

Abstract
APOBEC3F (A3F) is a member of the family of cytidine deaminases that is often coexpressed with APOBEC3G (A3G) in cells susceptible to HIV infection. A3F has been shown to have strong antiviral activity in transient-expression studies, and together with A3G, it is considered the most potent cytidine deaminase targeting HIV. Previous analyses suggested that the antiviral properties of A3F can be dissociated from its catalytic deaminase activity. We were able to confirm the deaminase-independent antiviral activity of exogenously expressed A3F; however, we also noted that exogenous expression was associated with very high A3F mRNA and protein levels. In analogy to our previous study of A3G, we produced stable HeLa cell lines constitutively expressing wild-type or deaminase-defective A3F at levels that were more in line with the levels of endogenous A3F in H9 cells. A3F expressed in stable HeLa cells was packaged into Vif-deficient viral particles with an efficiency similar to that of A3G and was properly targeted to the viral nucleoprotein complex. Surprisingly, however, neither wild-type nor deaminase-defective A3F inhibited HIV-1 infectivity. These results imply that the antiviral activity of endogenous A3F is negligible compared to that of A3G.
The human immunodeficiency virus type 1 (HIV-1) accessory protein Vif plays an important role in regulating virus infectivity. It is now well established that Vif counteracts the antiviral activity of several human cytidine deaminases, including APOBEC3G (A3G), APOBEC3F (A3F), and APOBEC3DE (reviewed in reference 10). However, in spite of Vif, hypermutated HIV-1 sequences have been identified in more than 40% of HIV-infected individuals (17). The accumulation of G-to-A hypermutations was first attributed to an error-prone HIV reverse transcriptase (31, 47). However, the identification of APOBEC3G as a major HIV host restriction factor targeting single-stranded viral DNA leading to G-to-A hypermutation on the plus-strand DNA has put the spotlight on cellular cytidine deaminases as factors possibly contributing to HIV hypermutation. It is interesting that, in patient samples, G-to-A hypermutations were observed in a preferred GG and GA context and that their appearance was independent of the normal accumulation of random mutations (17). It is also interesting that hypermutation in the GA dinucleotide context exceeded that in the GG context not only in HIV-1 sequences from infected human patients (17) but also in those from macaques experimentally infected with a simian HIV variant expressing a mutated Vif protein (43). Subsequent reports identified matching dinucleotide preferences for A3G (GG) and A3F (GA) (4, 13, 18, 52, 56), arguing for a role for these cytidine deaminases in HIV hypermutation. Such a role for host deaminases is further supported by the fact that A3G and A3F are expressed in a wide variety of cell types, including cells susceptible to HIV infection (4, 23, 28, 52).
Transient-expression studies have demonstrated that A3F potently inhibits HIV-1 replication in a Vif-sensitive manner (4, 8, 28, 52, 55, 58); overall, however, A3F appears to be less sensitive to HIV-1 Vif than A3G is (8, 28, 48, 55). Interestingly, whereas dose-response studies indicated that wild-type (WT) A3G had a stronger inhibitory effect on viral infectivity than its deaminase-defective variant, WT A3F and deaminase-defective A3F inhibited viral infectivity equally well (14). This observation implies that A3F-mediated inhibition of viral infectivity occurs through a predominantly deamination-independent mechanism. Indeed, the existence of a deamination-independent mechanism to inhibit viral infectivity has been widely reported for A3G and A3F (3, 7, 11, 12, 14, 16, 27, 29, 32-34, 36, 38, 53). Deaminase-independent inhibition by A3G was also reported for other viruses, such as HTLV-1 and hepatitis B virus (26, 37, 40, 42). However, most of these studies were done under conditions of experimentally elevated levels of A3G or A3F. Indeed, we and others have found that A3G-dependent inhibition of HIV-1 and inhibition of the yeast retrotransposon Ty1 and the murine endogenous retrovirus MusD require catalytic deaminase activity when A3G expression approaches endogenous levels (6, 34, 44).
