

Abstract
Prolonged cellular hypoxia results in energy failure and ultimately cell death. However, less-severe hypoxia can induce a cytoprotective response termed hypoxic preconditioning (HP). The unfolded protein response pathway (UPR) has been known for some time to respond to hypoxia and regulate hypoxic sensitivity; however, the role of the UPR, if any, in HP essentially has been unexplored. We have shown previously that a sublethal hypoxic exposure of the nematode Caenorhabditis elegans induces a protein chaperone component of the UPR (L. L. Anderson, X. Mao, B. A. Scott, and C. M. Crowder, Science 323:630-633, 2009). Here, we show that HP induces the UPR and that the pharmacological induction of misfolded proteins is itself sufficient to stimulate a delayed protective response to hypoxic injury that requires the UPR pathway proteins IRE-1, XBP-1, and ATF-6. HP also required IRE-1 but not XBP-1 or ATF-6; instead, GCN-2, which is known to suppress translation and induce an adaptive transcriptional response under conditions of UPR activation or amino acid deprivation, was required for HP. The phosphorylation of the translation factor eIF2α, an established mechanism of GCN-2-mediated translational suppression, was not necessary for HP. These data suggest a model where hypoxia-induced misfolded proteins trigger the activation of IRE-1, which along with GCN-2 controls an adaptive response that is essential to HP.
A lack of oxygen supply (hypoxia) poses serious challenges for cells that must then adapt to low oxygen until conditions improve or die. However, the precise cascade of events that control whether a cell adapts or dies in the face of hypoxia is unclear. Adaptive hypoxic protective mechanisms can be induced by brief sublethal exposures to hypoxia and/or ischemia. This phenomenon is called hypoxic or ischemic preconditioning and has been the subject of intense study to define intrinsic hypoxia protective mechanisms (6, 12, 30, 34). Two forms of hypoxic preconditioning (HP) have been described (12, 51). Immediate preconditioning appears within minutes after the sublethal hypoxic/ischemic episode and wanes within about 4 h; delayed preconditioning appears 12 to 24 h later and can last for days. Delayed preconditioning is thought to require changes in gene expression through new transcription (12).
The nematode Caenorhabditis elegans has been found to have delayed HP (10). A sublethal exposure of C. elegans to hypoxia induces the hypoxic protection of the animal as a whole and its myocytes and neurons with an onset of approximately 16 h and a duration of at least 36 h. As for delayed preconditioning in mammals, the mechanism in C. elegans for sensing hypoxia, transducing the signal, and inducing cytoprotection is unclear. We recently reported that a sublethal hypoxic exposure similar to that which produces HP induces a reporter of the unfolded protein response (UPR) in C. elegans (1). The C. elegans UPR consists of three defined branches: IRE-1-XPB-1, ATF-6, and PEK-1 (49). Misfolded proteins are sensed by IRE-1, resulting in homo-oligomerization, autophosphorylation, and activation. Activated IRE-1 cleaves XBP-1 mRNA with subsequent splicing to produce a new open reading frame that can be translated into the XBP-1 transcription factor. Similarly activated ATF-6 translocates to the Golgi apparatus in response to elevated misfolded proteins, where it is cleaved by proteases, producing a transcriptionally active form of ATF-6. Both XBP-1 and ATF-6 control the transcription of a large number of genes whose functions are crucial for maintaining endoplasmic reticulum (ER) homeostasis. PEK-1 (PERK-1 in mammals) acts more directly to phosphorylate translation initiation factor eIF2α and thereby suppress general protein translation, thus reducing the nascent unfolded protein load presented to the ER (44).
As mentioned above, we have shown previously that a UPR reporter is induced by hypoxia in C. elegans (1). We also found that a reduction-of-function mutation in ire-1 can decrease or increase the hypoxic survival of C. elegans depending on the presence or absence, respectively, of a second mutation that reduces global translation rates. Others have reported the activation of the UPR after ischemia (2, 11, 17, 18, 39, 43, 54, 57). Hypoxia also has been shown to activate the PERK-1 pathway in cell culture (4, 25). These findings together suggest that the misfolded proteins generated by hypoxia can trigger hypoxic preconditioning by the activation of the UPR that then can protect cells, perhaps by the activation of PERK-1 and translational suppression. Here, we test the various aspects of this hypothesis using C. elegans genetic tools.
MATERIALS AND METHODS
Strains.
C. elegans strains were obtained from the Caenorhabditis Genetics Center (CGC), except where noted, and outcrossed three times prior to testing. Mutations were confirmed after outcrossing by sequencing. All strains were maintained at 20°C on NGM agar seeded with OP50 bacteria as described previously (7, 52). The strains carrying ire-1(zc14) and xbp-1(zc12) were obtained through outcrossing SJ30 ire-1(zc14);hsp-4::GFP and SJ17 xbp-1(zc12);hsp-4::GFP with the N2 wild type to get rid of the green fluorescent protein (GFP) transgene. The balancer mIn1[dpy-10(e128) mIs14(p-myo-2::GFP)] (14) was used to generate ire-1 heterozygotes and transheterozygotes as follows. mIn1/+ males were crossed with ire-1(zc14) or ire-1(v33) hermaphrodites. GFP-positive progeny were kept and presumed to have the genotype zc14/mIn1 or v33/mIn1. zc14/mIn1 males were crossed with ire-1(v33) hermaphrodites, and non-GFP-expressing F1 hermaphrodites segregating from a cross that produced 50% male progeny were assumed to have the genotype zc14/v33 and were immediately tested. ire-1(tm400) and atf-6(tm1153) were from Shohei Mitani (Tokyo Women's Medical College, Tokyo, Japan) and the Japan National Bioresource Project (http://www.shigen.nig.ac.jp/c.elegans/index.jsp). tm400 is a relatively new ire-1 allele that was supplied as a heterozygote, since homozygotes initially were classified as lethal/sterile. However, after outcrossing a few generations, we identified tm400 homozygotes that were viable and fertile. By using primers encompassing the deletion region of tm400, three cDNA products from each primer pair were identified by RT-PCR from ire-1(tm400) animals (data not shown). After we sequenced these products, they were identified as novel ire-1 mRNA species, with a frameshift and early stop codon downstream of the deletion breakpoint. Thus, these mutant mRNAs likely would produce nonfunctional proteins. Given a weaker tunicamycin sensitivity phenotype than that of ire-1(v33), the putative null mutant, we deduce that tm400 mostly likely produces protein products generated by utilizing downstream in-frame ATGs (most likely Met228, which could produce a 740-amino-acid residue polypeptide with transmembrane, kinase, and riboendonuclease domains).
Imaging Phsp-4::GFP.
The detailed protocol for imaging Phsp-4::GFP was described previously (1).
Western blotting.
