis of total genomic DNA using primers NEO_F (CTTGGGTGGAGAGGCTATTC), NEO_R (AGGTGAGATGACAGGAGATC), TLR3-F (ACTCCTTTGGGGGACTTTTG), and TLR3-R (CAGGTTCGTGCAGAAGACAA) (forward primers and reverse primers indicated by F and R, respectively, at the end of the primer designation). After 10 generations, heterozygous mice were intercrossed, and congenic homozygous TLR3−/− mice on a C57BL/6 background were selected to maintain the colony. In all experiments where TLR3−/− mice were used, WT pups from 3 different litters served as controls.
Injection schedule. (i) Behavioral studies.
Mice received a single intraperitoneal (i.p.) injection on gestational day 16 of either polyinosinic-polycytidylic potassium salt [poly(I ⋅ C)] (Sigma-Aldrich, St. Louis, MO) (5 mg/kg of body weight) dissolved in phosphate-buffered saline (PBS) or PBS alone.
(ii) Birth dating and cell fate experiments.
Mice received three intraperitoneal injections on GD15, GD16, and GD17 of either 5 mg/kg of poly(I ⋅ C) or PBS alone. On GD16, poly(I ⋅ C)- or PBS-treated mice also received a single i.p. injection of the thymidine analog 5-bromo-2′-deoxyuridine (BrdU) (Sigma-Aldrich, St. Louis, MO) dissolved in 0.9% NaCl with 7 mM NaOH at a dosage of 50 mg/kg. To avoid possible litter effect bias, no more than two pups per litter were used in these experiments [both the PBS-treated group and the poly(I ⋅ C)-treated group contained 7 pups obtained from 4 different litters].
(iii) Cell cycle arrest experiments.
Mice were treated on GD16 with either poly(I ⋅ C) (5 mg/kg) or PBS followed by sequential injection with two different halogenated nucleotide analogs. Poly(I ⋅ C)- or PBS-treated pregnant mice first received a single injection of 5-iodo-2′-deoxyuridine (IdU) at 50 mg/kg and a single injection of 5-chloro-2′-deoxyuridine (CldU) at 50 mg/kg (both IdU and CldU from Sigma-Aldrich, St. Louis, MO) 10 hours later [the PBS-treated group contained 8 pups obtained from 4 different litters, while the poly(I ⋅ C)-treated group contained 4 pups obtained from 4 different litters].
(iv) Anti-inflammatory pretreatment studies.
On GD15, -16, and -17, mice were injected i.p. with either PBS or poly(I ⋅ C) 2 h after subcutaneous injection with either carprofen (Rimadyl; Pfizer Animal Health, New York, NY) (50 mg/kg) or PBS. On GD16, mice from all dose groups also received a single i.p. injection of BrdU (both the carprofen-treated group and the non-carprofen-treated group contained 8 pups obtained from 8 different litters).
Cortical cell isolation.
Pregnant mice were euthanized on GD18 through CO2 inhalation followed by decapitation. The brains from embryos were removed and immediately placed in ice-cold Hank’s buffered saline solution (HBSS) (Invitrogen, Carlsbad, CA). The cortices were trimmed and homogenized using the papain neural tissue dissociation kit (Miltenyi Biotec, Auburn, CA).
Antibodies.
The primary antibodies used included BrdU conjugated to fluorescein isothiocyanate (BrdU-FITC) (clone BU1/75 ICR1 with cross-reactivity to CldU; Abcam, Cambridge, MA), BrdU (clone B44 with cross-reactivity to IdU; Becton Dickinson, San Jose, CA), PAX6 (developed by Atsushi Kawakami and obtained from Development Studies Hybridoma Bank, University of Iowa, Iowa City, IA), TBR2 and TBR2 conjugated to phycoerythrin (TBR2-PE) (clone 21Mgs8; eBioscience, San Diego, CA), TLR3 (Sigma-Aldrich, St. Louis, MO), TLR3 (eBioscience, San Diego, CA), and CUX1 (Santa Cruz Biotechnology, Santa Cruz, CA). The secondary antibodies used included donkey anti-mouse antibody conjugated to allophycocyanin (APC), goat anti-rabbit antibody conjugated to PE, donkey anti-mouse antibody conjugated to FITC, donkey anti-rat antibody conjugated to FITC, goat anti-chicken antibody conjugated to Cy3, goat anti-rat conjugated to Cy2, and goat anti-mouse antibody conjugated to Cy5 (Jackson ImmunoResearch, West Grove, MA). The flow cytometry isotype controls were PE-conjugated rat IgG2a κ, FITC-conjugated rat IgG2a κ, FITC-conjugated mouse IgG1 κ, and APC-conjugated mouse IgG1 κ isotype (BD Biosciences, Bedford, MA).
