

Abstract
We have recently shown that Src induces the formation of podosomes and cell invasion by suppressing endogenous p53, while enhanced p53 strongly represses the Src-induced invasive phenotype. However, the mechanism by which Src and p53 play antagonistic roles in cell invasion is unknown. Here we show that the Stat3 oncogene is a required downstream effector of Src in inducing podosome structures and related invasive phenotypes. Stat3 promotes Src phenotypes through the suppression of p53 and the p53-inducible protein caldesmon, a known podosome antagonist. In contrast, enhanced p53 attenuates Stat3 function and Src-induced podosome formation by upregulating the tumor suppressor PTEN. PTEN, through the inactivation of Src/Stat3 function, also stabilizes the podosome-antagonizing p53/caldesmon axis, thereby further enhancing the anti-invasive potential of the cell. Furthermore, the protein phosphatase activity of PTEN plays a major role in the negative regulation of the Src/Stat3 pathway and represses podosome formation. Our data suggest that cellular invasiveness is dependent on the balance between two opposing forces: the proinvasive oncogenes Src-Stat3 and the anti-invasive tumor suppressors p53-PTEN.
p53 is a potent tumor suppressor that plays a critical role in the regulation of cell cycle progression, DNA repair, apoptosis, and senescence (3, 10, 32, 57). Approximately half of all human tumors have compromised p53 function (25, 62). Loss of p53 function has also been implicated in the evolution of aggressive and metastatic cancers (28, 33, 42, 43), suggesting an anti-invasive and migration role of p53. Recent studies have increasingly unveiled this relatively less known aspect of p53 function in the regulation of cell migration and invasion (19, 20, 45, 63, 66). We have recently shown that p53, acting downstream of Src, strongly suppresses the formation of podosomes (also called invadopodia in cancer cells) and extracellular matrix (ECM) digestion by upregulating the expression of caldesmon, a known antagonist of podosomes (44).
Src, a proto-oncogenic nonreceptor tyrosine kinase, induces migratory and invasive phenotypes in various cell types by initiating extensive cytoskeletal rearrangements (38, 51, 67). Activated Src induces the formation of podosomes and rosettes of podosomes, which are dynamic, actin-rich membrane protrusions (9, 24, 40), specialized in the degradation of the ECM by the recruitment and secretion of matrix metalloproteinases (MMPs) (8, 38, 60, 64). Although the collaboration of Src with other oncogene products has been implicated in cellular transformation (4, 6), involvement of other oncogenes in the Src pathway leading to the formation of podosomes and invadopodia has not been proposed. One possible link is the transcription factor Stat3, which is activatable by Src and has been implicated in oncogenesis and the development of invasive phenotypes (22, 49, 69). Stat3 is often found to be upregulated in many cancers and is implicated in the promotion of aggressive metastasis (1, 14) via the transactivation of MMPs (21). The majority of reports have emphasized the transcription-dependent function of Stat3 in the regulation of cell proliferation and in prosurvival and antiapoptotic signaling. Relatively little is known, however, about its role in modulating cytoskeletal rearrangements leading to cell migration and invasion.
Phosphatase and tensin homologue deleted on chromosome 10 (PTEN) is another important tumor suppressor that has been shown to be mutated in the majority of advanced, invasive tumors (55, 59, 70). PTEN is a dual lipid phosphatidylinositol-3,4,5-phosphate (PtdInsP3) and protein phosphatase (46, 47). The lipid phosphatase activity of PTEN has been shown to play the dominant role as a tumor suppressor by negatively modulating the phosphatidylinositol 3-kinase (PI3K)/Akt pathway (11, 55). Accumulating data, however, have implicated the protein phosphatase activity of PTEN in cell motility (29). Possible links between PTEN, p53, Stat3, and Src can be gleaned from previous reports that PTEN can be transactivated by p53 (58) and that PTEN acts as a negative (61, 71) or positive (12) regulator of Stat3. Furthermore, it has been shown recently that PTEN suppresses the Src family kinase Fyn (15).
The objective of this study is to determine whether Stat3 and PTEN are involved in the Src-p53-caldesmon pathway for the formation of podosomes and the degradation of the ECM. For this study we used primary rat aortic smooth muscle cells (SMC) and NIH 3T3 (3T3) fibroblasts stably transduced with a constitutively active mutant of Src (SrcY527F). These Src cells are endowed with a strong propensity to produce numerous podosomes and rosettes of podosomes, and they have been used widely as excellent study models of cell invasion. In addition, we wanted to determine whether similar regulatory mechanisms exist for the invasion of smooth muscle cells and fibroblasts. Here we show that Stat3, activated by Src, promotes Src-induced invasive phenotypes through its suppressive role in the p53-caldesmon pathway. In turn, p53, besides inducing caldesmon expression, also downregulates the function of Src, as well as that of Stat3, through the induction of PTEN. Our findings provide new evidence for the existence of complex interplays between the Src-Stat3 and p53-PTEN axes and have demonstrated that their mutual antagonism plays a critical role in determining the outcome of Src-induced invasive phenotypes.
MATERIALS AND METHODS
Plasmid constructs/shRNAs.
Constitutively active SrcY527F (44, 72), wild-type (wt) p53 (13), mouse constitutively active Stat3 (caStat3) (7), and the wild-type caldesmon expression plasmid (EGFP-Cald) (17) have been described previously. The expression constructs for rat wt Stat3 (MRN1768-98078699) and mouse wt PTEN (MMM1013-7511653) were purchased from Open Biosystems. Mutations for the PTEN-C124S mutant (catalytically inactive), the PTEN-G129E mutant (deficient in lipid phosphatase activity while retaining its tyrosine phosphatase activity), and the Stat3 A661C N663C mutant (constitutively active dimer form) were all made using the QuikChange II XL site-directed mutagenesis kit (Stratagene). The short hairpin RNAs (shRNAs) were designed and cloned as previously described (44, 52). shRNAs were designed such that they target both rat and mouse transcripts unless otherwise indicated. The shRNA against p53 has already been published (44); the target/sense sequences used for designing shRNAs are 5′-GCAGGTATCTTGAGAAGCCAA-3′ and 5′-GAGCTGCACCTGATCACCTTA-3′ for mouse/rat Stat3 and 5′-GAGATCGTTAGCAGAAACAAAA-3′ and 5′-CCACAGCTAGAACTTATCAA-3′ for PTEN. A control shRNA that is not homologous with any known human, mouse, or rat sequence was designed (5′-TAATAAATAATAAGCTAATAA-3′). Promoter-luciferase constructs for p21, Mdm2, Bax, and Puma were a generous gift from M. Oren, Weizmann Institute of Science, Rehovot, Israel.
siRNA.
Small interfering RNAs (siRNAs) targeting MMP1 at two different loci were designed and were designated MMP1-1 (5′-CACUAACAUUCGAAAGGGU-3′) and MMP1-2 (5′-GCAGGUUCUACAUUCGUGU-3′) (Integrated DNA Technologies). Predesigned siRNAs targeting MMP10 at two different loci were ordered from Invitrogen (MMP10-1 [catalogue no. RSS332026] and MMP10-2 [catalogue no. RSS332027]). Negative siRNA control 1 was ordered from Applied Biosystems. siRNAs were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol 20 h postseeding. Forty-eight hours posttransfection, cells were harvested for a Matrigel assay, ECM degradation, or semiquantitative reverse transcription-PCR (RT-PCR).
Cell culturing, retroviral transduction, and transient transfection.
All cell lines were grown, and retroviral infections/transfections were carried out, as previously described (44). After infection or transfection, stable cell lines were selected with 5 μg/ml of puromycin (Sigma), 100 to 200 μg/ml hygromycin (Sigma), and 1 mg/ml of neomycin (Sigma) wherever applicable.
Antibodies/chemical inhibitors and other specialty reagents.
Antibodies were procured from Millipore for Src (05-185), green fluorescent protein (GFP) (AB3080), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (MAB374); from Cell Signaling for p53 (2524 and 9282), Stat3 (9132), Stat3-pY705 (91385), and PTEN (9559); from Abcam for Stat3 (ab69513) and phospho-Src (the anti-Src-pY418 antibody was used to detect chicken Src-pY416) (ab4816-50); from Sigma-Aldrich for MDM2 (M4308) and paxillin (P1093); from Biosource for phospho-cortactin (CTN-pY421) (44-854G); and from BD Bioscience for caldesmon (610661). 4′,6-Diamidino-2-phenylindole (DAPI) (D3571) and phalloidin 350 (A22281) were from Invitrogen; tetramethyl rhodamine isocyanate (TRITC)-conjugated phalloidin (P1951) and fluorescein isothiocyanate (FITC)-conjugated phalloidin (P5282) were from Sigma-Aldrich. The Src inhibitor AG-1879 (PP2) was purchased from Calbiochem; the p53 inhibitor pifithrin-α (PFA) and the genotoxic drug doxorubicin, from Sigma-Aldrich.
Extracellular matrix digestion assay.