After treatment, worms were harvested with M9 buffer. For hypoxic treatments without recovery, worms were harvested inside the hypoxic chamber with deoxygenated M9 buffer (52). The worm pellets were collected, and 1% SDS buffer was added before freezing. After an overnight freeze at −80°C, worm pellets were briefly sonicated and insoluble debris pelleted by centrifugation at 16,000 × g for 15 min. Sample protein concentrations were determined by a NanoDrop spectrophotometer (Thermo Scientific, Wilmington, DE). Forty micrograms of total protein was loaded for each sample for SDS-PAGE. The detailed Western blotting protocol is available elsewhere (www.cellsignaling.com). All antibodies were from Cell Signaling (Beverly, MA): phospho-eIF2α (p-e1F2α) (ser51) (no. 9721), β-actin (13E5) (no. 4970), and anti-rat IgG horseradish peroxidase (HRP)-linked secondary antibody and detection system (no. 7077).
RT-PCR.
A synchronous population (1 day post-larval stage 4) of animals was treated under conditions described in Results and the figure legends. RNA was isolated by a TRIzol freeze-cracking method. cDNA was synthesized with a RETROscript random decamer kit (Applied Biosystems, Foster City, CA) with 1 μg of total RNA as the template. Quantitative real-time PCR was performed as described previously (1) with SYBR green PCR master mix (Applied Biosystems, Foster City, CA) in an Applied Biosystems 7500-fast reverse transcription-PCR (RT-PCR) instrument with a Rox passive-reference dye. Primers were constructed to amplify an approximately 100-bp fragment. Standard PCR amplification with the primer sets produced single bands migrating at the correct size. Primers for hsp-4 were AACGGAATATTGCACGTAAGCGCC (forward) and TGAGACGATTGTGGTCGTTGGTGA (reverse), and those for ire-1 were GCTGAACGTGGAGCCATTGCACCG (forward) and CATCCAAGTGAGAAGATATCAACTGG (reverse). Semiquantitative RT-PCR for xbp-1 was determined with a DNAEngine Peltier thermal cycler (Bio-Rad, Hercules, CA). The exponential phases of amplification for act-1 and xbp-1 were empirically determined by two-cycle increments from 14 to 28 cycles. Sixteen and 20 cycles were used for act-1 and xbp-1 amplifications, respectively. xbp-1 primers were GGAGAGGATCGCCGTGCCT (forward) and GATGGAGGTGGATCGGGCCTGTT (reverse). The primers created 81- and 58-bp products for unspliced and spliced forms of xbp-1, respectively. The products were resolved with an 8% PAGE gel.
Hypoxia and Tm incubations.
Hypoxia treatment and HP protocols were similar to those published previously, except no washes were performed inside the hypoxia chamber, as we have found these to be unnecessary (10, 47). Briefly, synchronized populations of young adult animals were transferred from NGM plates to 1.5-ml Eppendorf tubes with 1 ml M9 buffer; the buffer was removed down to 100 μl, and the tubes then were placed into the hypoxia chamber as described previously (47). For HP incubations, the tubes were removed after 4 h and the worms recovered for 20 h on fresh NGM plates before a hypoxic killing incubation. Normoxic preconditioned controls were treated identically, except the preconditioning incubation instead was performed at 26°C in a normoxic chamber. For hypoxic killing incubations, worms were placed in the hypoxic chamber as described above and incubated for 20 h. The temperature of the hypoxic incubations was 26°C, and the oxygen concentration was <0.2%. Animals were scored 20 to 24 h after the completion of the hypoxic killing incubations. Animals without pharyngeal pumping and without spontaneous or touch-evoked movement were scored as dead. For tunicamycin (Tm) pretreatments, 1 day post L4 worms were washed off plates and incubated with designated concentrations of Tm in M9 buffer for 4 h at 20°C. After Tm incubation, worms were returned to regular NGM plates with food for 20 h at 20°C before they were challenged with a lethal 20-h hypoxic exposure. Worms then were scored for survival as previously described (10, 47). The Tm growth arrest assay has been published previously (1). Briefly, eggs from animals of various genotypes were laid on NGM plates containing 1 μg/ml Tm. The fraction of worms reaching the adult stage was scored 3 days later.
RESULTS
Hypoxia induces the UPR.
We first asked whether hypoxic preconditioning induces the expression of components of the UPR. We measured two markers of UPR induction, HSP-4 induction and eIF2α phosphorylation. HSP-4 expression is transcriptionally induced in response to an increase in misfolded proteins by an IRE-1-dependent mechanism (8, 48). After 4 h of hypoxia (<0.2% O2) but no reoxygenation, expression from a GFP transgene driven by an hsp-4 promoter was unchanged from control levels (Fig. 1 A and B). However, 4 h of recovery and reoxygenation produced a marked increase in pHSP-4::GFP expression, which then returned to control levels after 20 h of recovery (Fig. 1A and B). To confirm that the transgene expression accurately reflects the native gene activity, we measured HSP-4 native transcript levels by quantitative RT-PCR (qRT-PCR) (Fig. 1C). Indeed, native HSP-4 transcript levels remained unchanged even after 8 h of hypoxia but were significantly increased after reoxygenation, with a return to control levels by 20 h. The translation factor eIF2α is phosphorylated by the UPR components PERK-1 and GCN-2. Phosphorylated eIF2α levels increased rapidly during hypoxic incubation but fell back to baseline within 1 h of reoxygenation (Fig. 1D). Thus, while eIF2α is phosphorylated in response to hypoxia in C. elegans, the time course is inconsistent with translational suppression by p-eIF2α contributing to the effector mechanism of hypoxic cytoprotection by HP.
FIG. 1.

Hypoxia and HP activate the UPR. HP activates an hsp-4 promoter-GFP fusion reporter (A and B) and the endogenous hsp-4 gene (C). (B) After HP, the level of GFP increased significantly after a 4-h recovery (*, P < 0.001, two-tailed t test), and it returned to the control level after 16 more hours. (C) The level of hsp-4 mRNA was measured by qRT-PCR and was significantly elevated after a 2- and 4-h recovery from 4 h of hypoxia (P < 0.05, two-tailed t test), while hypoxia alone (up to 8 h) or a shorter HP incubation had no effect. (D) Hypoxia induced eIF2α phosphorylation. The level of phosphorylated eIF2α increased after 1 h of hypoxia and remained high under hypoxic conditions but rapidly returned to baseline during normoxic recovery. Relative band intensities normalized to no hypoxia are given. β-Actin levels decreased relative to total protein during the hypoxic incubation, thus the p-eIF2α/β-actin ratio increased greatly. The 0-h hypoxia/0 recovery and the 4-h hypoxia/0 recovery conditions were repeated for a total of four trials, and the relative p-eIF2α induction (1.96 ± 0.29) was statistically significant (P < 0.01, paired t test).