Flow cytometry.
Cortical cell suspensions were obtained from the offspring of pregnant mice treated with PBS or poly(I ⋅ C) and injected with PBS or BrdU. Intracellular BrdU was measured using the BrdU flow kit (BD Biosciences, Bedford, MA). The cells were resuspended in 50 µl of staining buffer (PBS plus 1.0% fetal bovine serum [FBS]). The cells were fixed, permeabilized, and treated with DNase (30 µg per tube) to expose incorporated BrdU. The cells were resuspended in 50 µl of BD Perm/fix buffer containing anti-BrdU antibody conjugated to APC (1:50) and specific relevant antibodies to detect intracellular TBR2 (1:100), CUX1 (1:50), PAX6 (1:100), and TLR3 (1:500) antigens, followed by staining with secondary antibodies diluted 1:1,000 (for CUX1, anti-rabbit antibody conjugated to PE; for PAX6, anti-mouse antibody conjugated to APC or FITC; for TLR3, anti-rat antibody conjugated to FITC). The cells that were isolated from mice that were not injected with BrdU but stained with anti-BrdU antibody were used as controls for gating the BrdU-positive population. Intracellular IdU or CldU was detected using anti-BrdU antibodies that cross-react with each of the nucleotide analogs (1:50). Expression of intracellular activated caspase 3 was analyzed using the caspase 3 active form from the monoclonal antibody apoptosis kit (1:50; BD Biosciences, Bedford, MA). The cells were washed and resuspended in staining buffer and analyzed by multicolor flow cytometry on an LSRII analyzer (Becton Dickinson, Franklin Lakes, NJ). After gating to exclude dead cells and debris based on forward scatter and side scatter, data were analyzed using FACS DiVa acquisition software (Becton Dickinson, Franklin Lakes, NJ) and FlowJo6.1 (Tree Star, Ashland, OR).
Open-field locomotor activity.
An automated open-field testing arena system was used to quantitate protoambulatory locomotor activity in a 2-min test (Coulbourn Instruments, Allentown, PA). WT pregnant C57BL/6 mice were treated on GD16 with either poly(I ⋅ C) (5 mg/kg) or PBS, and locomotor activity was tested in the offspring of the treated dams on postnatal day 8 (PND8) [the PBS-treated group contained 37 pups obtained from 20 different litters, while the poly(I ⋅ C)-treated group contained 40 pups obtained from 20 different litters].
Startle and PPI session.
Prepulse inhibition (PPI), an operational measure of acoustic sensorimotor gating in which a weak prestimulus (prepulse) attenuates the startle response to a sudden, loud noise (59), is regarded as a robust measure of sensorimotor gating, attention, and distractibility (60). PPI was tested in adult offspring (postnatal [PN] weeks 15 to 17) of WT C57BL/6 mice that had been exposed to either poly(I ⋅ C) (5 mg/kg) or PBS [the PBS-treated group contained 16 mice obtained from 8 different litters, and the poly(I ⋅ C)-treated group contained 22 mice obtained from 12 different litters] on GD16. Mice were placed in acoustically isolated startle chambers (SR-Lab; San Diego Instruments, San Diego, CA). All PPI test sessions consisted of startle trials (pulse alone), prepulse trials (prepulse plus pulse), and no-stimulus trials with only background noise (nostim). Following a 5-min period for apparatus acclimatization, five consecutive pulse-alone trials were presented to allow animals to habituate to the startle stimulus (120 dB). Subsequently, animals had 30 blocks of PPI test trials, presented in pseudorandomized order (i.e., excluding any trials involving identical stimuli in consecutive trials [59]). Each block consisted of one trial of either the pulse alone or a prepulse plus pulse stimulus at one of four levels of prepulse intensity over background or a nostim trial. The pulse-alone trial consisted of 40-ms 120-dB pulse of broadband noise. The prepulse-plus-pulse trials consisted of a 15-ms noise prepulse, a 100-ms delay, and then a 40-ms 120-dB startle pulse. The prepulse intensities were 2, 4, 8, and 16 dB above the 60-dB background noise. The session concluded with five consecutive pulse-alone trials. The interval between successive trials was 12 s. A reduction in the startle magnitude in any prepulse-plus-pulse trial relative to that observed in pulse-alone trials defines PPI. The percent PPI was calculated for each of the prepulse intensities as [(mean of pulse-alone trials − mean of prepulse-plus-pulse trials)/(mean of pulse-alone trials) × 100%]. PPI values were averaged across the four prepulse intensities to produce the mean percent PPI. The startle magnitude was calculated as the average response to all of the pulse-alone trials, excluding the first and last blocks of five pulse-alone trials. The habituation of the startle response was investigated by grouping the startle trials into three blocks (5 pulse-alone trials each, in order of presentation) and measuring the decrease of startle magnitudes across blocks.