Our current study further investigated the functional importance of A3F catalytic activity for the inhibition of HIV-1 replication. We employed a strategy similar to the one used for the analysis of A3G (34). First, we performed a titration of exogenously expressed WT and deaminase-defective A3F. Consistent with published reports, we found that in such a setting, A3F had strong antiviral activity but that deaminase activity was not important for the inhibition of HIV-1 infectivity. We next established stable HeLa cell lines expressing WT or deaminase-defective A3F. We found that virus produced from these cells incorporated A3F with an efficiency similar to that with which it incorporated A3G. However, inhibition of A3F packaging by Vif was less efficient than inhibition of A3G packaging, consistent with the reported relative insensitivity of A3F to Vif. Surprisingly, neither WT nor deaminase-deficient A3F produced in the stable HeLa lines inhibited HIV-1 infectivity, and hypermutation of viral genomes was not detected. In conclusion, our data suggest that (i) A3F is not a major contributor to deaminase-dependent inhibition of viral infectivity and (ii) the deaminase-independent inhibition of viral infectivity is largely restricted to assay systems involving transient overexpression of A3F.
MATERIALS AND METHODS
Plasmids.
The full-length molecular clone of HIV-1 (pNL4-3) was used for the production of WT infectious virus (1). Construction of its vif-defective variant pNL4-3Vif(−) was described previously (21). A vector for the expression of myc-tagged A3G wt in the backbone of pcDNA3.1(−) (Invitrogen Corp., Carlsbad, CA) has been previously described (19). Vector pcDNA3.1-APOBEC3F-V5-6XHis was obtained from Matija Peterlin and Yong-Hui Zheng through the NIH AIDS Research and Reference Reagent Program (catalog no. 10100). To replace the V5 tag with a C-terminal myc tag, the A3F insert was subcloned into pcDNA3.1(−)MycHis using standard molecular biology techniques. Mutation of cysteine residues C280 and C283 in human A3F in the backbone of A3F-myc was accomplished by PCR-directed mutagenesis. The presence of the desired mutations was verified by sequence analysis.
Cell culture, transfections, and construction of HeLa-A3F cell lines.
HeLa cells were propagated in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum. For transient transfection of HeLa cells, cells were grown in 25-cm2 flasks to about 80% confluence. Cells were transfected using TransIT-LT1 (Mirus Corp., Madison, WI) by following the manufacturer's recommendations. Unless noted otherwise, a total of 5 μg of plasmid DNA per 25-cm2 flask (∼5 × 106 cells) was used. Cells were harvested at 24 to 48 h posttransfection. HeLa cell lines stably expressing C-terminally Myc-tagged WT A3F (A3F wt), deaminase-defective mutant A3F C280S/C283A (A3F mt), or WT A3G (A3G wt) or containing the empty vector (control [Ctrl]) were constructed by transfecting HeLa cells in a 10-cm dish with pcDNA-A3F-myc, pcDNA-A3F-myc C280S/C283A, pcDNA-APO3G-Myc, or empty pcDNA3.1(−), respectively. Two days following transfection, cells were treated with G418 (800 μg/ml) and cultured until G418-resistant colonies became apparent. Individual colonies were transferred to 24-well plates and maintained in DMEM containing 10% fetal bovine serum and 400 μg/ml G418. Cells were passaged three times and then screened by immunoblot analysis for A3F expression using a myc-specific antibody. Two clones each of WT (A3F wt) and deaminase-defective A3F C280S/C283A (A3F mt) and one clone each of WT A3G (A3G wt) and the empty vector (Ctrl) were selected for functional studies.
Monocyte-derived macrophages (MDM) and IFN-α treatment.
Elutriated monocytes from healthy donors were allowed to differentiate into macrophages in six-well plates (4 × 106 per well) in 2 ml of complete DMEM containing10% pooled human serum for 7 days (35). The cells were then treated with alpha 2c interferon (IFN-α2c; 0 to 10 ng/ml) for 24 h and analyzed for protein expression and mRNA levels by immunoblotting or Northern blot analysis, respectively.
Antibodies.
A peptide antibody to human A3G was prepared by immunizing rabbits with keyhole limpet hemocyanin-coupled peptides corresponding to residues 367 to 384 of human A3G (20). A monoclonal antibody (MAb) to Vif (MAb 319) and a polyclonal antibody to A3F were obtained from Michael Malim through the NIH AIDS Research and Reference Reagent Program (catalog no. 6459 and 11474) (14, 45). A MAb to c-myc (Sigma-Aldrich, Inc., St. Louis, MO) was used for detection of epitope-tagged A3F and A3G. For identification of tubulin and actin, commercial MAbs were used (Sigma-Aldrich, Inc., St. Louis, MO). An HIV-positive patient serum was used for identification of the HIV-1 capsid (CA) and matrix (MA) proteins.
Immunocytochemistry.