Extracellular matrix digestion assays were carried out using a previously described method (65). The area of digestion was determined in pixels and converted to square micrometers by using Image Pro Plus 6 software. For each condition, a minimum of 30 cells from each of three independent experiments were assayed. A cell was considered to be invasive if one or more digested cavities were formed in the TRITC-fibronectin-labeled matrix along the migration path of that cell.
Cell migration assay.
Cell migration assays were carried out by monitoring the healing of scratch-induced wounds, as previously described (44). A minimum of 20 cells from each of three independent experiments were tracked using Image J software (NIH) to determine the average speed of individual cells moving within the wound path. A minimum of four separate wound width measurements from each of three independent experiments were taken using Slidebook software to assay the speed of the closing wound front. Distances given in pixels were then converted to micrometers traveled and subsequently to velocities in micrometers per hour.
Matrigel invasion assay.
Matrigel (BD Bioscience) invasion assays were performed as previously described (44) and as suggested by the manufacturer. Ten separate fields were counted from each of three independent experiments for both Matrigel and control inserts using a Zeiss Axiovert S100 microscope outfitted with a Plan-Neofluar 10× (numerical aperture, 0.30) lens objective. The percentage of invasion was calculated by dividing the number of cells that had invaded the Matrigel insert by the number of cells that had invaded the control insert and multiplying by 100. The invasion index of a cell line is a relative representation of the percentage of invasion compared to the percentage of invasion of the control cell line.
Detection of proteins and mRNAs.
Cellular protein and mRNA levels were determined by Western blotting and semiquantitative RT-PCR assays using standard techniques. Additionally, the primers used to amplify MMP1 and MMP10 have been described previously (26). The protein/mRNA levels of GAPDH or TATA binding protein (TBP) were used as controls. The TBP primers for semiquantitative RT-PCR were 5′-GGCCTCTCAGAAGCATCACTA-3′ and 5′-GCCAAGCCCTGAGCATAA-3′. The primers used to PCR amplify rat and mouse MDM2, BAX, PUMA, and GAPDH have been described previously (44).
Promoter activity assay.
Relative promoter activities were determined as described previously (44) using a dual-luciferase assay kit (Promega, Madison, WI). Additional treatments of the cells are indicated wherever applicable. Samples were read using an Lmax microplate luminometer (Molecular Devices, Sunnyvale, CA) in a 96-well plate format, and data were acquired with SoftmaxPro software.
RESULTS
p53 suppresses and Stat3 promotes Src-induced invasive phenotypes.
We have recently shown that Src and p53 play antagonistic roles in the manifestation of the invasive phenotype in both rat aortic smooth muscle cells (SMC) and 3T3 cells, characterized by the formation of podosomes and rosettes, ECM digestion, cell migration, and invasion of Matrigel (44). We were not clear, however, about the connections between Src and p53 functions in the regulation of cell invasion. There is strong evidence suggesting that Stat3 is involved in cell migration and invasion (1, 14, 21), and it has been shown that Stat3 is activated by Src (22, 69). These data suggest that Stat3 is a strong candidate that may play a role in mediating the Src-p53 pathway in the regulation of the invasive phenotypes.
As shown in Fig. 1 a and b, primary rat aortic SMC and 3T3 fibroblasts stably expressing constitutively active Src (SrcY527F) have a propensity for producing podosomes and rosettes, with concomitant decreases in the levels of actin stress fibers and endogenous p53. On the other hand, expression of wild-type (wt) p53 inhibits podosome formation in these cells with the SrcY527F background (Fig. 1c, d, k, and l), as previously shown (44). Interestingly, the SrcY527F cells also express significantly higher levels of active, Tyr-phosphorylated Stat3 (Stat3-pY705) (Fig. 1e), suggesting that Stat3 is upregulated in SrcY527F cells and that this upregulation correlates directly with podosome/rosette formation (see also Fig. S1 and S2 in the supplemental material).
FIG. 1.

p53 is a suppressor and Stat3 is a promoter of SrcY527F-induced formation of podosomes in SMC and 3T3 cells. (a and b) Immunofluorescence microscopic images of SMC and 3T3 cells stably expressing constitutively active SrcY527F. Cells forming large numbers of podosomes and rosettes as a result of SrcY527F expression (arrowheads) also show lower levels of nuclear p53. (c and d) SrcY527F cells transduced with a retroviral vector expressing wt p53. Cells with more-intense nuclear p53 staining (arrowheads) show a reduced capacity to form podosomes. TRITC-phalloidin was used to label actin-rich podosomes in panels a to d. (e) Western blots showing upregulation of active Stat3 (Stat3-pY705) in SMC (left) and 3T3 (right) cells stably expressing either an empty vector (control) or SrcY527F. (f) Western blot analysis of SrcY527F cells to show the knockdown of Stat3 and pYStat3 by shStat3-1 and shStat3-2. GAPDH was used as a loading control. (g to j) SrcY527F cells coexpressing shStat3-GFP were immunostained for GFP and F-actin using TRITC-phalloidin. shRNA-mediated knockdown of Stat3 (cells marked with arrowheads) as identified by GFP expression resulted in a diminished ability of SrcY527F cells to produce podosomes and rosettes. Bars, 20 mm. (k and l) Statistical analyses of the effects of either gain or loss of function for p53 and Stat3 on the ability of SrcY527F cells to form podosomes and rosettes. SMC (k) and 3T3 (l) cells coexpressing SrcY527F and either p53 or Stat3 constructs, as indicated, were used. A minimum of 100 individual cells in three separate experiments for each cell type, as labeled, were counted to determine the percentage of cells bearing high-density podosomes (>50 and >10 podosomes per cell, respectively, for SMC and 3T3 cell derivatives) or at least 1 rosette per cell. Error bars represent standard deviations for three separate experiments. P values were determined using a two-sided, equal-variance Student t test. *, significant difference (P < 0.05) in the count from that for the respective control.
To investigate whether Stat3 is required for the Src-induced invasive phenotype, we knocked down Stat3 expression in SrcY527F cells by expressing two shRNAs, shStat3-1 and shStat3-2, that targeted rat and mouse Stat3. A high degree of Stat3 knockdown by shRNA causes apoptosis, as has been reported previously by others (23, 35). In the generation of stable shRNA-expressing cell lines in this study, only viable cells that had moderate knockdown survived the selection process and were selected for analyses. Although either Stat3 shRNA caused moderate knockdown of Stat3 protein and Stat3-pY705 (50% and 65%, respectively) in SMC, as well as in 3T3 cells (Fig. 1f), stable expression of these shRNAs significantly reduced the ability of SrcY527F cells to form podosomes and/or rosettes (Fig. 1g to j), and the level of Stat3 staining correlated with the degree of podosome and rosette formation (see Fig. S1a to d in the supplemental material). This finding is supported by statistics indicating that shStat3 caused a significant reduction in the percentage of SrcY527F cells that form high-density podosomes and rosettes and that, furthermore, those shStat3-harboring cells that did produce podosomes had considerably fewer podosomes per cell (Fig. 1k and l). In contrast, stable expression of wt Stat3 or constitutively active Stat3 (caStat3) augmented the ability of the SrcY527F cells to produce podosomes and rosettes (Fig. 1k and l).
We also observed that endogenous Stat3 and activated Stat3-pY705 were enriched in the actin columns of Src-induced podosomes and rosettes (see Fig. S2a and b in the supplemental material), which were also labeled with other known podosomal proteins, such as Src (see Fig. S2c and d), paxillin (see Fig. S2e and f), and phospho-Tyr cortactin (CTN-pY421) (see Fig. S2g and h). Although these data strongly suggest that Src induces the translocation of Stat3 to podosomes and rosettes, the Stat3-binding partner(s) in podosomes remains to be identified.
Next, we determined if Stat3 knockdown also affects SrcY527F-induced digestion of ECM and cell invasion in vitro. As shown in Fig. 2c to f and in Fig. S1e to h in the supplemental material, by imaging the digestion of fibronectin-containing substrates using cells expressing various levels of shStat3s, we observed that expression levels of Stat3 correlated positively with the ability of cells to digest the ECM in vitro. This is confirmed by statistical analyses showing that the ECM-degrading capacity of SrcY527F cells was reduced by about 70% as a result of Stat3 knockdown (Fig. 2g). As shown in Fig. 2h, Stat3 knockdown also reduced Src-induced Matrigel invasion in vitro by >50% in both SMC and 3T3 cells.
FIG. 2.