Statistical analysis.
StatView version 5.0.1 software (Windows version; SAS Institute, Cary, NC) was used for all statistical analyses. Group comparisons were carried out using analysis of variance (ANOVA) for normally distributed data or data that became Gaussian when transformed (one-way ANOVA using the dose group as the between-subject independent variable for the primary analyses); Fisher’s protected least-significant difference (PLSD) test was used for post hoc comparisons. Mann-Whitney U tests (independent variable, dose group) were used for group comparisons requiring nonparametric analytic approaches. For PPI test, sex effects were also examined using a 2 × 2 × 4 (sex × dose group × prepulse intensities) split-plot ANOVA. For all tests, statistical significance was assumed where P < 0.05.
ACKNOWLEDGMENTS
This work was supported by awards from the National Institutes of Health (NIH-NIAID 5 U01 AI070411-Lipkin), the Department of Defense, and Google.org.
We thank Vishal Kapoor for technical assistance. We are grateful to Katrina Ciraldo for helpful comments and editing the manuscript.
Footnotes
Citation De Miranda, J., K. Yaddanapudi, M. Hornig, G. Villar, R. Serge, et al. 2010. Induction of Toll-like receptor 3-mediated immunity during gestation inhibits cortical neurogenesis and causes behavioral disturbances. mBio 1(4):e00176-10. doi:10.1128/mBio.00176-10.
SUPPLEMENTAL MATERIAL
REFERENCES
- 1. Brown A. S., Vinogradov S., Kremen W. S., Poole J. H., Deicken R. F., Penner J. D., McKeague I. W., Kochetkova A., Kern D., Schaefer C. A. 2009. Prenatal exposure to maternal infection and executive dysfunction in adult schizophrenia. Am. J. Psychiatry 166:683–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Patterson P. H. 2007. Neuroscience. Maternal effects on schizophrenia risk. Science 318:576–577 [DOI] [PubMed] [Google Scholar]
- 3. Schmitz C., van Kooten I. A., Hof P. R., van Engeland H., Patterson P. H., Steinbusch H. W. 2005. Autism: neuropathology, alterations of the GABAergic system, and animal models. Int. Rev. Neurobiol. 71:1–26 [DOI] [PubMed] [Google Scholar]
- 4. Meyer U., Engler A., Weber L., Schedlowski M., Feldon J. 2008. Preliminary evidence for a modulation of fetal dopaminergic development by maternal immune activation during pregnancy. Neuroscience 154:701–709 [DOI] [PubMed] [Google Scholar]
- 5. Shi L., Smith S. E., Malkova N., Tse D., Su Y., Patterson P. H. 2009. Activation of the maternal immune system alters cerebellar development in the offspring. Brain Behav. Immun. 23:116–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Meyer U., Spoerri E., Yee B. K., Schwarz M. J., Feldon J. 2010. Evaluating early preventive antipsychotic and antidepressant drug treatment in an infection-based neurodevelopmental mouse model of schizophrenia. Schizophr. Bull. 36:607–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Winter C., Djodari-Irani A., Sohr R., Morgenstern R., Feldon J., Juckel G., Meyer U. 2009. Prenatal immune activation leads to multiple changes in basal neurotransmitter levels in the adult brain: implications for brain disorders of neurodevelopmental origin such as schizophrenia. Int. J. Neuropsychopharmacol. 12:513–524 [DOI] [PubMed] [Google Scholar]
- 8. Atladottir H. O., Thorsen P., Ostergaard L., Schendel D. E., Lemcke S., Abdallah M., Parner E. T. Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J. Autism Dev. Disord., in press [DOI] [PubMed] [Google Scholar]
- 9. Ciaranello A. L., Ciaranello R. D. 1995. The neurobiology of infantile autism. Annu. Rev. Neurosci. 18:101–128 [DOI] [PubMed] [Google Scholar]