Cells were grown in LabTek eight-well chambers (Nalge Nunc International, Naperville, IL) overnight and fixed with 1% paraformaldehyde. The samples were rinsed in phosphate-buffered saline (PBS), blocked with methanol-hydrogen peroxide, rinsed, and blocked with PBS and normal goat serum. The primary antibody, rabbit polyclonal APO3F (preabsorbed with HeLa cells at 4°C overnight) was diluted in PBS and applied to the samples at 1:200 for 1 h, followed by Dako EnVision goat anti-rabbit IgG horseradish peroxidase (HRP)-labeled polymer (K4002; Dako). The HRP label was reacted with diaminobenzidine, rinsed, and counterstained with hematoxylin. For a negative control, we omitted the primary antibody. The labeled samples were viewed and photographed with a Zeiss Axio Imager Z1.
Deamination assay.
For the analysis of virus-associated deaminase activity, virus-containing supernatants (7 ml) were precleared by filtration (0.45 μm) and pelleted through 20% sucrose (4 ml). Concentrated virions were directly suspended in 100 μl of deaminase buffer (40 mM Tris, pH 8.0, 40 mM KCl, 50 mM NaCl, 5 mM EDTA, 1 mM dithiothreitol, 2% [vol/vol] glycerol, 0.1% [vol/vol] Triton X-100) and incubated with a 32P-labeled oligonucleotide substrate (5′-ATTATTATTATTATACCCAATTCTTTATTTATTTATTTATTT-3′) containing A3G and A3F target sites (underlined). After overnight incubation at 37°C with occasional agitation, the oligonucleotides were purified on G25 Quick Spin columns (Roche) and reacted with uracil-DNA glycosylase (Roche) for 2 h at 37°C in uracil-DNA glycosylase buffer (60 mM Tris-HCl, pH 8.0, 1 mM EDTA, 1 mM dithiothreitol, 0.001% bovine serum albumin) to remove the uracil bases generated by deamination. For cleavage at the abasic site, the samples were treated for 5 min at 37°C with 0.15 M NaOH. Samples were then neutralized by the addition of 0.15 M HCl. The cleaved products were separated on 15% acrylamide-7 M urea gels and detected by autoradiography.
Immunoblotting.
For immunoblot analysis of intracellular proteins, whole-cell lysates were prepared as follows. Cells were washed once with PBS, suspended in PBS, and mixed with an equal volume of sample buffer (4% sodium dodecyl sulfate [SDS], 125 mM Tris-HCl, pH 6.8, 10% 2-mercaptoethanol, 10% glycerol, 0.002% bromophenol blue). To analyze virus-associated proteins, cell-free filtered supernatants from transfected HeLa cells (3.5 ml) were pelleted (75 min, 35,000 rpm) through a 20% sucrose cushion (5.5 ml) in an SW41 rotor. The concentrated virus pellet was suspended in PBS and mixed with an equal volume of sample buffer. Proteins were solubilized by heating for 10 to 15 min at 95°C. Cell and virus lysates were subjected to SDS-polyacrylamide gel electrophoresis; proteins were transferred to polyvinylidene difluoride membranes and reacted with appropriate antibodies as described in the text. Membranes were then incubated with horseradish peroxidase-conjugated secondary antibodies (Amersham Biosciences, Piscataway, NJ), and proteins were visualized by enhanced chemiluminescence (Amersham Biosciences).
Northern blot analysis.
Total RNA was prepared using the RNeasy mini kit according to the manufacturer's instructions (Qiagen, Valencia, CA). Total RNA (5 to 20 μg, as indicated) was electrophoresed on a denaturing 1% agarose gel and capillary blotted onto a nylon membrane using a Turbo blotter (Schleicher & Schuell, Inc., Keene, NH). After UV cross-linking, the membrane was stained for rRNA. For that purpose, the membrane was soaked in 5% acetic acid for 15 min at ambient temperature and then incubated in a solution of 0.5 M sodium acetate (pH 5.2) and 0.04% methylene blue for 5 min at ambient temperature. The membrane was rinsed in water for 10 min, and the stained membrane was photographed. Following staining, the membrane was prehybridized with 10 ml of QuickHyb hybridization solution (Stratagene, La Jolla, CA) for 1 h at 65°C. The membrane was then hybridized with probes for 5 h at 65°C. The probe for A3F mRNA was an 807-bp EcoRI/HindIII restriction fragment isolated from pcDNA-A3F. Probes were labeled with [32P]dTTP and a random primer using a Ladderman labeling kit (TakaRa, Madison, WI), and 1 × 107 cpm of the probe was added after premixing with 100 μl sonicated salmon sperm DNA (Stratagene, La Jolla, CA), heating at 94°C for 5 min, and chilling on ice. Following hybridization, membranes were washed twice with wash buffer (2× SSPE [1× SSPE is 0.18 M NaCl, 10 mM NaH2PO4, and 1 mM EDTA, pH 7.7]-0.1% SDS) for 15 min at ambient temperature, followed by a 15-min wash in 0.2× SSPE-0.1% SDS at 60°C. RNA bands were visualized by autoradiography.