Stat3 knockdown adversely affects ECM degradation, Matrigel invasion, and cell migration induced by SrcY527F in SMC and 3T3 cells. (a to f) SrcY527F cells stably transduced with either a control shRNA-GFP (a and b) or a Stat3-targeted shRNA-GFP (c to f) were used to study the effect of Stat3 knockdown on Src-induced ECM digestion. Cells were cultured on gelatin-coated coverslips layered with TRITC-fibronectin (TRITC-FN) for 7 h. Cells expressing shStat3 (marked with arrowheads) were monitored by GFP expression. Actin was labeled with phalloidin 350. Bars, 20 μm. (g) Quantitative measurements were performed to determine the area of ECM digestion for each cell type. Thirty cells for each category were measured for the area of digestion (in μm2) in the migration path of the cell after 7 h. (h) SMC- or 3T3-derived cell lines expressing the indicated constructs were examined in order to determine their ability to invade Matrigel. The number of cells that migrated through the screen was counted, and the percentage of invasion was determined by dividing the invasion number (Matrigel-coated screen) by the migrating number (uncoated screen). (i and j) A scratch-induced wound-healing assay was performed to determine the average migration rate of individual cells as well as the speed of wound front migration (wound closure) in SMC-SrcY527F (i) and 3T3-SrcY527F (j) cells expressing either an empty vector (control) or a shRNA against Stat3 (shStat3-1 or shStat3-2). Error bars represent standard deviations for three separate experiments. P values were determined using a two-sided, equal-variance Student t test. *, significant difference (P < 0.05) from the value for the respective control.
To determine whether knockdown of Stat3 by shRNA also affects cell migration, we carried out wound-healing assays. As shown in Fig. 2i and j and in Fig. S3 in the supplemental material, there is a significant reduction in the rate of migration of individual cells at the wound fronts, as well as in the rate of wound closure of shStat3-expressing cells. Together, these results strongly suggest that Stat3 function is a required downstream effector of Src in inducing invasive and migratory phenotypes in both vascular smooth muscle cells and 3T3 fibroblasts.
Stat3 promotes Src-induced invasive phenotypes through the suppression of p53-caldesmon.
We have recently shown that the ability of Src to induce full-blown invasive phenotypes hinges on Src-induced suppression of p53 function (44). We have noticed that cells expressing higher levels of Src also had increases in nuclear Stat3 (Fig. 3a and b) and active Stat3-pY705 (Fig. 1e) levels. Furthermore, there was a distinct inverse relationship between the nuclear staining of Stat3 and that of p53 in both SMC and 3T3 cells (Fig. 3c and d). These data suggest to us that Stat3 may mediate the suppression of p53 by Src.
FIG. 3.

Src suppresses the podosome inhibitors p53/caldesmon through the activation of Stat3. (a to d) SMC and 3T3 cells stably transduced with SrcY527F were subjected to immunofluorescence microscopy. Cells were labeled with combinations of either Src and Stat3 (a and b) or Stat3 and p53 (c and d). DAPI was used as a nuclear stain. Arrowheads indicate cells expressing either higher Src/Stat3 levels (a and b) or contrasting levels of Stat3/p53 (c and d). (e to g) Stat3 knockdown results in upregulation of the level/function of p53/caldesmon. Proteins/mRNAs were isolated from SMC-SrcY527F cells stably infected either with an empty vector (control) or with shStat3 expression constructs (shStat3-1 and -2). The levels of the indicated proteins (e) and mRNAs (f) were analyzed by Western blotting and semiquantitative RT-PCR methods, respectively. GAPDH protein/mRNA levels were used as equal loading/amplification controls. (g) The relative promoter activities for the genes in the SMC-SrcY527F cell line were determined by a dual-luciferase assay. Error bars represent the standard deviations for three replicate measurements. Asterisks indicate significant differences (P < 0.05) from the respective controls. (h to k) Stat3 negatively regulates the podosome inhibitors p53 and caldesmon. SMC-SrcY527F cells were transiently transfected with either a shStat3-2 (coexpressing GFP) (h and i) or a wt Stat3 (cotransfected with GFP) (j and k) expression construct. The comparative expression patterns of p53 and caldesmon in transfected (GFP-positive) (indicated by arrowheads) and nontransfected (GFP-negative) cells were visualized by immunofluorescence microscopy. Bar, 20 μm.
To determine whether Stat3 is required for the suppression of p53 expression by SrcY527F, we examined the effects of two independent shStat3s, shStat3-1 and shStat3-2, on p53 expression and function in SMC-SrcY527F cells by biochemical analyses (Fig. 3e to g) and imaging (Fig. 3h to k). As shown in Fig. 3e, cells expressing shStat3-1 or -2 showed increases in the expression of p53 (5.5- and 6-fold, respectively), the widely known p53 target gene product MDM2 (3.7- and 9.5-fold, respectively), and the p53-inducible negative regulator of podosomes, caldesmon (8.4- and 6.7-fold, respectively) (17, 44, 68) (see also Fig. S4 in the supplemental material). Expression of shStat3-1 and shStat3-2 also led to increases in the mRNA levels of bona fide p53 targets: p21 (2.2- and 1.7-fold, respectively), BAX (2.4- and 1.8-fold, respectively), and PUMA (2.3- and 1.5-fold, respectively) (Fig. 3f). In agreement with the RT-PCR data, a dual-luciferase assay also revealed that Stat3 knockdown led to increases in the promoter activities of p53 target genes, namely, p21, MDM2, BAX, and PUMA, indicative of definite enhancement of p53 activity (Fig. 3g). As shown in Fig. 3h to k, immunofluorescence microscopy of SMC showed that cells expressing shStat3 also expressed higher levels of p53 and caldesmon (Fig. 3h and i), while overexpression of wt Stat3 in these cells showed a decrease in p53 and caldesmon staining (Fig. 3j and k). It has been shown that Stat3 binds to the TP53 gene promoter and represses the transcription of p53 mRNA (50); this suggests that Stat3 exerts its effect mainly on the transcription of p53 and consequently on the level of p53 protein and its function in the cell.
To ascertain that the SrcY527F effect is due to a direct increase in Src activity, we treated SMC-SrcY527F cells with the specific Src inhibitor PP2. As shown in Fig. S4 in the supplemental material, PP2 treatment restored the formation of actin stress fibers with reduced podosome structures (Fig. S4a to d), which correlated with increased levels of p53 (Fig. S4c and d) and caldesmon (compare Fig. S4g and h) expression, but a reduction in the level of nuclear Stat3 (compare Fig. S4e and f). These results indicate that inhibition of Src kinase activity in smooth muscle cells by PP2 reversed SrcY527F-induced podosome formation and Stat3 activation, on the one hand, and suppression of p53 and caldesmon, on the other.
Taken together, the data from Fig. 3 and from Fig. S4 in the supplemental material strongly suggest that Stat3 plays a major role in promoting Src-induced invasive phenotypes through the suppression of p53 and thereby the suppression of the p53-inducible podosome antagonist caldesmon.
Constitutively active Stat3 abrogates the ability of p53 to suppress Src-invasive phenotypes.
If Stat3 suppresses p53 expression, can overexpression of Stat3 abrogate p53-imposed suppression of Src-induced invasive phenotypes? To address this question, we expressed exogenously a constitutively active mutant of Stat3 (7), which does not require phosphorylation at Tyr705 to be active, in cells coexpressing SrcY527F and wt p53. As shown in Fig. 4a, about 25% of SrcY527F SMC and 3T3 cells produce high densities of podosomes and/or rosettes, and coexpression of wt p53 caused about a 50% reduction in podosome/rosette formation in both cell types. However, ectopic expression of caStat3 in SrcY527F/wt p53 cells largely abolished the p53-induced suppression of podosome/rosette formation. This is also illustrated by images (Fig. 4g to j) showing that cells coexpressing SrcY527F and wt p53 contain many actin stress fibers but fewer podosomes, whereas cells harboring caStat3-GFP produce prominent rosettes of podosomes. A similar trend of antagonism between wt p53 and caStat3 is also observed in ECM digestion (Fig. 4b), cell migration (Fig. 4c; see also Fig. S3 in the supplemental material), and in vitro invasion (Fig. 4d).
FIG. 4.

caStat3 abrogates the suppressive effect of p53 on Src invasive phenotypes. (a to d) caStat3 rescues Src phenotypes from suppression by exogenously overexpressed p53. Stable cell lines expressing SrcY527F (control), SrcY527F-wt p53, or SrcY527F-wt p53-caStat3 were generated. (a) A minimum of 100 individual cells in three separate experiments for each cell type were counted in order to determine the percentage of cells bearing high-density podosomes (>50 and >10 podosomes per cell, respectively, for SMC and 3T3 cell derivatives) or at least 1 rosette per cell for both cell types. (b) Cells as indicated were cultured on gelatin-coated coverslips layered with TRITC-fibronectin for 7 h. Quantitative measurements were performed to determine the area of ECM digestion for each cell type. Thirty cells for each category were measured for the area of digestion (in μm2) in the migration path of the cell. (c and d) Stable SMC- or 3T3-derived cell lines as indicated were analyzed for their abilities to migrate (c) or invade (d) in vitro by wound-healing and Matrigel invasion assays, respectively. Asterisks in panels a to d indicate significant differences (P < 0.05) between control (SrcY527F) cells and SrcY527F-wt p53 cells or between SrcY527F-wt p53 and SrcY527F-wt p53-caStat3 cells. (e and f) SMC and 3T3 cells stably expressing SrcY527F (control) or SrcY527F-caStat3, as indicated, were treated with either dimethyl sulfoxide (−) or 500 ng/ml doxorubicin (+) in order to activate endogenous p53; then their abilities to form podosomes/rosettes and to digest ECM were assessed. In all cases, error bars represent standard deviations for three separate experiments. Asterisks indicate significant differences (P < 0.05) between control (SrcY527F) and SrcY527F-doxorubicin cells or between SrcY527F-doxorubicin and SrcY527F-doxorudicin-caStat3 cells. (g to j) Stable SrcY527F cells also expressing wt p53 (SrcY527F-wt p53) were transfected with a caStat3 expression construct (coexpressing GFP). These cells were immunostained for either GFP (g and h) or Stat3 (i and j) along with F-actin by using TRITC-phalloidin. Cells expressing caStat3, identifiable by either GFP or strong Stat3 staining (marked with arrowheads), regain the ability to produce large numbers of podosomes/rosettes. Bars, 20 μm.