- 10. Fatemi S. H., Folsom T. D. 2009. The neurodevelopmental hypothesis of schizophrenia, revisited. Schizophr. Bull. 35:528–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lintas C., Altieri L., Lombardi F., Sacco R., Persico A. M. 2010. Association of autism with polyomavirus infection in postmortem brains. J. Neurovirol. 16:141–149 [DOI] [PubMed] [Google Scholar]
- 12. Pearce B. D. 2001. Schizophrenia and viral infection during neurodevelopment: a focus on mechanisms. Mol. Psychiatry 6:634–646 [DOI] [PubMed] [Google Scholar]
- 13. Sorensen H. J., Mortensen E. L., Reinisch J. M., Mednick S. A. 2009. Association between prenatal exposure to bacterial infection and risk of schizophrenia. Schizophr. Bull. 35:631–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yolken R. H., Torrey E. F. 2008. Are some cases of psychosis caused by microbial agents? A review of the evidence. Mol. Psychiatry 13:470–479 [DOI] [PubMed] [Google Scholar]
- 15. Akira S. 2006. TLR signaling. Curr. Top. Microbiol. Immunol. 311:1–16 [DOI] [PubMed] [Google Scholar]
- 16. Parker L. C., Prince L. R., Sabroe I. 2007. Translational mini-review series on Toll-like receptors: networks regulated by Toll-like receptors mediate innate and adaptive immunity. Clin. Exp. Immunol. 147:199–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. De Miranda J., Yaddanapudi K., Hornig M., Lipkin W. I. 2009. Astrocytes recognize intracellular polyinosinic-polycytidylic acid via MDA-5. FASEB J. 23:1064–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Babcock A. A., Wirenfeldt M., Holm T., Nielsen H. H., Dissing-Olesen L., Toft-Hansen H., Millward J. M., Landmann R., Rivest S., Finsen B., Owens T. 2006. Toll-like receptor 2 signaling in response to brain injury: an innate bridge to neuroinflammation. J. Neurosci. 26:12826–12837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lehnardt S., Lachance C., Patrizi S., Lefebvre S., Follett P. L., Jensen F. E., Rosenberg P. A., Volpe J. J., Vartanian T. 2002. The Toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. J. Neurosci. 22:2478–2486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ma Y., Li J., Chiu I., Wang Y., Sloane J. A., Lu J., Kosaras B., Sidman R. L., Volpe J. J., Vartanian T. 2006. Toll-like receptor 8 functions as a negative regulator of neurite outgrowth and inducer of neuronal apoptosis. J. Cell Biol. 175:209–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tang S. C., Arumugam T. V., Xu X., Cheng A., Mughal M. R., Jo D. G., Lathia J. D., Siler D. A., Chigurupati S., Ouyang X., Magnus T., Camandola S., Mattson M. P. 2007. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc. Natl. Acad. Sci. U. S. A. 104:13798–13803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rolls A., Shechter R., London A., Ziv Y., Ronen A., Levy R., Schwartz M. 2007. Toll-like receptors modulate adult hippocampal neurogenesis. Nat. Cell Biol. 9:1081–1088 [DOI] [PubMed] [Google Scholar]
- 23. Bayer S., Altman J. 1991. Neocortical development. Raven, New York, NY. [Google Scholar]
- 24. Noctor S. C., Martinez-Cerdeno V., Ivic L., Kriegstein A. R. 2004. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat. Neurosci. 7:136–144 [DOI] [PubMed] [Google Scholar]
- 25. McConnell S. K., Kaznowski C. E. 1991. Cell cycle dependence of laminar determination in developing neocortex. Science 254:282–285 [DOI] [PubMed] [Google Scholar]
- 26. Takahashi T., Goto T., Miyama S., Nowakowski R. S., Caviness V. S., Jr. 1999. Sequence of neuron origin and neocortical laminar fate: relation to cell cycle of origin in the developing murine cerebral wall. J. Neurosci. 19:10357–10371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Acosta M. T., Pearl P. L. 2003. The neurobiology of autism: new pieces of the puzzle. Curr. Neurol. Neurosci. Rep. 3:149–156 [DOI] [PubMed] [Google Scholar]