Virus preparation.
Virus stocks were prepared by transfection of HeLa cells with appropriate plasmid DNAs. Virus-containing supernatants were harvested 24 to 48 h after transfection. Cellular debris was removed by centrifugation (5 min, 1,500 rpm), and clarified supernatants were filtered (0.45 μm) to remove residual cellular contaminants. For immunoblot analyses, filtered virus stocks were concentrated by pelleting through a 20% sucrose cushion (75 min at 4°C and 35,000 rpm in an SW41 rotor).
Viral infectivity assay.
Viral infectivity was determined in a single-cycle assay by measuring the virus-induced luciferase activity in infected TZM-bl cells. TZM-bl cells are derived from HeLa cells and contain the CXCR4 coreceptor and a luciferase indicator gene under the control of the HIV-1 long terminal repeat (LTR). Those cells were obtained from John Kappes through the NIH AIDS Research and Reference Reagent Program (catalog no. 8129). Cells were maintained in complete DMEM supplemented with 10% fetal bovine serum. For infection, TZM-bl cells (5 × 104) were plated into 24-well plates and allowed to adhere overnight. Virus-containing supernatants were then added, and cells were incubated for 24 h at 37°C. Cells were washed twice with 1 ml of cold PBS and lysed in 300 μl of Reporter Lysis Buffer (Promega Corp., Madison, WI). To determine luciferase activity, 20 μl of each lysate was combined with luciferase substrate (Promega) by automatic injection and light emission was measured for 10 s at room temperature in a luminometer (Optocomp II; MGM Instruments, Hamden, CT).
Detergent stripping.
To prepare sucrose step gradients, 2.0 ml of a 60% sucrose solution was placed into the bottom of an SW55 centrifuge tube and overlaid with 2.1 ml of a 20% sucrose solution. Immediately prior to the addition of concentrated virus stocks (500 μl), the step gradients were overlaid with 100 μl of a protease inhibitor cocktail (Complete; Roche Diagnostics, Indianapolis, IN) and 50 μl of either PBS or 1% Triton X-100. Samples were then centrifuged in an SW55 Ti rotor for 60 min at 35,000 rpm, 4°C. Four fractions of 1.1 ml each were collected from the top (see diagram in Fig. 7). Aliquots from each fraction were combined with sample buffer, incubated at 95°C for 10 min, and then processed for immunoblot analysis.
RESULTS
Transient expression of WT and deaminase-defective A3F reveals antiviral activity.