Induction of the UPR produces a delayed hypoxic protective response.
We next asked whether the induction of misfolded proteins is sufficient to induce a hypoxic protective response in a manner similar to that of HP. Tunicamycin (Tm) inhibits the N-glycosylation of proteins and thereby increases the load of misfolded proteins transiting the endoplasmic reticulum (29). In C. elegans, Tm reliably induces the UPR, presumably through its known activity to increase protein misfolding (8, 48). We pretreated wild-type C. elegans for 4 h with various concentrations of Tm and then allowed the animals to recover for 20 h before beginning a normally lethal hypoxic incubation. Tm induced a significant protection from hypoxia in a concentration-dependent manner (Fig. 2 A and B). The onset and duration of the tunicamycin preconditioning (TmP) after recovery from Tm incubation was delayed (Fig. 2C). Significant protection compared to control incubations in buffer was not seen until 14 h after the Tm incubation. Maximal TmP was seen after a recovery time of 16 h, and the protection persisted for at least 24 h, although the magnitude of the protection was waning at that point. Notably, Tm present during the hypoxic incubation was not protective (data not shown). Thus, the protective effect was a delayed reaction to the Tm and not due to the Tm itself. Buffer incubations also induced a hypoxic protection but with a time course distinct from that of TmP. The buffer-induced protection peaked at 4 h and fell thereafter. We speculate that the buffer induces a starvation stress or that the act of swimming in buffer for 4 h induces a brief protective stress response.
FIG. 2.

Tunicamycin-induced hypoxic protection. (A) Tunicamycin (Tm) pretreatment induced protection from subsequent hypoxic injury. Worms were treated with the indicated concentrations of Tm for 4 h before being recovered for 16 h. After recovery, Tm-pretreated worms were challenged with hypoxia for 22 h, and survival was scored after another 24-h recovery. Values are means ± standard deviations (SD) from three trials (* P < 0.001, paired t test, Tm versus buffer control). (C) Time course of Tm-induced hypoxia protection. The experiment was performed as described above with 10 μg/ml Tm or buffer only with various recovery times prior to the 22-h hypoxic exposure. The control value is for animals receiving no pretreatment as opposed to buffer pretreatment. Values are means ± SD from three trials (*, P < 0.01, paired t test, Tm versus buffer). (D) Wild-type (N2) or mutant animals were tested for Tm (10 μg/ml)-induced hypoxia protection. Animals were exposed for 4 h to Tm or buffer control and then recovered for 20 h prior to a 22-h hypoxic exposure, and then they were scored 24 h later for survival. Net survival (Tm survival − buffer survival) was calculated for each genotype. Tm induced significant hypoxic protection compared to that by buffer (P < 0.01, paired t test) in all strains except for the ire-1, atf-6, and xbp-1 mutants. Each bar represents the means ± SD from a minimum of three independent trials with at least 30 animals/trial. *, P < 0.01, paired t test, Tm versus buffer.
To determine whether TmP was mediated by the induction of the UPR, we measured TmP in reduction-of-function mutations in UPR genes (Fig. 3) with the hypothesis that one or more pathways within the UPR are necessary for the induction of TmP. A large deletion mutation in pek-1 or gcn-2 did not block Tm preconditioning (Fig. 2D and 3C, E). On the other hand, three loss-of-function mutant alleles of ire-1 (Fig. 3B), two alleles of atf-6 (Fig. 3D), and an allele of xbp-1 (Fig. 3F) all were defective for Tm preconditioning (Fig. 2D). Indeed, in the xbp-1(lf) allele, Tm preincubation significantly reduced survival from hypoxia. These data indicate that the ire-1, xbp-1, and atf-6 branches but not the pek-1 or gcn-2 branches of the UPR are essential for TmP. GRP78/Bip is a family of ER chaperones that regulate the UPR by binding to misfolded proteins, resulting in the disinhibition of UPR components, including homologs of IRE-1 and ATF-6 (42). C. elegans has two Bip homologs, hsp-3 and hsp-4, both of which are activated by ER stress (48). A reasonable hypothesis is that an early step in tunicamycin preconditioning is the activation of HSP-3 or HSP-4, which then disinhibit IRE-1 and ATF-6 and promote hypoxic protection. However, null mutations in hsp-3 or hsp-4 (Fig. 3G, H) neither blocked nor enhanced Tm preconditioning (Fig. 2D).
FIG. 3.

UPR pathway and mutants. (A) Schematic of UPR pathways. PEK-1, IRE-1, and ATF-6 are activated in the presence of unfolded proteins in the ER lumen. These pathways can promote adaptation to unfolded proteins via translational suppression or through a transcriptional response. GCN-2 functions along with activated PEK-1 to suppress translation. (B) ire-1 mutations. ire-1(v33) has an N-terminal 878-bp deletion resulting in a frameshift and stop and is a presumptive null mutation (48). ire-1(ok799) has a 2,093-bp deletion and 409-bp insertion and also should represent a null mutation (50). ire-1(zc14) has a missense mutation in a conserved residue in the kinase domain (8). ire-1(tm400) has a 600-bp deletion and 1-bp insertion that ends in an intron (see Wormbase.org and Materials and Methods). The mutant product is unclear. (C) pek-1(ok275) has a 2,073-bp deletion that produces a frameshift and stop codon (48). (D) Proteolysis of ATF-6 produces ATF-6s with only the maroon domain that is truncated by both mutations. ok551 has a 1,900-bp deletion (49); tm1153 has a 643-bp frameshift deletion (Wormbase.org). (E) gcn-2(ok871) has a 1,481-bp in-frame deletion starting and ending in exons (33); gcn-2(ok886) has a 1,179-bp in-frame deletion that starts and ends in exons (33). (F) xbp-1(zc12) has an early stop (8). (G) hsp-3(ok1083) has a 1,422-bp deletion that starts and ends in exons, causing frameshift (22). (H) hsp-4(gk514) has a 752-bp deletion that starts and ends in exons, causing frameshift (46). TM, transmembrane domain. The RWD domain was named after three major RWD-containing proteins: RING finger-containing proteins, WD-repeat-containing proteins, and yeast DEAD (DEXD)-like helicases. ΨPK, degenerate kinase domain; PK, kinase domain; HisRS, histidyl-tRNA synthetase; RB/DD, ribosome-binding and dimerization domain.
ire-1 and gcn-2 are required for hypoxic preconditioning.