We also wanted to know whether caStat3 could alleviate endogenous-p53-induced suppression of Src phenotypes. To this end we introduced caStat3 into SrcY527F cells that did not express exogenous wt p53; instead, endogenous p53 in these cells was activated with doxorubicin. As shown in Fig. 4e and f, activation of p53 by doxorubicin caused significant suppression of Src-induced podosome/rosette formation (Fig. 4e) as well as of ECM degradation (Fig. 4f) for both SMC and 3T3 cells. However, in spite of doxorubicin treatment, the ability of SrcY527F to induce podosome/rosette formation and ECM digestion was significantly enhanced when these cells were transfected with a caStat3 expression construct. Thus, these data clearly show that Stat3 reverses the suppression of the Src invasive phenotype by p53.
p53 and Stat3 are mutually antagonistic: activation of p53 downregulates functional Stat3 and overcomes the Src-induced invasive phenotype.
Next, we asked if Stat3 and p53 are mutually antagonistic in the manifestation of the Src invasive phenotype. To this end, we investigated whether forced gain of function of p53 may overcome the proinvasive effects of Src by downregulating the expression of functional Stat3. As shown in Fig. 5 a and b, either activation of endogenous p53 with the genotoxic drug doxorubicin (Fig. 5a) or overexpression of wt p53 in SrcY527F cells (Fig. 5b), as shown by an increase in either p53-inducible PTEN/caldesmon (Fig. 5a) or MDM2 (Fig. 5b) expression, caused a significant decrease in the active species of Stat3 (Stat3-pY705) (54% and 70% for SMC and 3T3, respectively).
FIG. 5.

Enhanced p53 deactivates the Src effector Stat3 with corresponding suppression of the Src phenotype. (a) Progressive activation of endogenous p53 leads to a corresponding decrease in active Stat3 (Stat3-pY705) levels. SMC and 3T3 cells were serum starved 24 h prior to treatment with the p53-activating drug doxorubicin (500 ng/ml) for different durations of time, as indicated. Cell lysates were immunoblotted for Stat3-pY705 and total Stat3. Levels of the p53-inducible proteins PTEN/caldesmon served as indicators of p53 activation. GAPDH levels served as a loading control. (b) Regulation of Stat3 activation by Src and p53. The Stat3-pY705 status as a result of expression of either SrcY527F or SrcY527F-wt p53 was determined by a Western blot assay. The level of MDM2 protein was used to monitor cellular p53 function, while the GAPDH protein level served as a loading control. (c to f) Effects of p53 function on expression levels of nuclear p53/Stat3 and the formation of actin-rich podosomes in SrcY527F-wt p53 cells. p53 activation with doxorubicin (c and d) results in strong nuclear p53 staining (arrowheads), with concomitant reduction of functional, nuclear Stat3 staining. Inhibition of p53 with pifithrin-α (e and f) causes an increase in the level of Stat3 nuclear staining but a decrease in nuclear p53. Bars, 20 μm. (g) Semiquantitative RT-PCR assay to show that wt p53 suppresses the expression of Stat3-regulated MMP1 and MMP10 in SrcY527F cells. Cellular p53 function was reflected by MDM2 mRNA levels. GAPDH mRNA served as an internal control. (h) Knockdown of MMP1 and MMP10 expression by siRNAs. TBP was used an as internal control. (i) Effects of MMP1 and MMP10 knockdown on ECM digestion and Matrigel invasion. Error bars represent standard deviations for three independent experiments. Asterisks indicate significant differences (P < 0.05). (j) Schematic representation of the process by which Src leads to the formation of podosome/invadopodial structures and the role of Stat3-p53 antagonism, downstream of Src, in modulating the Src-invasive phenotype. Src suppresses p53 through Stat3-mediated transcriptional repression. p53, on the other hand, deactivates Stat3, likely through a p53-inducible tyrosine phosphatase such as PTEN.
The mutually antagonistic relationship between p53 and Stat3 functions was further demonstrated by direct imaging. As shown in Fig. 5c and d, doxorubicin-treated cells with strong nuclear p53 staining had weak Stat3 staining. In contrast, inhibition of p53 functions with pifithrin-α (PFA), as expected, resulted in strong nuclear Stat3 staining (Fig. 5e and f). It is worth mentioning here that although PFA abolishes the transcription-dependent function of p53, paradoxically, the level of p53 increases due to the absence of p53-induced negative feedback through MDM2 and p21 (27). Importantly, podosome-bearing capability correlates inversely with the level of nuclear p53 (Fig. 5c and d) but positively with that of Stat3 (Fig. 5c to f).
We next determined whether expression of the Stat3-regulated matrix metalloproteinases MMP1 and MMP10 (22) was also affected by wt p53 overexpression. As shown in Fig. 5g, SrcY527F-treated cells had significant increases in the mRNA levels of both MMP1 and MMP10. However, overexpression of wt p53 in SrcY527F SMC reduced the mRNA levels of MMP1 by about 35% and those of MMP10 to an almost undetectable level. These results were mirrored by SrcY527F 3T3 cells, where exogenous wt p53 suppressed MMP1 and MMP10 mRNA levels by 65% and 41%, respectively. Next, we investigated whether MMP1 and MMP10 contributed to Src-induced ECM degradation. As shown in Fig. 5h and i, siRNA knockdown of MMP1, but not of MMP10, reduced Src-induced ECM digestion as well as in vitro invasion of Matrigel. This finding suggests that p53 may also contribute to the suppression of ECM invasion by downregulating MMP1.
Loss-of-function p53 mutants have been shown to promote cell invasion (45, 63), suggesting that a p53 mutant may fail to suppress the Src-Stat3 proinvasion axis. To determine if a p53 mutant is able to suppress Stat3 activation, we compared the expression of a p53 mutant and pYStat3 in metastatic MDA-MB-231 breast cancer and Du145 prostate cancer cells with those in their noninvasive counterparts, MCF7 and LNCaP cells, which express wild-type p53. As shown in Fig. S5 in the supplemental material, both MDA-MB-231 and Du145 cells tolerate overexpression of the p53 mutant due to its inability to cause apoptosis; however, the p53 mutant fails to suppress the activation of Stat3.
As summarized schematically in Fig. 5j, the data presented in Fig. 5 show that p53 opposes Src function partly through the inactivation of the Src effector Stat3. This is also supported by the data presented in Fig. 4, where we have seen that the caStat3 mutant, which could not be inactivated by dephosphorylation (of Tyr705), almost completely reversed the suppression of Src phenotypes by both exogenously overexpressed (Fig. 4a to d and g to j) and endogenously overactivated (Fig. 4e and f) p53. Thus, p53-Stat3 antagonism downstream of Src likely determines the aggressiveness of Src phenotypes. However, this raises the question of how the p53 transcription factor induces the deactivation (dephosphorylation) of Stat3.
PTEN is a mediator of p53-induced suppression of Src phenotypes: PTEN suppresses Src invasive phenotypes by downregulation of Src/Stat3 function and stabilization of the p53/caldesmon axis.
How does p53 downregulate Stat3? We hypothesized that PTEN, which is a known p53-inducible tumor suppressor (58) and antimotility protein (30, 55), is a possible candidate. We showed above, in Fig. 5a, that activation of endogenous p53 by doxorubicin increases PTEN expression and decreases the level of Stat3-pY705 in both SMC and 3T3 cells, indicating that PTEN is a downstream effector of p53. Additionally, Western blots showed that knockdown of PTEN by shRNA (shPTEN) in smooth muscle cells coexpressing SrcY527F and wt p53 resulted in large increases in the levels of active species of Src (Src-pY416) and Stat3 (Stat3-pY705), whereas the levels of p53 and p53-inducible caldesmon and MDM2 were decreased significantly in the same cells (Fig. 6a). Images of shPTEN-transfected SMC-SrcY527F-wt p53 cells show that cells expressing shPTEN-GFP expressed a higher level of nuclear Stat3 (Fig. 6b) and a lower level of p53 (Fig. 6c) than their nontransfected counterparts. Interestingly, PTEN knockdown also led to abrogation of the suppression of the Src-induced invasive phenotype by p53, as evidenced by the presence of large numbers of podosomes/rosettes in shPTEN-expressing cells (Fig. 6d and e). In contrast, we used SMC-SrcY527F cells to investigate whether the overexpression of wt PTEN alone may reverse the Src-induced effect on p53 and Stat3 expression and the corresponding invasive phenotypes (Fig. 6f to i). Western blots show that overexpression of wt PTEN led to diminished levels of active Src (Src-pY416) and Stat3 (Stat3-pY705) and to elevated levels of p53 and its inducible gene products caldesmon and MDM2 (Fig. 6f). This finding is further illustrated by fluorescence microscopy images, showing that wt PTEN-expressing cells have a greatly reduced nuclear Stat3 level (Fig. 6g), an enhanced level of p53 (Fig. 6h), and consequently reduced podosome/rosette counts (Fig. 6i). Statistical analysis of these cells also shows that overexpression of wt PTEN impairs the ability of SMC-SrcY527F cells to form podosomes (Fig. 7h).