APOBEC3G (A3G) and APOBEC3F (A3F) are considered the most potent cytidine deaminases in humans to target retroviral infections (4). We and others have found that catalytic activity of A3G is critical for its antiviral effect (6, 34, 44). In contrast, WT and deaminase-defective A3F proteins were reported to have similar antiviral activities (3, 14). In an effort to confirm this observation, we constructed a deaminase-defective A3F protein in the backbone of a C-terminally myc-tagged A3F vector. Two mutations (C280S/C283A) were introduced into the C-terminal catalytic domain of A3F, in analogy to our previous deaminase-defective A3G protein (38). Virus encapsidation and loss of catalytic activity of the resulting mutant protein (referred to as A3F mt for the remainder of this report) were first tested by transiently transfecting HeLa cells with vif-deficient pNL4-3 together with a vector encoding WT A3G (A3G wt), WT A3F (A3F wt), or deaminase-defective A3F (A3F mt). Vif-deficient virus produced in the absence of APOBEC was included as a control (Ctrl). Virus-containing supernatants were collected at 48 h after transfection and used for immunoblot analysis (Fig. 1 A) or for analysis of virus-associated deaminase activity (Fig. 1B). Samples were processed for immunoblotting as described in Materials and Methods, and the blot was probed first with a myc-specific antibody to identify virus-associated A3G and A3F proteins (Fig. 1A, top). The same blot was then stripped and reprobed with an HIV-positive patient serum to detect the viral CA protein (Fig. 1A, bottom). Virus-associated A3G and A3F deaminase activities were measured as described in Materials and Methods. A synthetic 5′-end-labeled single-stranded oligonucleotide containing both A3G and A3F signature motifs (CC and TC, respectively) was used. Deamination of the oligonucleotide is evident from the resulting cleavage of the probe (Fig. 1B). The difference in mobility between the products produced by A3G- and A3F-specific deamination corresponds to the relative distance of the deamination site from the labeled 5′ end of the probe (16 or 17 bases for A3G, 22 bases for A3F). As expected, the A3G and A3F proteins were efficiently incorporated into Vif-deficient particles (Fig. 1A, lanes 2 to 4). Moreover, virus-associated A3G wt and A3F wt were catalytically active in the in vitro deaminase assay (Fig. 1B, lanes 2 and 3). In contrast, A3F mt was unable to deaminate the substrate (Fig. 1B, lane 4) despite the presence of equivalent amounts of A3F protein in the virus preparation (Fig. 1A, compare lanes 3 and 4). These results confirm that A3F mt is catalytically inactive but is efficiently encapsidated into HIV-1 virions.
FIG. 1.

Transiently expressed A3F wt and A3F mt are packaged into Vif-deficient HIV-1 virions and have antiviral activity. (A) HeLa cells were transfected with vif-defective pNL4-3 (4 μg) together with 1 μg of a human A3G wt (lane 2)-, A3F wt (lane 3)-, or A3F mt (lane 4)-expressing vector. Lane 1 is a control consisting of vif-defective pNL4-3 without APOBEC. Virus-containing supernatants were collected at 48 h posttransfection and analyzed by immunoblotting for the presence of APOBEC proteins using a myc-specific antibody (A3G/A3F). The same blot was subsequently reprobed with an HIV-positive patient serum to identify the CA protein. (B) Virus-associated deaminase activity was determined on viruses from panel A using a 5′-end 32P-labeled single-stranded DNA template with signature motifs for A3G and A3F. Oligonucleotides were separated by 15% acrylamide-7 M urea gel electrophoresis. The positions of the cleaved products and the uncleaved probe are indicated on the right. (C) HeLa cells were cotransfected with 4 μg of pNL4-3Vif(−) (lanes 3 to 15) and increasing amounts of A3G wt (lanes 4 to 7), A3F wt (lanes 8 to 11), or A3F mt (lanes 12 to 15). The amounts of transfected APOBEC DNA used were 30 ng (lanes 4, 8, and 12), 100 ng (lanes 5, 9, and 13), 300 ng (lanes 6, 10, and 14), and 1 μg (lanes 7, 11, and 15). Lane 3 is a control consisting of HeLa cells transfected with 4 μg of pNL4-3Vif(−) in the absence of APOBEC. Lane 2 shows HeLa cells transfected with 4 μg of pNL4-3 wt in the absence of APOBEC. The total DNA amount in all samples was adjusted to 5 μg with the appropriate amount of empty vector DNA. Lane 1 is a mock-treated control consisting of HeLa cells transfected with 5 μg of empty vector DNA. Cells and virus-containing supernatants were harvested at 24 h following transfection. Whole-cell lysates were subjected to immunoblot analysis using a myc-specific antibody (A3G/A3F) or an antibody to Vif or HIV-1 Gag (CA). (D) The infectivity of viruses produced in panel G was determined by infecting TZM-bl indicator cells. Virus input was normalized by reverse transcriptase activity. Virus-induced luciferase activity was determined at 24 h after infection in a standard luciferase assay. The infectivity of virus produced in the absence of APOBEC was defined as 100% (lane 3). The infectivity of the other viruses was expressed as a percentage of that of the control virus. Error bars reflect standard deviations from three independent infections.