We next tested whether the same UPR components were required for hypoxic preconditioning (HP). As for Tm preconditioning, HP consistently provided protection from subsequent harsh hypoxic exposure for wild-type animals (Fig. 4 A and B). Again, the pek-1 deletion mutation had no effect on HP; pek-1(ok275) animals were strongly protected by HP (Fig. 4B). However, unlike for TmP, gcn-2(ok871) completely blocked HP (Fig. 4C). gcn-2(ok886), an allele with a smaller deletion that removes less of the kinase and tRNA-binding domains (Fig. 3E), also failed to exhibit a significant increase in survival after HP, although there was a trend toward protection (Fig. 4C). Two ire-1 alleles (v33 and ok799) blocked HP (Fig. 4C); however, unlike the case for Tm preconditioning, ire-1(zc14), a missense mutation in the kinase domain that is thought to abolish the XBP-1 endonuclease activity of IRE-1 and behaves as a reduction-of-function allele (Fig. 3B) (8), did not block HP (Fig. 4C). Also, unlike the case for TmP, neither atf-6 nor xbp-1 mutation blocked HP (Fig. 4C). As for TmP, the mutation of hsp-3 or hsp-4 had no effect on HP (Fig. 4C). These data show that Tm preconditioning and hypoxic preconditioning both require an intact UPR but that the mechanisms are not identical. In common to both TmP and HP is a requirement for IRE-1.
FIG. 4.

UPR components required for hypoxic preconditioning (HP). (A and B) Wild-type (N2) animals were exposed to hypoxia (HP) or normoxia (control) incubations for 4 h and then allowed to recover for 20 h prior to a 20-h hypoxic incubation. Survival was scored after another 24-h recovery. (C) Survival from HP in the wild type and UPR mutants. Net survival (survival of HP-treated animals − survival of control animals) is plotted for each genotype. Each bar represents the means ± standard deviations from a minimum of three independent trials with at least 30 animals/trial. *, P < 0.01, paired t test, HP versus control.
Role of ire-1 in hypoxic injury.
Given the unique role among UPR components of IRE-1 in both HP and Tm preconditioning, we wanted to compare the native hypoxic sensitivity of ire-1 alleles to that of the other UPR mutants. As previously reported (1), the missense allele ire-1(zc14) was significantly hypoxia resistant (Fig. 5 A). In addition, ire-1(tm400), a deletion allele that has the potential for an alternative translation start site downstream of the deletion (see Materials and Methods), also was hypoxia resistant. However, the two other ire-1 deletion alleles were not hypoxia resistant. Likewise, none of the other UPR mutants were hypoxia resistant. The apparent less-severe phenotype of ire-1(v33) compared to those of zc14 and tm400 was particularly surprising given that the v33 deletion mutation results in a frameshift and an early stop codon and is presumably a null mutation (Fig. 3B) (48). Three mechanisms might explain this result. First, other unknown mutations in the zc14 and tm400 mutant strains might be responsible for the resistance. Second, IRE-1 might have both hypoxic sensitivity promoting and blocking activities and zc14 and tm400 only disrupt the promoting activity. Third, hypoxic sensitivity might have a biphasic response to the level of activity of IRE-1 so that the complete absence of IRE-1 function is deleterious but a partially reduced activity can protect from hypoxia death. To distinguish between these mechanisms, we measured the hypoxic sensitivity of heterozygous and transheterozygous ire-1(zc14), tm400, and v33 mutants (Fig. 5B). zc14/balancer heterozygous animals had a hypoxic sensitivity similar to that of wild-type and balancer/+ animals. However, v33 and tm400 heterozygotes were strongly hypoxia resistant. zc14/v33 transheterozygotes had a hypoxic sensitivity similar to that of zc14 homozygous animals. These data are most consistent with the third hypothesis that hypoxic sensitivity is reduced with partial but not complete loss of ire-1 function.
FIG. 5.

Effect of UPR mutants on hypoxic and tunicamycin sensitivity. (A) Animals with the indicated alleles were exposed to hypoxia for 20 h without any pretreatment, and survival was scored after a 24-h recovery (*, P < 0.0001 versus N2 by unpaired t test). (B) Animals with different ire-1 genetic backgrounds were tested for hypoxic survival without any pretreatment. (*, P < 0.001 versus N2; #, P < 0.05 versus zc14/zc14 or zc14/v33, unpaired t test). (C) Animals with different ire-1 genetic backgrounds were tested for sensitivity to Tm toxicity. Eggs were laid on the plates with 1 μg/ml Tm. After 3 days, the percentage of adult worms was scored (*, P < 0.01 for results greater than those for N2; #, P < 0.01 for results less than those for N2, unpaired t test). (D) The levels of ire-1 mRNA from the wild type and mutants were determined by quantitative RT-PCR by using a primer pair annealed 5′ of ire-1 cDNA. {*, P < 0.01, versus N2, zc14/zc14, or zc14/mIn1[dpy-10(e128) mIs14(p-myo-2::GFP)], unpaired t test}.
We then tested if the tunicamycin sensitivities of the ire-1 allelic combinations mirrored their hypoxic sensitivity. Indeed, in an assay of Tm-induced developmental arrest, both zc14 and tm400 homozygotes and v33 heterozygotes were Tm resistant, whereas v33 homozygotes were Tm hypersensitive, as had been reported previously (Fig. 5C) (48). Consistent with an early stop mutation and putative null phenotype of ire-1(v33), the transcript levels of v33 homozygotes were about 10-fold lower than that in wild-type or zc14 mutant animals. v33 heterozygotes had significantly reduced ire-1 mRNA levels as well, which is consistent with a haploinsufficient phenotype seen in v33 heterozygotes (Fig. 5D). This correspondence of Tm and hypoxic sensitivity phenotypes is consistent with the hypothesis that the biphasic effect of reducing IRE-1 activity on hypoxic sensitivity is due to the response to unfolded protein stress.
Mechanism of gcn-2- and ire-1-mediated HP and hypoxia resistance.
The best-established target of the GCN-2 kinase is the translation factor eIF2α. To determine whether the increase in p-eIF2α during the hypoxic preconditioning incubation (Fig. 1D) is required for the subsequent induction of HP, we measured p-eIF2α levels in the wild type and the HP-defective mutant, gcn-2(ok871). In both strains, p-eIF2α levels were similarly and significantly increased relative to that of β-actin during the 4-h hypoxic preconditioning incubation (Fig. 6 A and B). On the other hand, the significant hypoxic induction of p-eIF2α was blocked in pek-1(ok275), a mutant with normal HP (Fig. 6C). Thus, the phosphorylation of eIF2a is neither necessary nor sufficient for HP, and the relevant GCN-2 target is unknown.
FIG. 6.