FIG. 6.

PTEN is a mediator of p53-induced suppression of Src phenotypes. (a to e) Effect of PTEN knockdown on the Src/Stat3/p53/caldesmon/podosome expression status in SMC-SrcY527F-wt p53 cells. (a) SMC-SrcY527F-wt p53 cells retrovirally transduced with anti-PTEN shRNAs. One clone each for shPTEN-1 and shPTEN-2 was analyzed by Western blotting. GAPDH was used as a loading control. (b to d) A stable SMC-SrcY527F-wt p53 cell line was transfected with one of the shPTEN expression constructs (coexpressing GFP) and was analyzed by immunofluorescence microscopy as indicated. TRITC-phalloidin was used for actin/podosome staining. Bars, 20 μm. (e) A stable SMC-SrcY527F-wt p53 cell line was transfected with either an empty vector (control) or one of two shPTEN expression constructs (shPTEN-1 and shPTEN-2) and was then assayed for podosome/rosette formation. Error bars represent standard deviations for three independent measurements. Asterisks indicate significant differences (P < 0.05) from the control. (f to i) Effect of PTEN overexpression on the Src/Stat3/p53/caldesmon/podosome expression status in SMC-SrcY527F cells. (f) SMC-SrcY527F cells were transiently transfected with increasing amounts of a wt PTEN expression construct (0, 4, and 8 μg DNA per 10-cm-diameter plate at about 80% confluence) using the Lipofectamine Plus protocol. Forty-eight hours posttransfection, samples were analyzed by Western blotting for proteins/phosphoproteins as shown. GAPDH protein levels indicate equal loading. (g to i) SMC-SrcY527F cells were cotransfected with wt PTEN and a GFP expression construct (pBabe-GFP) prior to analysis by fluorescence imaging. TRITC-phalloidin was used for actin/podosome staining. Bars, 20 μm. (j) PTEN mediates the suppressive effect of p53 on invasive phenotypes by inactivating Src/Stat3, which also serves to stabilize the podosome-antagonizing p53-caldesmon axis.
FIG. 7.

Role of the lipid phosphatase/protein phosphatase activity of PTEN in the regulation of Src/Stat3 function and podosome formation. (a to f) SMC-SrcY527F cells were transiently transfected with various PTEN expression constructs either alone (a and b) or in combination with a GFP expression vector (pBabe-GFP) (c to f). Cells overexpressing either wt PTEN (a) or PTEN-G129E (b) form fewer podosomes than nontransfected cells. Fluorescent microscopic images show the effect of overexpressed PTEN-G129E (c and e) or PTEN-C124S (d and f) on Stat3 and caldesmon (Cald) expression. Bars, 20 μm. (g) SMC-SrcY527F cells were transiently transfected with either an empty vector (control) or various PTEN expression constructs (8 μg DNA per 10-cm-diameter plate) using the Lipofectamine Plus protocol. The cell lysates were prepared 48 h posttransfection and were Western blotted for proteins/phosphoproteins as shown. GAPDH protein levels were used as a loading control. (h) SMC-SrcY527F cells were transfected with pBabe-GFP either alone (control) or along with wild-type/mutant PTEN expression constructs as indicated and were assayed for podosome/rosette formation. Error bars represent standard deviations of three independent measurements. Asterisks indicate significant differences (P < 0.05) from control values.
p53 stabilization has been shown to be an important mechanism through which PTEN executes its tumor-suppressive function (18, 55). The data presented in Fig. 6 (diagramed in Fig. 6j) indicate that PTEN-mediated inactivation of proinvasive Src-pY416/Stat3-pY705 also leads to stabilization of the anti-invasive p53/caldesmon axis. These results strongly implicate PTEN as the mediator of the antagonistic effect of p53 on Src/Stat3 function and Src/Stat3-induced invasive phenotypes.
The protein phosphatase activity of PTEN plays a dominant role in mediating the suppression of Src/Stat3 function and podosome formation.
PTEN is a dual lipid and protein phosphatase. Although the lipid phosphatase activity is well documented to play a major role in tumor suppression (11, 55), recent data have implicated the protein phosphatase activity of PTEN, through a largely unknown substrate or pathway, in the regulation of cell motility (29). To determine the contribution of the protein phosphatase activity of PTEN to the downregulation of Src-induced podosome formation, we generated two mutants, PTEN-G129E and PTEN-C124S; the former lacks lipid phosphatase activity but retains protein phosphatase activity, while the latter is deficient in both lipid and protein phosphatase activities (46, 47). As shown in Fig. 7h, PTEN-G129E suppresses Src-induced formation of podosomes/rosettes at a level comparable to that achieved by wt PTEN; this is further illustrated by fluorescent microscopic images (Fig. 7a and b). However, the phosphatase-inactive mutant PTEN-C124S has no significant effect on Src-induced podosome formation (Fig. 7h). Thus, these results show that the protein phosphatase activity of PTEN plays the dominant role in mediating the suppression of Src-induced podosome formation.
We next investigated whether PTEN acts by inactivating Stat3 and/or Src via dephosphorylation at Tyr705 and Tyr416, respectively. As shown by the Western blot analyses in Fig. 7g, wt PTEN reduces the Stat3-pY705 level, which is, interestingly, further reduced by PTEN-G129E. However, the phosphatase-inactive PTEN-C124S mutant also decreases the Stat3-pY705 level as effectively as wt PTEN, suggesting that although the protein phosphatase activity has a major negative effect on the phosphorylation of Stat3, the lipid phosphatase activity may play a positive role, perhaps in fine tuning the activity of Stat3. In a parallel manner, wt PTEN and PTEN-G129E have comparable negative effects on Src phosphorylation at Tyr416 (Fig. 7g), and PTEN-C124S restores Src-pY416 levels partially. Thus, these results strongly indicate that the protein phosphatase component of PTEN function is required and sufficient to inactivate the proinvasive Src/Stat3 function and the related invasive phenotype.
DISCUSSION
Oncogenic signaling has been shown to be a major stimulus of p53 activation, which protects the cells against a proliferative and invasive phenotype (16, 41). However, when overwhelmed with a consistent oncogenic insult, such as stable expression of SrcY527F, as used in our study model, the affected cells fail to upregulate p53 and succumb to an invasive phenotype (44). In this study, we have provided novel data to show that perturbation of the balance between the proinvasive Src pathway and the anti-invasive p53-caldesmon axis dictates the outcome of the expressed phenotype. We have identified Stat3 as a downstream effector of Src and the protein phosphatase activity of PTEN as a p53 collaborator. A delicate balance of the Src-Stat3 and p53-PTEN pathways is maintained by mutual antagonistic regulation and cross-checking between Stat3 and p53. Furthermore, these data also suggest a commonality in the mechanisms that regulate cell invasion in cancer and vascular smooth muscle cells in atherosclerosis.
We have shown in this study that Stat3 acts downstream of Src and promotes the formation of podosomes and related invasive phenotypes (Fig. 1 and 2). Interestingly, Stat3 and Stat3-pY705 localize in Src-induced podosomes (see Fig. S2 in the supplemental material). One possible advantage is that translocation of Stat3 to Src-enriched podosomes allows phosphorylation and activation of Stat3, which then relocates to the nucleus and promotes Src-associated invasive phenotypes through its transcriptional functions, such as suppression of p53/caldesmon (Fig. 3). This is in line with a previous report that Stat3 can be phosphorylated and activated by cytoplasmic Src kinase (22, 69). Stat3 may also be involved in promoting ECM degradation by regulating its known MMP targets, MMP1 and MMP10 (21). Here we have shown that p53 suppresses the expression of Stat3-regulated MMP1 and MMP10. However, only MMP1 may be involved in Src-induced ECM degradation and in vitro invasion of Matrigel (Fig. 5) suggesting that Src-Stat3 may induce ECM invasion via activation of MMP1. We do not, however, rule out a role for transcription-independent functions of Stat3 in modulating the kinetics of podosome formation, in a manner similar to its role in microtubule organization and cell migration (48), or the involvement of other Stats, such as phospho-Stat5, which has been shown to be associated with podosomes in Hck-transformed cells (53).