- 28. Englund C., Fink A., Lau C., Pham D., Daza R. A., Bulfone A., Kowalczyk T., Hevner R. F. 2005. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J. Neurosci. 25:247–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hodge R. D., Kowalczyk T. D., Wolf S. A., Encinas J. M., Rippey C., Enikolopov G., Kempermann G., Hevner R. F. 2008. Intermediate progenitors in adult hippocampal neurogenesis: Tbr2 expression and coordinate regulation of neuronal output. J. Neurosci. 28:3707–3717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sessa A., Mao C. A., Hadjantonakis A. K., Klein W. H., Broccoli V. 2008. Tbr2 directs conversion of radial glia into basal precursors and guides neuronal amplification by indirect neurogenesis in the developing neocortex. Neuron 60:56–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tarabykin V., Stoykova A., Usman N., Gruss P. 2001. Cortical upper layer neurons derive from the subventricular zone as indicated by Svet1 gene expression. Development 128:1983–1993 [DOI] [PubMed] [Google Scholar]
- 32. Aten J. A., Stap J., Hoebe R., Bakker P. J. 1994. Application and detection of IdUrd and CldUrd as two independent cell-cycle markers. Methods Cell Biol. 41:317–326 [PubMed] [Google Scholar]
- 33. Cubelos B., Sebastian-Serrano A., Kim S., Moreno-Ortiz C., Redondo J. M., Walsh C. A., Nieto M. 2008. Cux-2 controls the proliferation of neuronal intermediate precursors of the cortical subventricular zone. Cereb. Cortex 18:1758–1770 [DOI] [PubMed] [Google Scholar]
- 34. Zimmer C., Tiveron M. C., Bodmer R., Cremer H. 2004. Dynamics of Cux2 expression suggests that an early pool of SVZ precursors is fated to become upper cortical layer neurons. Cereb. Cortex 14:1408–1420 [DOI] [PubMed] [Google Scholar]
- 35. Babulas V., Factor-Litvak P., Goetz R., Schaefer C. A., Brown A. S. 2006. Prenatal exposure to maternal genital and reproductive infections and adult schizophrenia. Am. J. Psychiatry 163:927–929 [DOI] [PubMed] [Google Scholar]
- 36. Brown A. S., Schaefer C. A., Wyatt R. J., Goetz R., Begg M. D., Gorman J. M., Susser E. S. 2000. Maternal exposure to respiratory infections and adult schizophrenia spectrum disorders: a prospective birth cohort study. Schizophr. Bull. 26:287–295 [DOI] [PubMed] [Google Scholar]
- 37. Urakubo A., Jarskog L. F., Lieberman J. A., Gilmore J. H. 2001. Prenatal exposure to maternal infection alters cytokine expression in the placenta, amniotic fluid, and fetal brain. Schizophr. Res. 47:27–36 [DOI] [PubMed] [Google Scholar]
- 38. Chess S. 1977. Follow-up report on autism in congenital rubella. J. Autism Child. Schizophr. 7:69–81 [DOI] [PubMed] [Google Scholar]
- 39. Bonthius D. J., Perlman S. 2007. Congenital viral infections of the brain: lessons learned from lymphocytic choriomeningitis virus in the neonatal rat. PLoS Pathog. 3:e149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hotchin J., Seegal R. 1977. Virus-induced behavioral alteration of mice. Science 196:671–674 [DOI] [PubMed] [Google Scholar]
- 41. Bautista J. R., Rubin S. A., Moran T. H., Schwartz G. J., Carbone K. M. 1995. Developmental injury to the cerebellum following perinatal Borna disease virus infection. Brain Res. Dev. Brain Res. 90:45–53 [DOI] [PubMed] [Google Scholar]
- 42. Hirano N., Kao M., Ludwig H. 1983. Persistent, tolerant or subacute infection in Borna disease virus-infected rats. J. Gen. Virol. 64(Pt. 7):1521–1530 [DOI] [PubMed] [Google Scholar]