eIF2α phosphorylation and XBP-1 splicing after HP. (A to C) N2, pek-1(ok275), and gcn-2(ok871) animals were treated with hypoxia for 4 h and recovered under normoxia. Protein samples from various recovery time points were subjected to Western blotting and were probed by an antibody against p-eIF2α (ser51). The same blots were stripped and reprobed with a β-actin antibody. Intensity values for the p-eIF2α bands normalized to the control are indicated along with the ratio of the normalized intensities of the p-eIF2α bands to β-actin. Four independent trials of the control and 0-h recovery time point gave normalized p-eIF2α intensities of 1.96 ± 0.29 for N2, 1.80 ± 0.07 for gcn-2(ok871), and 1.14 ± 0.15 for pek-1(ok275). P values for the change in intensities (paired t test) were the following: for N2, 0.00531; for gcn-2(ok871), 0.00028; and for pek-1(ok275), 0.399. (D and E) The unspliced and spliced forms of xbp-1 mRNA were amplified by RT-PCR in N2 and ire-1 mutant animals under control conditions (D) and after a 2-h recovery from a 4-h HP incubation (E). Spliced xbp-1 was undetectable in all three ire-1 mutant alleles under both conditions.
While IRE-1 has other known downstream targets (15, 20, 21, 27), XBP-1 is the best characterized. XBP-1 clearly is not required for HP, as an xbp-1(lf) mutant exhibits a normal HP response (Fig. 4C). However, this result does not rule out the possibility that XBP-1 acts redundantly to induce HP or to regulate hypoxic sensitivity in general. Thus, we asked whether ire-1 allelic differences for HP and hypoxic sensitivity phenotypes correlated with XBP-1 splicing. All three ire-1 alleles failed to produce detectable levels of spliced XBP-1 under normal conditions or after an HP incubation (Fig. 6D and E). These data, along with the wild-type hypoxia/HP phenotypes of xbp-1(lf), indicate that IRE-1 controls HP and baseline hypoxic sensitivity through an XBP-1-independent mechanism.
DISCUSSION
We showed that hypoxic preconditioning in C. elegans induces unfolded protein response pathways. We also found that preincubation with tunicamycin, a drug that promotes protein misfolding, is capable of producing a delayed hypoxia protection similar to that of delayed HP. Finally, we showed that distinct but overlapping components of the unfolded protein response are required for hypoxic preconditioning and tunicamycin preconditioning. These results suggest a model for hypoxic preconditioning where misfolded proteins serve as early hypoxic sensors that then signal through IRE-1 to induce an adaptive hypoxia protective response along with essential signaling from GCN-2 (Fig. 7). We now would like to place our results in the context of previous studies of protein misfolding, the UPR, and hypoxic injury/preconditioning.
FIG. 7.
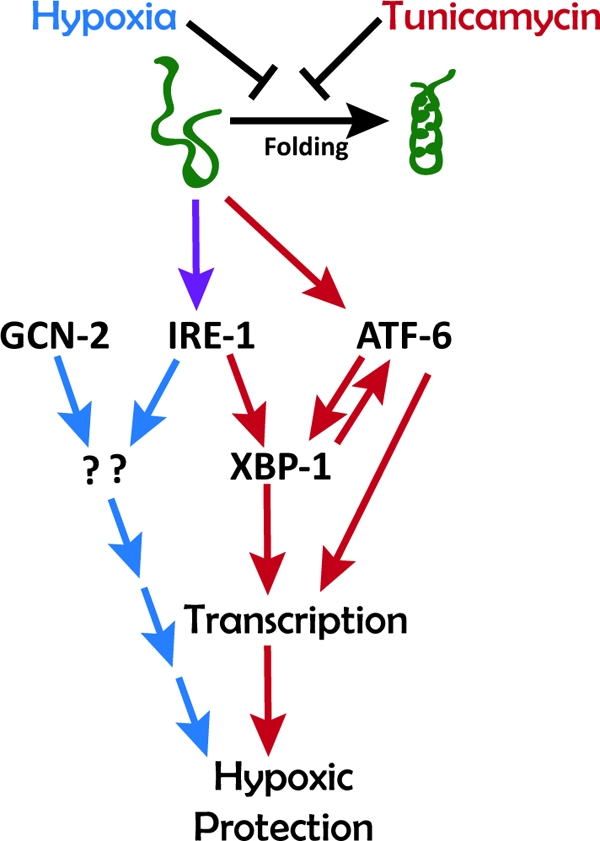
Working model for the role of the UPR in HP and TmP in C. elegans. Both hypoxia and tunicamycin inhibit protein folding and thereby activate signaling through IRE-1 and ATF-6 pathways. IRE-1 is required for both HP and TmP. GCN-2 is required for HP only, and ATF-6 is required for TmP only. The mechanisms downstream of IRE-1 and GCN-2 to induce HP are unknown.
A number of studies have suggested a role for the UPR in the hypoxic/ischemic injury of mammalian cells. Many studies have demonstrated an increase in UPR transcripts and protein in models of ischemic injury and preconditioning (2, 17, 39, 43, 54, 57). The induction of the UPR by hypoxia is consistent with studies of hypoxic tumor cells in which the UPR is activated (24). However, only a few studies have attempted to modulate the UPR to determine its functional role, if any, in hypoxic/ischemic injury. Tajiri et al. showed that hippocampal neurons from CHOP knockout mice were resistant to hypoxia-induced apoptosis (53). CHOP (C/EBP homologous protein) is a transcription factor induced by ER stress and is a target of ATF-6 and IRE-1; CHOP regulates the transcription of a number of genes that in general promote apoptosis (38). However, C. elegans does not have a known CHOP homolog. In the mouse kidney, pretreatment with tunicamycin was found to produce a long-lasting protection from ischemic injury (41). The mechanism of the protection was undetermined, although tunicamycin was shown to increase XBP-1 and GRP78 mRNA levels. Most recently, the modulation of ATF-6 has proven to be an important determinant of hypoxic/ischemic injury. The cardiac- specific expression of an inducible form of ATF6 in transgenic mice was found to markedly reduce cardiac myocyte death after ischemia in isolated hearts (28). On the other hand, the short hairpin RNA knockdown of ATF6 expression in primary rat myocyte cultures increased cell death after a severe hypoxic incubation (13). In C. elegans, we find that a deletion mutant of atf-6 blocks the induction of protection from hypoxia by tunicamycin, which is consistent with a role of ATF-6 in promoting hypoxic protection.
The requirements for the ATF-6 and IRE-1-XBP-1 pathways but not PEK-1 in Tm-induced hypoxic protection are intriguing. First, the requirements of both XBP-1 and ATF-6 are consistent with previous reports showing that these two transcription factors can target the same promoter elements and therefore coregulate the expression of certain ER stress response genes (59). XBP-1 and ATF-6 can heterodimerize as a prerequisite for binding to some promoter elements (58). Additionally, XBP-1 and ATF-6 are coordinately regulated by IRE-1, which is required for the activation of both proteins (26, 60). As for the role of pek-1, despite the rich literature showing that PEK-1 activation is prosurvival in ischemic/hypoxic cell death (4, 11, 25), we found no evidence for a role of PEK-1 in hypoxic cell death or preconditioning. In general in C. elegans, PEK-1 has been found to be dispensable for the UPR. For example, the inductions of apy-1 and Rho subfamily member crp-1 are controlled by ire-1, xbp-1, and atf-6 but not pek-1 (9, 55). Pore-forming toxins also activate ire-1, xbp-1, and atf-6 but not pek-1 in worms (5). However, we did find that the hypoxia-induced phosphorylation of eIF2α required functional PEK-1.