Although Src and Jak kinases are the important modulators of Stat3 function, other members of the Src family of kinases have also been shown to activate Stat3 (39, 56). Overexpression of a constitutively active mutant of Hck (53) led to the formation of podosomes in fibroblasts; however, it is not clear whether Hck acts on the Stat3 pathway. Since endogenous Src or even overexpression of wt Src (c-Src) in a normal cell system, such as fibroblasts or smooth muscle cells, fails to induce podosomes, the observed invasive phenotypes were induced primarily by ectopically expressed constitutively active mutant Src (SrcY527F). Thus, the contribution of endogenous levels of c-Src or other Src family members, in the present context, is likely to be negligible. Therefore, the PP2-mediated reversal of invasive phenotypes is attributable to the ability of PP2 to block the function of SrcY527F rather than that of endogenous Src or other Src family members. However, a definitive answer must await extensive detailed studies involving different non-Src tyrosine protein kinase (TPK) members.
The evidence for a mutually antagonistic regulation of Stat3 and p53 in Src-induced cell invasion was provided by data in Fig. 3 to 5 and Fig. S4 in the supplemental material. These data show that the ability of Src to induce podosome formation and ECM invasion depends on both the upregulation of Stat3 and the suppression of the p53-caldesmon pathway (Fig. 3 and 4). In turn, the upregulation of p53 is able to countervail the ability of Src to induce invasive phenotypes by downregulation of Stat3 (Fig. 4 and 5). The severity of Src phenotypes is likely determined by a balance between these two opposing forces, p53 and Stat3. Our findings agree with previous reports that Stat3 transcriptionally represses p53 expression (50) and that p53 can downregulate Stat3 in breast and prostate cancer cells (36, 37).
We have further identified the tumor suppressor PTEN as a mediator in p53 suppression of the Src-Stat3 axis in podosome formation and cell invasion. Progressive activation of p53 by doxorubicin increases PTEN expression, with a concomitant decrease in the level of Stat3-pY705 (Fig. 5). This is in agreement with earlier reports that PTEN is transactivatable by p53 (58) and is a negative regulator of Stat3 (61, 71). Additionally, knockdown of PTEN with shRNA (Fig. 6a) and overexpression of wt PTEN (Fig. 6f) effected, respectively, a large increase and a decrease in the Stat3-pY705 level. These data indicate that PTEN, while acting downstream of p53 as a negative regulator of Stat3 and Src, also acts as a positive regulator of p53 and the p53-inducible podosome antagonist caldesmon. Stabilization of the podosome-inhibiting p53-caldesmon axis by PTEN, as shown in Fig. 6 and 7, reveals a new component of the anti-invasive function of PTEN, i.e., to restrain the ability of Src to induce podosome formation. Stabilization of p53 expression and function by PTEN, either via the suppression of the Akt-MDM2 pathway or through direct interaction between PTEN and p53, has been reported previously (18, 34). Here we propose a novel mechanism by which p53 is stabilized by PTEN indirectly, by virtue of the ability of PTEN to downregulate Src and Stat3 (Fig. 6j). Thus, PTEN, acting as a Src-Stat3 negative regulator, also stabilizes the p53-caldesmon axis, reinforcing the anti-invasive function.
PTEN is a dual lipid PtdInsP3 and protein phosphatase, although the PtdInsP3-dependent activity of PTEN has been shown to play a dominant role as an inhibitor of the PI3K/Akt pathway (11). Recent studies, however, have invoked a strong argument for a significant role of the protein phosphatase activity in the regulation of cell migration (29). This is consistent with our finding that the PTEN-G129E mutant, which lacks lipid phosphatase activity but retains its protein phosphatase activity, was as efficient as wt PTEN in downregulating Src-pY416 and Stat3-pY705 (Fig. 7g), as well as podosome formation (Fig. 7h), suggesting that the protein phosphatase activity of PTEN plays a major role in the suppression of the Src-Stat3 axis in cell invasion. Whether Stat3 is a substrate of PTEN is not clear. In vivo PTEN protein substrates have not been positively identified, except for the autodephosphorylation site at the C2 inhibitory domain (31, 54), and a recent report shows that in Caenorhabditis elegans, the Eph kinase is a substrate of PTEN (5). We have not been able to coimmunoprecipitate Stat3 and PTEN (data not shown), suggesting that the PTEN-Stat3 interaction is either too weak or transient. Alternatively, Stat3 inactivation by PTEN is an indirect event requiring the dephosphorylation of yet unknown protein substrates, leading to inactivation of Src, which in turn fails to phosphorylate and activate Stat3. This possibility is consistent with our data showing that Src-pY416 levels closely parallel those of Stat3-pY705 in cells expressing different levels of PTEN (Fig. 6a and f and 7g) and is in line with reports that Stat3 is a substrate of Src (22, 69) and that PTEN inactivates another member of the Src family of kinases, Fyn (15).
It has been shown recently that p53 mutants promote cell invasion (45, 63). These data are consistent with our results; together, they point to a general description of p53 as a suppressor of tumor cell invasion and metastasis. Interestingly, p53 acts via multiple pathways in the regulation of cell invasion, including the stabilization of Slug, the invasion promoter (63), integrin and epidermal growth factor receptor (EGFR) trafficking (45), and suppression of Src/Stat3 activity as shown here. Furthermore, we have shown in Fig. S5 in the supplemental material that the p53 mutant in MDA-MB-231 breast cancer and Du145 prostate cancer cells fails to suppress Stat3 activation, which contributes to the invasive potential of these cancer cells. It has been shown that MDA-MB-231 cells harboring mutant p53 have a limited ability to form podosomes/invadopodia, which are strongly induced only after the introduction of SrcY527F (2). This shows that mutant p53 alone is a weak promoter of podosome formation in the absence of oncogenic insult by Src.
In conclusion, we propose that two opposing teams regulate the outcome of Src-induced podosome formation and the Src-induced invasive phenotype, as depicted in Fig. 8. On one side, the two oncogenes Src and Stat3 cooperate to induce the formation of podosomes and the manifestation of the invasive phenotype. On the other side, p53, in partnership with the PTEN tumor suppressor, acts against the oncogenic impact of Src/Stat3. A positive feedback loop between PTEN and p53/caldesmon serves to strengthen the anti-invasive pathway. Mutually antagonistic cross talk between the pro- and anti-invasive pathways involving Src/Stat3 and p53/PTEN, respectively, serves as a check and balance that dictates the outcome of either an invasive or a noninvasive phenotype. Lastly, similar regulatory mechanisms appear to exist in invasion of immortalized fibroblasts and invasion of vascular smooth muscle cells. Strategies to combat cell migration and invasion-related pathologies such as cancer cell metastasis and vascular smooth muscle cell invasion in atherosclerosis should include both blockage of the proinvasive oncogenes Src-Stat3 and empowerment of the anti-invasive guardians p53 and PTEN.
FIG. 8.
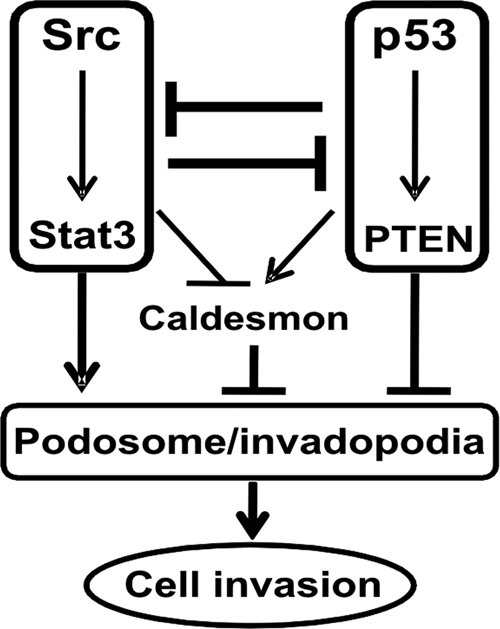
Schematic representation of the mutual antagonism of the Src-Stat3 and p53-PTEN axes in the regulation of cellular invasion. Two opposing forces, constituted by the proinvasive oncogenes Src-Stat3 and the anti-invasive tumor suppressors p53-PTEN, antagonize each other, affecting cellular invasive phenotypes. The dominant force likely determines the level of caldesmon expression and thereby the formation of invasive podosomes/invadopodia.
Supplementary Material
Acknowledgments
This work was supported by the Canadian Institute of Health Research and Ontario Heart and Stroke Foundation (CIHR MOP78468 and HSFO T5829, to A.S.M.).
We acknowledge the help of Chris Moyes, Christine Genge, and Melanie Fortner with fluorometry.