Next, we tested the antiviral activity of A3F mt. HeLa cells were transfected with NL4-3vif(−) DNA together with various amounts of A3G wt (Fig. 1C, lanes 4 to 7), A3F wt (lanes 8 to 11), or A3F mt (lanes 12 to 15). WT NL4-3 (lane 2) or NL4-3vif(−) in the absence of APOBEC (lane 3) was included as a control. A mock-transfected sample (lane 1) was also included as a background control. Immunoblot analysis was performed as described in Materials and Methods to identify cellular and virus-associated A3G, A3F, Vif, and CA. Consistent with the results in Fig. 1A, the A3G and A3F proteins were expressed and packaged into Vif-deficient HIV-1 virions with similar efficiency and in a dose-dependent manner. Viral supernatants from Fig. 1C were then used to test viral infectivity in a single-round infectivity assay using TZM-bl target cells. TZM-bl cells contain an integrated HIV-1 LTR-driven luciferase gene and express CD4 and CXCR4 receptors (50). Virus-induced luciferase activity was measured 24 h after infection as described in Materials and Methods. Luciferase activity observed for NL4-3Vif(−) virus (Fig. 1D, lane 3) was defined as 100% relative infectivity. As expected, expression of increasing amounts of A3G wt and A3F wt resulted in a concomitant loss of viral infectivity (Fig. 1D, lanes 4 to 11). Interestingly, A3F mt had antiviral activity very similar to that of its WT counterpart (Fig. 1D, lanes 12 to 15). These results confirm the previous observations that A3F has potent antiviral activity and that A3F's antiviral activity is largely deaminase independent (14).
Analysis of endogenous A3F expression in MDM and T-cell lines.
We next studied the expression of endogenous A3F in MDM and T-cell lines. MDM were previously shown to express A3F in an IFN-inducible manner (15, 23, 39, 54). All of the previous studies assessed A3F expression on the level of mRNA only. In contrast, we compared A3F expression in MDM, H9, and Jurkat cells both on the mRNA level (Fig. 2 A) and on the protein level (Fig. 2B). The A3F-specific antibody used in this study had not previously been used for the detection of endogenous A3F protein. Consistent with earlier studies, we found that A3F mRNA levels in MDM increased in response to IFN treatment (Fig. 2A, lanes 1 to 3). rRNA served as a loading control (Fig. 2A, bottom). Interestingly, Jurkat cells expressed very low levels of A3F mRNA (Fig. 2A, lane 5). In contrast, A3F mRNA levels in H9 cells were comparable to those in MDM treated with the highest level of IFN stimulation (Fig. 2A, compare lanes 3 and 4). Unstimulated MDM expressed only low levels of A3F mRNA (Fig. 2A, lane 1).
FIG. 2.
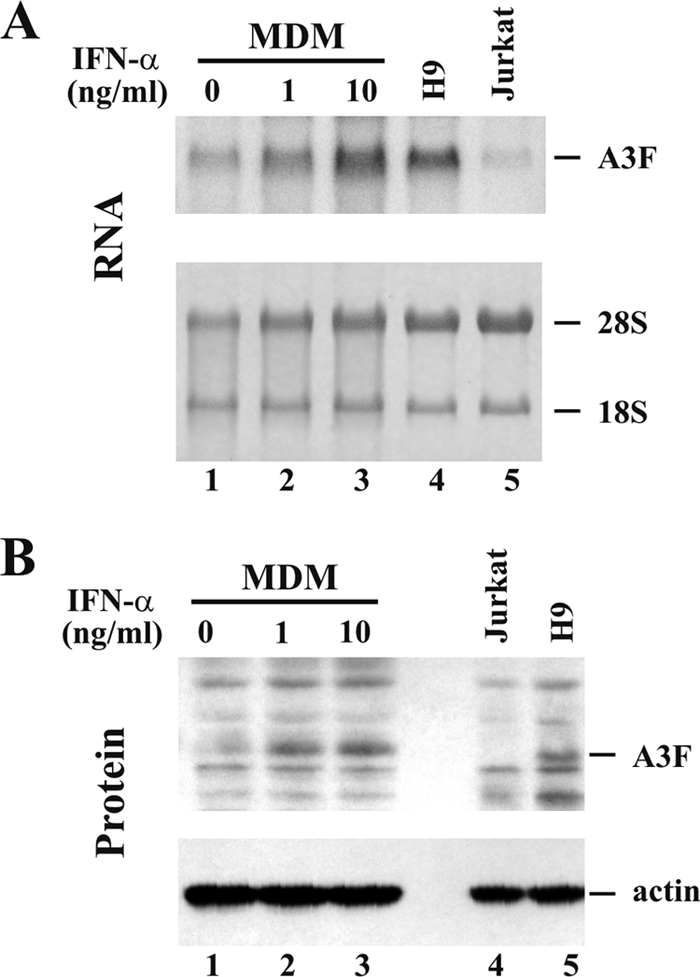
Analysis of endogenous A3F expression in MDM and T-cell lines. (A) Northern blot analysis to determine mRNA expression. MDM were treated for 24 h with IFN-α at 0 ng/ml (lane 1), 1 ng/ml (lane 2), or 10 ng/ml (lane 3). The H9 (lane 4) and Jurkat (lane 5) T-cell lines were analyzed in parallel. Each lane contained 5 μg of total RNA. Samples were separated by denaturing 1% agarose gel electrophoresis and capillary blotted onto a nylon membrane as described in Materials and Methods. The membrane was then probed with an A3F-specific 32P-labeled probe (top). Equal sample loading was verified by staining membranes with methylene blue to identify rRNA species (18S and 28S, bottom). (B) Endogenous expression of A3F protein (top). MDM were treated with IFN-α as for panel A (lanes 1 to 3). The Jurkat (lane 4) and H9 (lane 5) T-cell lines were included for reference. An A3F-specific polyclonal antibody was used for the detection of A3F protein (top). The same blot was stripped and reprobed with an actin-specific antibody (bottom) to control sample loading.