The finding that GCN-2 is required for HP was surprising. GCN-2 is homologous to the only eIF2α kinase found in yeast. In yeast, the gcn-2 homolog has been found to be essential for UPR function, so it also has been known as a super-UPR component in yeast (40). However, GCN-2 was not required for hypoxia-induced eIF2α phosphorylation (Fig. 6B) nor for TmP (Fig. 2D). Also, GCN-2 doesn't appear to be directly activated by ER stress in mammalian cells (16). The target of GCN-2 signaling in the context of HP is unknown; however, the yeast bZIP transcription factor Gcn4, a homolog of mammalian ATF4, functions downstream of Gcn2 and independently of eIF2α to regulate yeast UPR target genes (40). In yeast, Gcn2 appears to be required for the basal expression of Gcn4, which is further activated by Ire1 during ER stress to promote UPR gene transcription. Our data are consistent with cooperativity between GCN-2 and IRE-1, but the downstream pathways in C. elegans are undefined.
IRE-1 was unique among the UPR genes in having essential roles in both tunicamycin and hypoxic preconditioning. However, the transduction pathway downstream of IRE-1 was distinct for the two preconditioning conditions; tunicamycin preconditioning required XBP-1, whereas hypoxic preconditioning did not. IRE-1 is classically thought to function in a linear pathway with its downstream target being the transcription factor XBP-1. However, potential XBP-1-independent functions of IRE-1 have been reported and fall into two broad classes, mRNA degradation and protein-protein interactions (19). Regulated Ire1-dependent decay of mRNAs (RIDD) was defined originally in Drosophila melanogaster cells (21) and subsequently demonstrated in mammalian cells (15, 20, 37). Whether a RIDD mechanism functions in C. elegans is unknown. Various aspects of the RIDD pathway are similar to the role of the UPR in HP in C. elegans. Like HP, RIDD appears not to require XBP-1 (20). Second, in the context of ER stress, RIDD mechanisms can be induced pharmacologically and bypass the requirement for the kinase activity of IRE1 in mouse embryonic fibroblasts (20). The normal HP phenotype of the ire-1 kinase domain mutant zc14 suggests that IRE-1 kinase activity also is not essential for HP, although the kinase activity of zc14 has not been directly assayed. However, Han et al. found in an HEK293-derived cell line that IRE1 kinase activity was required for RIDD (15), so the resemblance of HP and RIDD with regard to IRE-1 kinase activity is unclear. Another issue with RIDD and its role in HP is the timing. Protection after hypoxic preconditioning in C. elegans occurs about 16 h after preconditioning (10), whereas RIDD is thought to act fully within 4 to 8 h to degrade certain RNAs (15, 21). These disparate time courses suggest that RIDD itself is not the effector of protection after precondition; rather, if involved in HP, RIDD would act earlier and upstream of the effector mechanism, perhaps in transducing the preconditioning stimulus.
Alternatively, IRE-1 could control HP in C. elegans via proteins that have been found to interact with IRE1 (27). Mammalian IRE1 has been shown to form a complex with TRAF2, ASK-1, JNK, and ASK1-interacting protein (AIP1), which together promote apoptotic cell death in models of ER stress (23, 27, 32, 56). This IRE1 pathway is thought to be independent of XBP1 because an endonuclease-deficient IRE1 was competent to interact with TRAF2 and activate JNK (56). Paradoxically, in C. elegans, the overexpression of the C. elegans homolog of JNK, JNK-1, increases life span and thermal and oxidative stress resistance (36). Thus, a plausible hypothesis is that limited hypoxia activates IRE-1, stimulating JNK-1, which promotes the transcription of proadaptive gene products, perhaps similar to those that increase life span and stress resistance. More prolonged hypoxia also could act through an IRE-1/JNK-1 pathway to promote cell death, the more typical output of the JNK1 pathway in mammalian models. One output of the JNK pathway that might reasonably regulate HP is macroautophagy. Autophagy has been shown to protect against ER stress (3, 35) and is activated by ER stress by an IRE1-, JNK-, and TRAF2-dependent mechanism (35). We have previously shown that macroautophagy is activated by hypoxia and protects against hypoxic injury in C. elegans (45). Thus, the activation of autophagy is a plausible candidate as the effector of IRE-1-dependent HP. Another IRE1-interacting protein is USP14. USP14, a ubiquitin-specific protease, has been shown to interact directly with IRE1α in HEK293 cells (31). Kinase-dead IRE1α is capable of recruiting USP14 to a complex that includes members of the ER-associated protein degradation (ERAD) machinery. The association of UPR14 with kinase-dead IRE1α correlated with the inhibition of ERAD, whereas autophosphorylated IRE1α did not bind UPR14 and was incompetent for UPR14-mediated ERAD inhibition. Although not directly tested, this IRE1 function presumably would be independent of XBP-1 and therefore have characteristics consistent with the mediation of HP. Future studies will be aimed at elucidating the mechanism(s) whereby IRE-1 and GCN-2 regulate HP in C. elegans and determining whether this mechanism is operant in mammalian cells.
Acknowledgments
This work was supported by R01-NS045905 from the National Institute of Neurological Disorders and Stroke, the McKnight Endowment Fund for Neuroscience, the American Heart Association, and the International Anesthesia Research Society.
Footnotes
Published ahead of print on 23 August 2010.