Footnotes
Published ahead of print on 23 August 2010.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Abdulghani, J., L. Gu, A. Dagvadorj, J. Lutz, B. Leiby, G. Bonuccelli, M. P. Lisanti, T. Zellweger, K. Alanen, T. Mirtti, T. Visakorpi, L. Bubendorf, and M. T. Nevalainen. 2008. Stat3 promotes metastatic progression of prostate cancer. Am. J. Pathol. 172:1717-1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Artym, V. V., Y. Zhang, F. Seillier-Moiseiwitsch, K. M. Yamada, and S. C. Mueller. 2006. Dynamic interactions of cortactin and membrane type 1 matrix metalloproteinase at invadopodia: defining the stages of invadopodia formation and function. Cancer Res. 66:3034-3043. [DOI] [PubMed] [Google Scholar]
- 3.Aylon, Y., and M. Oren. 2007. Living with p53, dying of p53. Cell 130:597-600. [DOI] [PubMed] [Google Scholar]
- 4.Bowman, T., M. A. Broome, D. Sinibaldi, W. Wharton, W. J. Pledger, J. M. Sedivy, R. Irby, T. Yeatman, S. A. Courtneidge, and R. Jove. 2001. Stat3-mediated Myc expression is required for Src transformation and PDGF-induced mitogenesis. Proc. Natl. Acad. Sci. U. S. A. 98:7319-7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brisbin, S., J. Liu, J. Boudreau, J. Peng, M. Evangelista, and I. Chin-Sang. 2009. A role for C. elegans Eph RTK signaling in PTEN regulation. Dev. Cell 17:459-469. [DOI] [PubMed] [Google Scholar]
- 6.Bromberg, J. F., C. M. Horvath, D. Besser, W. W. Lathem, and J. E. Darnell, Jr. 1998. Stat3 activation is required for cellular transformation by v-src. Mol. Cell. Biol. 18:2553-2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bromberg, J. F., M. H. Wrzeszczynska, G. Devgan, Y. Zhao, R. G. Pestell, C. Albanese, and J. E. Darnell, Jr. 1999. Stat3 as an oncogene. Cell 98:295-303. [DOI] [PubMed] [Google Scholar]
- 8.Buccione, R., G. Caldieri, and I. Ayala. 2009. Invadopodia: specialized tumor cell structures for the focal degradation of the extracellular matrix. Cancer Metastasis Rev. 28:137-149. [DOI] [PubMed] [Google Scholar]
- 9.Buccione, R., J. D. Orth, and M. A. McNiven. 2004. Foot and mouth: podosomes, invadopodia and circular dorsal ruffles. Nat. Rev. Mol. Cell Biol. 5:647-657. [DOI] [PubMed] [Google Scholar]
- 10.Campisi, J. 2005. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 120:513-522. [DOI] [PubMed] [Google Scholar]
- 11.Chalhoub, N., and S. J. Baker. 2009. PTEN and the PI3-kinase pathway in cancer. Annu. Rev. Pathol. 4:127-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de la Iglesia, N., G. Konopka, S. V. Puram, J. A. Chan, R. M. Bachoo, M. J. You, D. E. Levy, R. A. DePinho, and A. Bonni. 2008. Identification of a PTEN-regulated STAT3 brain tumor suppressor pathway. Genes Dev. 22:449-462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Stanchina, E., E. Querido, M. Narita, R. V. Davuluri, P. P. Pandolfi, G. Ferbeyre, and S. W. Lowe. 2004. PML is a direct p53 target that modulates p53 effector functions. Mol. Cell 13:523-535. [DOI] [PubMed] [Google Scholar]
- 14.Devarajan, E., and S. Huang. 2009. STAT3 as a central regulator of tumor metastases. Curr. Mol. Med. 9:626-633. [DOI] [PubMed] [Google Scholar]
- 15.Dey, N., H. E. Crosswell, P. De, R. Parsons, Q. Peng, J. D. Su, and D. L. Durden. 2008. The protein phosphatase activity of PTEN regulates SRC family kinases and controls glioma migration. Cancer Res. 68:1862-1871. [DOI] [PubMed] [Google Scholar]
- 16.Efeyan, A., and M. Serrano. 2007. p53: guardian of the genome and policeman of the oncogenes. Cell Cycle 6:1006-1010. [DOI] [PubMed] [Google Scholar]
- 17.Eves, R., B. A. Webb, S. T. Zhou, and A. S. Mak. 2006. Caldesmon is an integral component of podosomes in smooth muscle cells. J. Cell Sci. 119:1691-1702. [DOI] [PubMed] [Google Scholar]
- 18.Freeman, D. J., A. G. Li, G. Wei, H. H. Li, N. Kertesz, R. Lesche, A. D. Whale, H. Martinez-Diaz, N. Rozengurt, R. D. Cardiff, X. Liu, and H. Wu. 2003. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell 3:117-130. [DOI] [PubMed] [Google Scholar]
- 19.Gadea, G., M. de Toledo, C. Anguille, and P. Roux. 2007. Loss of p53 promotes RhoA-ROCK-dependent cell migration and invasion in 3D matrices. J. Cell Biol. 178:23-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gadéa, G., L. Lapasset, C. Gauthier-Rouviere, and P. Roux. 2002. Regulation of Cdc42-mediated morphological effects: a novel function for p53. EMBO J. 21:2373-2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao, S. P., and J. F. Bromberg. 2006. Touched and moved by STAT3. Sci. STKE 2006:pe30. [DOI] [PubMed] [Google Scholar]
- 22.Garcia, R., T. L. Bowman, G. Niu, H. Yu, S. Minton, C. A. Muro-Cacho, C. E. Cox, R. Falcone, R. Fairclough, S. Parsons, A. Laudano, A. Gazit, A. Levitzki, A. Kraker, and R. Jove. 2001. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene 20:2499-2513. [DOI] [PubMed] [Google Scholar]
- 23.Geletu, M., C. Chaize, R. Arulanandam, A. Vultur, C. Kowolik, A. Anagnostopoulou, R. Jove, and L. Raptis. 2009. Stat3 activity is required for gap junctional permeability in normal rat liver epithelial cells. DNA Cell Biol. 28:319-327. [DOI] [PubMed] [Google Scholar]
- 24.Gimona, M., R. Buccione, S. A. Courtneidge, and S. Linder. 2008. Assembly and biological role of podosomes and invadopodia. Curr. Opin. Cell Biol. 20:235-241. [DOI] [PubMed] [Google Scholar]
- 25.Hollstein, M., D. Sidransky, B. Vogelstein, and C. C. Harris. 1991. p53 mutations in human cancers. Science 253:49-53. [DOI] [PubMed] [Google Scholar]
- 26.Itoh, M., T. Murata, T. Suzuki, M. Shindoh, K. Nakajima, K. Imai, and K. Yoshida. 2006. Requirement of STAT3 activation for maximal collagenase-1 (MMP-1) induction by epidermal growth factor and malignant characteristics in T24 bladder cancer cells. Oncogene 25:1195-1204. [DOI] [PubMed] [Google Scholar]
- 27.Kruse, J. P., and W. Gu. 2009. Modes of p53 regulation. Cell 137:609-622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ku, T. K., D. C. Nguyen, M. Karaman, P. Gill, J. G. Hacia, and D. L. Crowe. 2007. Loss of p53 expression correlates with metastatic phenotype and transcriptional profile in a new mouse model of head and neck cancer. Mol. Cancer Res. 5:351-362. [DOI] [PubMed] [Google Scholar]
- 29.Leslie, N. R., H. Maccario, L. Spinelli, and L. Davidson. 2009. The significance of PTEN′s protein phosphatase activity. Adv. Enzyme Regul. 49:190-196. [DOI] [PubMed] [Google Scholar]
- 30.Leslie, N. R., X. Yang, C. P. Downes, and C. J. Weijer. 2005. The regulation of cell migration by PTEN. Biochem. Soc. Trans. 33:1507-1508. [DOI] [PubMed] [Google Scholar]
- 31.Leslie, N. R., X. Yang, C. P. Downes, and C. J. Weijer. 2007. PtdIns(3,4,5)P(3)-dependent and -independent roles for PTEN in the control of cell migration. Curr. Biol. 17:115-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levine, A. J. 1997. p53, the cellular gatekeeper for growth and division. Cell 88:323-331. [DOI] [PubMed] [Google Scholar]
- 33.Lewis, B. C., D. S. Klimstra, N. D. Socci, S. Xu, J. A. Koutcher, and H. E. Varmus. 2005. The absence of p53 promotes metastasis in a novel somatic mouse model for hepatocellular carcinoma. Mol. Cell. Biol. 25:1228-1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li, A. G., L. G. Piluso, X. Cai, G. Wei, W. R. Sellers, and X. Liu. 2006. Mechanistic insights into maintenance of high p53 acetylation by PTEN. Mol. Cell 23:575-587. [DOI] [PubMed] [Google Scholar]
- 35.Li, G. H., H. Wei, Z. T. Chen, S. Q. Lv, C. L. Yin, and D. L. Wang. 2009. STAT3 silencing with lentivirus inhibits growth and induces apoptosis and differentiation of U251 cells. J. Neurooncol. 91:165-174. [DOI] [PubMed] [Google Scholar]
- 36.Lin, J., X. Jin, K. Rothman, H. J. Lin, H. Tang, and W. Burke. 2002. Modulation of signal transducer and activator of transcription 3 activities by p53 tumor suppressor in breast cancer cells. Cancer Res. 62:376-380. [PubMed] [Google Scholar]