Immunoblot analysis of MDM (Fig. 2B, lanes 1 to 3), as well as Jurkat and H9 T-cell lines (Fig. 2B, lanes 4 and 5), revealed A3F protein levels that closely mirrored the mRNA expression profiles. Thus, the A3F antibody is indeed capable of identifying endogenous A3F protein. A3F protein levels were undetectable in Jurkat cells (Fig. 2B, lane 4) and near the limit of detection in unstimulated MDM (Fig. 2B, lane 1). In contrast, A3F protein levels in MDM increased following IFN treatment in a dose-dependent manner (Fig. 2B, lanes 2 to 3) and were comparable to A3F protein levels in H9 cells (Fig. 2B, lane 5). These results indicate that A3F expression in the H9 and Jurkat T-cell lines mimics A3G expression profiles and correlates with the Vif dependence of virus replication in these cells.
Comparison of endogenous levels of A3F to transiently expressed A3F.
We previously observed that transient expression of A3G from cytomegalovirus promoter-based vectors can lead to severe overexpression (34). To determine if this is also true for A3F, we used Northern blot analysis to compare mRNA levels of transiently expressed A3F in HeLa cells to endogenous A3F in H9 cells. H9 cells were used here as a source of endogenous A3F since these cells were previously reported to express A3F mRNA (52). HeLa cells were transiently transfected with various amounts (0 to 1 μg) of A3F-myc expression vector, and cells were processed 24 h later for Northern blot analysis as described in Materials and Methods. Exogenously expressed A3F produces an ∼1.2-kb mRNA which migrates faster in the gel than the 2.4- to 2.7-kb endogenous A3F mRNA due to the absence of 3′ noncoding sequences in the exogenous mRNA (28). As expected, A3F mRNA was expressed in H9 cells (Fig. 3, lane 1) at higher levels than in HeLa cells, in which A3F mRNA levels approached the limit of detection (Fig. 3, lane 2). As expected, levels of transiently expressed A3F-myc mRNA increased in a dose-dependent manner (Fig. 3, lanes 3 to 6). Of note, even the smallest amount of transiently transfected A3F-myc vector produced A3F mRNA levels higher than those found in H9 cells (Fig. 3, compare lanes 1 and 3). Thus, transient transfection of HeLa cells leads to overexpression of A3F mRNA. Aside from the main 1.2-kb mRNA species, transiently transfected HeLa cells produced a second A3F mRNA species migrating slightly slower than the endogenous A3F mRNA (Fig. 3, arrow). The nature of this mRNA species is not clear; however, it is conceivable that the larger mRNA represents a transcriptional readthrough product that terminates at a distal poly(A) site in the pcDNA3.1 vector. However, this issue was not further investigated.
FIG. 3.

A3F mRNA expressed in transiently transfected HeLa cells very much exceeds endogenous A3F levels. HeLa cells were transfected with increasing amounts of the exogenous A3F-myc vector (lane 3, 30 ng; lane 4, 100 ng; lane 5, 300 ng; lane 6, 1 μg). The total amount of transfected DNA was adjusted to 5 μg in all samples by adding the appropriate amount of empty vector DNA. Untransfected HeLa cells (lane 2) and H9 cells (lane 1) were analyzed in parallel. Samples were processed for Northern blot analysis. Equal amounts (20 μg) of total RNA were separated by denaturing 1% agarose gel electrophoresis and transferred to a nylon membrane. Equal sample loading was verified by staining membranes with methylene blue to identify rRNA species (bottom). The membranes were then probed with an A3F-specific 32P-labeled probe (top). The positions of A3F-Myc mRNA and endogenous A3F mRNA are indicated. The arrow points to a second A3F-Myc mRNA species that is slightly larger than the endogenous mRNA and whose intensity increases with increasing amounts of transfected A3F-Myc vector.