REFERENCES
- 1.Anderson, L. L., X. Mao, B. A. Scott, and C. M. Crowder. 2009. Survival from hypoxia in C. elegans by inactivation of aminoacyl-tRNA synthetases. Science 323:630-633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Azfer, A., J. Niu, L. M. Rogers, F. M. Adamski, and P. E. Kolattukudy. 2006. Activation of endoplasmic reticulum stress response during the development of ischemic heart disease. Am. J. Physiol. Heart Circ. Physiol. 291:H1411-H1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bernales, S., K. L. McDonald, and P. Walter. 2006. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 4:e423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bi, M., C. Naczki, M. Koritzinsky, D. Fels, J. Blais, N. Hu, H. Harding, I. Novoa, M. Varia, J. Raleigh, D. Scheuner, R. J. Kaufman, J. Bell, D. Ron, B. G. Wouters, and C. Koumenis. 2005. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J. 24:3470-3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bischof, L. J., C. Y. Kao, F. C. Los, M. R. Gonzalez, Z. Shen, S. P. Briggs, F. G. van der Goot, and R. V. Aroian. 2008. Activation of the unfolded protein response is required for defenses against bacterial pore-forming toxin in vivo. PLoS Pathog. 4:e1000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bolli, R. 2007. Preconditioning: a paradigm shift in the biology of myocardial ischemia. Am. J. Physiol. Heart Circ. Physiol. 292:H19-H27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brenner, S. 1974. The genetics of Caenorhabditis elegans. Genetics 77:71-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calfon, M., H. Zeng, F. Urano, J. H. Till, S. R. Hubbard, H. P. Harding, S. G. Clark, and D. Ron. 2002. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415:92-96. [DOI] [PubMed] [Google Scholar]
- 9.Caruso, M. E., S. Jenna, M. Bouchecareilh, D. L. Baillie, D. Boismenu, D. Halawani, M. Latterich, and E. Chevet. 2008. GTPase-mediated regulation of the unfolded protein response in Caenorhabditis elegans is dependent on the AAA+ ATPase CDC-48. Mol. Cell. Biol. 28:4261-4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dasgupta, N., A. M. Patel, B. A. Scott, and C. M. Crowder. 2007. Hypoxic preconditioning requires the apoptosis protein CED-4 in C. elegans. Curr. Biol. 17:1954-1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeGracia, D. J. 2004. Acute and persistent protein synthesis inhibition following cerebral reperfusion. J. Neurosci. Res. 77:771-776. [DOI] [PubMed] [Google Scholar]
- 12.Dirnagl, U., K. Becker, and A. Meisel. 2009. Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol. 8:398-412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doroudgar, S., D. J. Thuerauf, M. C. Marcinko, P. J. Belmont, and C. C. Glembotski. 2009. Ischemia activates the ATF6 branch of the endoplasmic reticulum (ER) stress response. J. Biol. Chem. 284:29735-29745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edgley, M. L., and D. L. Riddle. 2001. LG II balancer chromosomes in Caenorhabditis elegans: mT1(II;III) and the mIn1 set of dominantly and recessively marked inversions. Mol. Genet. Genomics 266:385-395. [DOI] [PubMed] [Google Scholar]
- 15.Han, D., A. G. Lerner, L. Vande Walle, J.-P. Upton, W. Xu, A. Hagen, B. J. Backes, S. A. Oakes, and F. R. Papa. 2009. IRE1α kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 138:562-575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harding, H. P., I. Novoa, Y. Zhang, H. Zeng, R. Wek, M. Schapira, and D. Ron. 2000. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6:1099-1108. [DOI] [PubMed] [Google Scholar]
- 17.Hayashi, T., A. Saito, S. Okuno, M. Ferrand-Drake, and P. H. Chan. 2003. Induction of GRP78 by ischemic preconditioning reduces endoplasmic reticulum stress and prevents delayed neuronal cell death. J. Cereb. Blood Flow Metab. 23:949-961. [DOI] [PubMed] [Google Scholar]
- 18.Hayashi, T., A. Saito, S. Okuno, M. Ferrand-Drake, R. L. Dodd, T. Nishi, C. M. Maier, H. Kinouchi, and P. H. Chan. 2003. Oxidative damage to the endoplasmic reticulum is implicated in ischemic neuronal cell death. J. Cereb. Blood Flow Metab. 23:1117-1128. [DOI] [PubMed] [Google Scholar]
- 19.Hetz, C., and L. H. Glimcher. 2009. Fine-tuning of the unfolded protein response: assembling the IRE1α interactome. Mol. Cell 35:551-561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hollien, J., J. H. Lin, H. Li, N. Stevens, P. Walter, and J. S. Weissman. 2009. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 186:323-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hollien, J., and J. S. Weissman. 2006. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313:104-107. [DOI] [PubMed] [Google Scholar]
- 22.Kapulkin, W. J., B. G. Hiester, and C. D. Link. 2005. Compensatory regulation among ER chaperones in C. elegans. FEBS Lett. 579:3063-3068. [DOI] [PubMed] [Google Scholar]
- 23.Kim, I., C. W. Shu, W. Xu, C. W. Shiau, D. Grant, S. Vasile, N. D. Cosford, and J. C. Reed. 2009. Chemical biology investigation of cell death pathways activated by endoplasmic reticulum stress reveals cytoprotective modulators of ASK1. J. Biol. Chem. 284:1593-1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koumenis, C., M. Bi, J. Ye, D. Feldman, and A. C. Koong. 2007. Hypoxia and the unfolded protein response. Methods Enzymol. 435:275-293. [DOI] [PubMed] [Google Scholar]
- 25.Koumenis, C., C. Naczki, M. Koritzinsky, S. Rastani, A. Diehl, N. Sonenberg, A. Koromilas, and B. G. Wouters. 2002. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol. Cell. Biol. 22:7405-7416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee, K., W. Tirasophon, X. Shen, M. Michalak, R. Prywes, T. Okada, H. Yoshida, K. Mori, and R. J. Kaufman. 2002. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 16:452-466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo, D., Y. He, H. Zhang, L. Yu, H. Chen, Z. Xu, S. Tang, F. Urano, and W. Min. 2008. AIP1 is critical in transducing IRE1-mediated endoplasmic reticulum stress response. J. Biol. Chem. 283:11905-11912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martindale, J. J., R. Fernandez, D. Thuerauf, R. Whittaker, N. Gude, M. A. Sussman, and C. C. Glembotski. 2006. Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ. Res. 98:1186-1193. [DOI] [PubMed] [Google Scholar]
- 29.Merksamer, P. I., A. Trusina, and F. R. Papa. 2008. Real-time redox measurements during endoplasmic reticulum stress reveal interlinked protein folding functions. Cell 135:933-947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murry, C. E., R. B. Jennings, and K. A. Reimer. 1986. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 74:1124-1136. [DOI] [PubMed] [Google Scholar]