- 43. Hornig M., Weissenbock H., Horscroft N., Lipkin W. I. 1999. An infection-based model of neurodevelopmental damage. Proc. Natl. Acad. Sci. U. S. A. 96:12102–12107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rubin S. A., Pletnikov M., Taffs R., Snoy P. J., Kobasa D., Brown E. G., Wright K. E., Carbone K. M. 2000. Evaluation of a neonatal rat model for prediction of mumps virus neurovirulence in humans. J. Virol. 74:5382–5384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shi L., Tu N., Patterson P. H. 2005. Maternal influenza infection is likely to alter fetal brain development indirectly: the virus is not detected in the fetus. Int. J. Dev. Neurosci. 23:299–305 [DOI] [PubMed] [Google Scholar]
- 46. Patterson P. H. 2009. Immune involvement in schizophrenia and autism: etiology, pathology and animal models. Behav. Brain Res. 204:313–321 [DOI] [PubMed] [Google Scholar]
- 47. Cardon M., Ron-Harel N., Cohen H., Lewitus G. M., Schwartz M. 2009. Dysregulation of kisspeptin and neurogenesis at adolescence link inborn immune deficits to the late onset of abnormal sensorimotor gating in congenital psychological disorders. Mol. Psychiatry 15:415–425 [DOI] [PubMed] [Google Scholar]
- 48. Kriegstein A., Noctor S., Martinez-Cerdeno V. 2006. Patterns of neural stem and progenitor cell division may underlie evolutionary cortical expansion. Nat. Rev. Neurosci. 7:883–890 [DOI] [PubMed] [Google Scholar]
- 49. Clancy B., Darlington R. B., Finlay B. L. 2001. Translating developmental time across mammalian species. Neuroscience 105:7–17 [DOI] [PubMed] [Google Scholar]
- 50. Merkle F. T., Alvarez-Buylla A. 2006. Neural stem cells in mammalian development. Curr. Opin. Cell Biol. 18:704–709 [DOI] [PubMed] [Google Scholar]
- 51. Akira S., Uematsu S., Takeuchi O. 2006. Pathogen recognition and innate immunity. Cell 124:783–801 [DOI] [PubMed] [Google Scholar]
- 52. Alexopoulou L., Holt A. C., Medzhitov R., Flavell R. A. 2001. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413:732–738 [DOI] [PubMed] [Google Scholar]
- 53. Cai Z., Pan Z. L., Pang Y., Evans O. B., Rhodes P. G. 2000. Cytokine induction in fetal rat brains and brain injury in neonatal rats after maternal lipopolysaccharide administration. Pediatr. Res. 47:64–72 [DOI] [PubMed] [Google Scholar]
- 54. Smith S. E., Li J., Garbett K., Mirnics K., Patterson P. H. 2007. Maternal immune activation alters fetal brain development through interleukin-6. J. Neurosci. 27:10695–10702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gandhi R., Hayley S., Gibb J., Merali Z., Anisman H. 2007. Influence of poly I:C on sickness behaviors, plasma cytokines, corticosterone and central monoamine activity: moderation by social stressors. Brain Behav. Immun. 21:477–489 [DOI] [PubMed] [Google Scholar]
- 56. Lathia J. D., Okun E., Tang S. C., Griffioen K., Cheng A., Mughal M. R., Laryea G., Selvaraj P. K., ffrench-Constant C., Magnus T., Arumugam T. V., Mattson M. P. 2008. Toll-like receptor 3 is a negative regulator of embryonic neural progenitor cell proliferation. J. Neurosci. 28:13978–13984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Moore P. A., Acs G., Hargreaves J. A. 1985. Postextraction pain relief in children: a clinical trial of liquid analgesics. Int. J. Clin. Pharmacol. Ther. Toxicol. 23:573–577 [PubMed] [Google Scholar]
- 58. Rotondo D., Abul H. T., Milton A. S., Davidson J. 1988. Pyrogenic immunomodulators increase the level of prostaglandin E2 in the blood simultaneously with the onset of fever. Eur. J. Pharmacol. 154:145–152 [DOI] [PubMed] [Google Scholar]
- 59. Geyer M. A., Dulawa S. C. 2003. Assessment of murine startle reactivity, prepulse inhibition, and habituation. Curr. Protoc. Neurosci. Chapter 8, unit 8.17 [DOI] [PubMed] [Google Scholar]
- 60. Swerdlow N. R., Geyer M. A. 1998. Using an animal model of deficient sensorimotor gating to study the pathophysiology and new treatments of schizophrenia. Schizophr. Bull. 24:285–301 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.