- 37.Lin, J., H. Tang, X. Jin, G. Jia, and J. T. Hsieh. 2002. p53 regulates Stat3 phosphorylation and DNA binding activity in human prostate cancer cells expressing constitutively active Stat3. Oncogene 21:3082-3088. [DOI] [PubMed] [Google Scholar]
- 38.Linder, S. 2007. The matrix corroded: podosomes and invadopodia in extracellular matrix degradation. Trends Cell Biol. 17:107-117. [DOI] [PubMed] [Google Scholar]
- 39.Lund, T. C., C. Coleman, E. Horvath, B. M. Sefton, R. Jove, M. M. Medveczky, and P. G. Medveczky. 1999. The Src-family kinase Lck can induce STAT3 phosphorylation and DNA binding activity. Cell. Signal. 11:789-796. [DOI] [PubMed] [Google Scholar]
- 40.Marx, J. 2006. Cell biology—podosomes and invadopodia help mobile cells step lively. Science 312:1868-1869. [DOI] [PubMed] [Google Scholar]
- 41.Meek, D. W. 2009. Tumour suppression by p53: a role for the DNA damage response? Nat. Rev. Cancer 9:714-723. [DOI] [PubMed] [Google Scholar]
- 42.Morton, J. P., D. S. Klimstra, M. E. Mongeau, and B. C. Lewis. 2008. Trp53 deletion stimulates the formation of metastatic pancreatic tumors. Am. J. Pathol. 172:1081-1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moskovits, N., A. Kalinkovich, J. Bar, T. Lapidot, and M. Oren. 2006. p53 attenuates cancer cell migration and invasion through repression of SDF-1/CXCL12 expression in stromal fibroblasts. Cancer Res. 66:10671-10676. [DOI] [PubMed] [Google Scholar]
- 44.Mukhopadhyay, U. K., R. Eves, L. Jia, P. Mooney, and A. S. Mak. 2009. p53 suppresses Src-induced podosome and rosette formation and cellular invasiveness through the upregulation of caldesmon. Mol. Cell. Biol. 29:3088-3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Muller, P. A., P. T. Caswell, B. Doyle, M. P. Iwanicki, E. H. Tan, S. Karim, N. Lukashchuk, D. A. Gillespie, R. L. Ludwig, P. Gosselin, A. Cromer, J. S. Brugge, O. J. Sansom, J. C. Norman, and K. H. Vousden. 2009. Mutant p53 drives invasion by promoting integrin recycling. Cell 139:1327-1341. [DOI] [PubMed] [Google Scholar]
- 46.Myers, M. P., I. Pass, I. H. Batty, J. Van der Kaay, J. P. Stolarov, B. A. Hemmings, M. H. Wigler, C. P. Downes, and N. K. Tonks. 1998. The lipid phosphatase activity of PTEN is critical for its tumor suppressor function. Proc. Natl. Acad. Sci. U. S. A. 95:13513-13518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Myers, M. P., J. P. Stolarov, C. Eng., J. Li, S. I. Wang, M. H. Wigler, R. Parsons, and N. K. Tonks. 1997. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc. Natl. Acad. Sci. U. S. A. 94:9052-9057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ng, D. C., B. H. Lin, C. P. Lim, G. Huang, T. Zhang, V. Poli, and X. Cao. 2006. Stat3 regulates microtubules by antagonizing the depolymerization activity of stathmin. J. Cell Biol. 172:245-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Niu, G., T. Bowman, M. Huang, S. Shivers, D. Reintgen, A. Daud, A. Chang, A. Kraker, R. Jove, and H. Yu. 2002. Roles of activated Src and Stat3 signaling in melanoma tumor cell growth. Oncogene 21:7001-7010. [DOI] [PubMed] [Google Scholar]
- 50.Niu, G., K. L. Wright, Y. Ma, G. M. Wright, M. Huang, R. Irby, J. Briggs, J. Karras, W. D. Cress, D. Pardoll, R. Jove, J. Chen, and H. Yu. 2005. Role of Stat3 in regulating p53 expression and function. Mol. Cell. Biol. 25:7432-7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oikawa, T., T. Itoh, and T. Takenawa. 2008. Sequential signals toward podosome formation in NIH-src cells. J. Cell Biol. 182:157-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paddison, P. J., M. Cleary, J. M. Silva, K. Chang, N. Sheth, R. Sachidanandam, and G. J. Hannon. 2004. Cloning of short hairpin RNAs for gene knockdown in mammalian cells. Nat. Methods 1:163-167. [DOI] [PubMed] [Google Scholar]
- 53.Poincloux, R., C. Cougoule, T. Daubon, I. Maridonneau-Parini, and V. Le Cabec. 2007. Tyrosine-phosphorylated STAT5 accumulates on podosomes in Hck-transformed fibroblasts and chronic myeloid leukemia cells. J. Cell. Physiol. 213:212-220. [DOI] [PubMed] [Google Scholar]
- 54.Raftopoulou, M., S. Etienne-Manneville, A. Self, S. Nicholls, and A. Hall. 2004. Regulation of cell migration by the C2 domain of the tumor suppressor PTEN. Science 303:1179-1181. [DOI] [PubMed] [Google Scholar]
- 55.Salmena, L., A. Carracedo, and P. P. Pandolfi. 2008. Tenets of PTEN tumor suppression. Cell 133:403-414. [DOI] [PubMed] [Google Scholar]
- 56.Schreiner, S. J., A. P. Schiavone, and T. E. Smithgall. 2002. Activation of STAT3 by the Src family kinase Hck requires a functional SH3 domain. J. Biol. Chem. 277:45680-45687. [DOI] [PubMed] [Google Scholar]
- 57.Sharpless, N. E., and R. A. DePinho. 2002. p53: good cop/bad cop. Cell 110:9-12. [DOI] [PubMed] [Google Scholar]
- 58.Stambolic, V., D. MacPherson, D. Sas, Y. Lin, B. Snow, Y. Jang, S. Benchimol, and T. W. Mak. 2001. Regulation of PTEN transcription by p53. Mol. Cell 8:317-325. [DOI] [PubMed] [Google Scholar]
- 59.Stiles, B. L. 2009. Phosphatase and tensin homologue deleted on chromosome 10: extending its PTENtacles. Int. J. Biochem. Cell Biol. 41:757-761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stylli, S. S., A. H. Kaye, and P. Lock. 2008. Invadopodia: at the cutting edge of tumour invasion. J. Clin. Neurosci. 15:725-737. [DOI] [PubMed] [Google Scholar]
- 61.Sun, S., and B. M. Steinberg. 2002. PTEN is a negative regulator of STAT3 activation in human papillomavirus-infected cells. J. Gen. Virol. 83:1651-1658. [DOI] [PubMed] [Google Scholar]
- 62.Vogelstein, B., D. Lane, and A. J. Levine. 2000. Surfing the p53 network. Nature 408:307-310. [DOI] [PubMed] [Google Scholar]
- 63.Wang, S. P., W. L. Wang, Y. L. Chang, C. T. Wu, Y. C. Chao, S. H. Kao, A. Yuan, C. W. Lin, S. C. Yang, W. K. Chan, K. C. Li, T. M. Hong, and P. C. Yang. 2009. p53 controls cancer cell invasion by inducing the MDM2-mediated degradation of Slug. Nat. Cell Biol. 11:694-704. [DOI] [PubMed] [Google Scholar]
- 64.Weaver, A. M. 2006. Invadopodia: specialized cell structures for cancer invasion. Clin. Exp. Metastasis 23:97-105. [DOI] [PubMed] [Google Scholar]
- 65.Webb, B. A., L. Jia, R. Eves, and A. S. Mak. 2007. Dissecting the functional domain requirements of cortactin in invadopodia formation. Eur. J. Cell Biol. 86:189-206. [DOI] [PubMed] [Google Scholar]
- 66.Xia, M., and H. Land. 2007. Tumor suppressor p53 restricts Ras stimulation of RhoA and cancer cell motility. Nat. Struct. Mol. Biol. 14:215-223. [DOI] [PubMed] [Google Scholar]
- 67.Yeatman, T. J. 2004. A renaissance for SRC. Nat. Rev. Cancer 4:470-480. [DOI] [PubMed] [Google Scholar]
- 68.Yoshio, T., T. Morita, Y. Kimura, M. Tsujii, N. Hayashi, and K. Sobue. 2007. Caldesmon suppresses cancer cell invasion by regulating podosome/invadopodium formation. FEBS Lett. 581:3777-3782. [DOI] [PubMed] [Google Scholar]
- 69.Yu, C. L., D. J. Meyer, G. S. Campbell, A. C. Larner, C. Carter-Su, J. Schwartz, and R. Jove. 1995. Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science 269:81-83. [DOI] [PubMed] [Google Scholar]
- 70.Zbuk, K. M., and C. Eng. 2007. Cancer phenomics: RET and PTEN as illustrative models. Nat. Rev. Cancer 7:35-45. [DOI] [PubMed] [Google Scholar]
- 71.Zhou, J., J. Wulfkuhle, H. Zhang, P. Gu, Y. Yang, J. Deng, J. B. Margolick, L. A. Liotta, E. Petricoin III, and Y. Zhang. 2007. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc. Natl. Acad. Sci. U. S. A. 104:16158-16163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou, S. T., B. A. Webb, R. Eves, and A. S. Mak. 2006. Effects of tyrosine phosphorylation of cortactin on podosome formation in A7r5 vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 290:C463-C471. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.