- 31.Nagai, A., H. Kadowaki, T. Maruyama, K. Takeda, H. Nishitoh, and H. Ichijo. 2009. USP14 inhibits ER-associated degradation via interaction with IRE1alpha. Biochem. Biophys. Res. Commun. 379:995-1000. [DOI] [PubMed] [Google Scholar]
- 32.Nishitoh, H., A. Matsuzawa, K. Tobiume, K. Saegusa, K. Takeda, K. Inoue, S. Hori, A. Kakizuka, and H. Ichijo. 2002. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 16:1345-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nukazuka, A., H. Fujisawa, T. Inada, Y. Oda, and S. Takagi. 2008. Semaphorin controls epidermal morphogenesis by stimulating mRNA translation via eIF2α in Caenorhabditis elegans. Genes Dev. 22:1025-1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Obrenovitch, T. P. 2008. Molecular physiology of preconditioning-induced brain tolerance to ischemia. Physiol. Rev. 88:211-247. [DOI] [PubMed] [Google Scholar]
- 35.Ogata, M., S. Hino, A. Saito, K. Morikawa, S. Kondo, S. Kanemoto, T. Murakami, M. Taniguchi, I. Tanii, K. Yoshinaga, S. Shiosaka, J. A. Hammarback, F. Urano, and K. Imaizumi. 2006. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 26:9220-9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oh, S. W., A. Mukhopadhyay, N. Svrzikapa, F. Jiang, R. J. Davis, and H. A. Tissenbaum. 2005. JNK regulates lifespan in Caenorhabditis elegans by modulating nuclear translocation of forkhead transcription factor/DAF-16. Proc. Natl. Acad. Sci. U. S. A. 102:4494-4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oikawa, D., M. Tokuda, and T. Iwawaki. 2007. Site-specific cleavage of CD59 mRNA by endoplasmic reticulum-localized ribonuclease, IRE1. Biochem. Biophys. Res. Commun. 360:122-127. [DOI] [PubMed] [Google Scholar]
- 38.Oyadomari, S., and M. Mori. 2004. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 11:381-389. [DOI] [PubMed] [Google Scholar]
- 39.Paschen, W., C. Aufenberg, S. Hotop, and T. Mengesdorf. 2003. Transient cerebral ischemia activates processing of xbp1 messenger RNA indicative of endoplasmic reticulum stress. J. Cereb. Blood Flow Metab. 23:449-461. [DOI] [PubMed] [Google Scholar]
- 40.Patil, C. K., H. Li, and P. Walter. 2004. Gcn4p and novel upstream activating sequences regulate targets of the unfolded protein response. PLoS Biol. 2:E246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prachasilchai, W., H. Sonoda, N. Yokota-Ikeda, S. Oshikawa, C. Aikawa, K. Uchida, K. Ito, T. Kudo, K. Imaizumi, and M. Ikeda. 2008. A protective role of unfolded protein response in mouse ischemic acute kidney injury. Eur. J. Pharmacol. 592:138-145. [DOI] [PubMed] [Google Scholar]
- 42.Quinones, Q. J., G. G. de Ridder, and S. V. Pizzo. 2008. GRP78: a chaperone with diverse roles beyond the endoplasmic reticulum. Histol. Histopathol. 23:1409-1416. [DOI] [PubMed] [Google Scholar]
- 43.Roberts, G. G., M. J. Di Loreto, M. Marshall, J. Wang, and D. J. DeGracia. 2007. Hippocampal cellular stress responses after global brain ischemia and reperfusion. Antioxid. Redox. Signal 9:2265-2275. [DOI] [PubMed] [Google Scholar]
- 44.Rzymski, T., and A. L. Harris. 2007. The unfolded protein response and integrated stress response to anoxia. Clin. Cancer Res. 13:2537-2540. [DOI] [PubMed] [Google Scholar]
- 45.Samokhvalov, V., B. A. Scott, and C. M. Crowder. 2008. Autophagy protects against hypoxic injury in C. elegans. Autophagy 4:1034-1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sasagawa, Y., K. Yamanaka, and T. Ogura. 2007. ER E3 ubiquitin ligase HRD-1 and its specific partner chaperone BiP play important roles in ERAD and developmental growth in Caenorhabditis elegans. Genes Cells 12:1063-1073. [DOI] [PubMed] [Google Scholar]
- 47.Scott, B. A., M. S. Avidan, and C. M. Crowder. 2002. Regulation of hypoxic death in C. elegans by the insulin/IGF receptor homolog DAF-2. Science 296:2388-2391. [DOI] [PubMed] [Google Scholar]
- 48.Shen, X., R. E. Ellis, K. Lee, C. Y. Liu, K. Yang, A. Solomon, H. Yoshida, R. Morimoto, D. M. Kurnit, K. Mori, and R. J. Kaufman. 2001. Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell 107:893-903. [DOI] [PubMed] [Google Scholar]
- 49.Shen, X., R. E. Ellis, K. Sakaki, and R. J. Kaufman. 2005. Genetic interactions due to constitutive and inducible gene regulation mediated by the unfolded protein response in C. elegans. PLoS Genet. 1:e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shim, J., T. Umemura, E. Nothstein, and C. Rongo. 2004. The unfolded protein response regulates glutamate receptor export from the endoplasmic reticulum. Mol. Biol. Cell 15:4818-4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sommerschild, H. T., and K. A. Kirkeboen. 2002. Preconditioning-endogenous defence mechanisms of the heart. Acta Anaesthesiol. Scand. 46:123-137. [DOI] [PubMed] [Google Scholar]
- 52.Stiernagle, T. 11 February 2006, posting date. Maintenance of C. elegans. In D. Fay (ed.), Wormbook. The C. elegans Research Community. doi: 10.1895/wormbook.1.101.1. [DOI] [PMC free article] [PubMed]
- 53.Tajiri, S., S. Oyadomari, S. Yano, M. Morioka, T. Gotoh, J. I. Hamada, Y. Ushio, and M. Mori. 2004. Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ. 11:403-415. [DOI] [PubMed] [Google Scholar]
- 54.Truettner, J. S., K. Hu, C. L. Liu, W. D. Dietrich, and B. Hu. 2009. Subcellular stress response and induction of molecular chaperones and folding proteins after transient global ischemia in rats. Brain Res. 1249:9-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Uccelletti, D., A. Pascoli, F. Farina, A. Alberti, P. Mancini, C. B. Hirschberg, and A. C. Palleschi. 2008. APY-1, a novel Caenorhabditis elegans apyrase involved in unfolded protein response signalling and stress responses. Mol. Biol. Cell 19:1337-1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Urano, F., X. Wang, A. Bertolotti, Y. Zhang, P. Chung, H. P. Harding, and D. Ron. 2000. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287:664-666. [DOI] [PubMed] [Google Scholar]
- 57.Wang, S., F. M. Longo, J. Chen, M. Butman, S. H. Graham, K. G. Haglid, and F. R. Sharp. 1993. Induction of glucose regulated protein (grp78) and inducible heat shock protein (hsp70) mRNAs in rat brain after kainic acid seizures and focal ischemia. Neurochem. Int. 23:575-582. [DOI] [PubMed] [Google Scholar]
- 58.Yamamoto, K., T. Sato, T. Matsui, M. Sato, T. Okada, H. Yoshida, A. Harada, and K. Mori. 2007. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell 13:365-376. [DOI] [PubMed] [Google Scholar]
- 59.Yamamoto, K., H. Yoshida, K. Kokame, R. J. Kaufman, and K. Mori. 2004. Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J. Biochem. 136:343-350. [DOI] [PubMed] [Google Scholar]
- 60.Yoshida, H., T. Matsui, A. Yamamoto, T. Okada, and K. Mori. 2001. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107:881-891. [DOI] [PubMed] [Google Scholar]