Construction and characterization of stable HeLa cell lines expressing A3F wt and A3F mt.
In our previous study, we found that the deaminase-defective A3G C288S/C291A variant had antiviral activity when transiently overexpressed. The same mutant protein, however, when expressed in stable HeLa cells, had no antiviral activity (34). From that study, we concluded that efficient inhibition of Vif-deficient HIV-1 requires catalytically active A3G.
To investigate whether the antiviral effect of A3F observed in Fig. 2B was similarly due to transient A3F overexpression, we created stable cell lines expressing WT and deaminase-defective A3F proteins. Cell clones were prescreened by Northern blot analysis, and two clones of each set were picked to match as closely as possible the A3F mRNA levels in H9 cells. A cell clone transduced with the empty vector but subjected to the same selection procedure was selected and used as a negative control in subsequent analyses. Since mRNA levels are not necessarily predictive of APOBEC protein levels (34), we compared A3F protein expression in our stable HeLa lines to that in H9 cells using the A3F-specific antibody. Stable expression of A3F wt (Fig. 4, lanes 7 and 8) and A3F mt (lanes 9 and 10) was compared to A3F wt expression in HeLa cells transiently transfected with increasing amounts of A3F wt DNA (Fig. 4, lanes 2 to 5). Mock-transfected HeLa cells (Fig. 4, lane 1), as well as H9 and Jurkat cells (Fig. 4, lanes 11 to 12), were also included for reference. Finally, a stable HeLa clone containing the empty vector was added as a negative control (Fig. 4, Ctrl, lane 6). Transfection of HeLa cells, preparation of cell lysates, and processing of lysates for immunoblotting were done as described in Materials and Methods. The blot was probed first with the A3F-specific antibody (Fig. 4, top), followed by an antibody to α-tubulin (bottom). The results show that the stable HeLa cell lines expressing A3F wt expressed levels of protein comparable to or higher than those in H9 cells (Fig. 4, compare lanes 7 and 8 to lane 11). Levels of A3F mt were slightly higher than those of A3F wt (Fig. 4, lanes 9 and 10).
FIG. 4.

Construction of stable HeLa cell lines expressing A3F wt and A3F mt. A comparison of the expression of transiently transfected, stably transfected, and endogenously expressed A3F protein is shown. To assess exogenous expression, HeLa cells were transiently transfected with increasing amounts (0, 0.03, 0.1, 0.3, and 1 μg) of A3F wt as described in the legend to Fig. 1C. Stable cell lines (A3F wt1, wt2, mt1, and mt2) were analyzed in parallel. HeLa cells containing empty vector DNA and subjected to the same selection process as the A3F lines were analyzed in parallel (Ctrl). H9 cell lysates were used to visualize endogenous A3F, and Jurkat cell lysates were used as a negative control. The A3F-specific antibody was used for the detection of A3F (top). The same blot was stripped and reprobed with a tubulin-specific antibody (bottom) to verify equal sample loading.
Stable HeLa cell lines uniformly express A3F.
To accurately measure the functional properties of A3F wt and A3F mt in stable cell lines, it was critical to demonstrate that 100% of the cells express A3F. This was done by immunocytochemistry (Fig. 5). Cells were grown in chamber flasks and stained with an A3F-specific polyclonal antibody as described in Materials and Methods. HeLa cells containing empty vector DNA showed low-level A3F staining (Fig. 5, Ctrl) relative to the negative control lacking the primary antibody (Fig. 5, no 10). It is unclear whether this is due to low-level A3F expression in HeLa cells or reflects inherent nonspecific background. Importantly, however, all A3F cell lines exhibited staining that was significantly above the level observed in normal HeLa cells; even more importantly, all cells of each culture were A3F positive. We therefore conclude that all of the stable cell lines used in our analysis uniformly express A3F wt or A3F mt.
FIG. 5.

Stable HeLa cell lines uniformly express A3F. Stable transduced HeLa cells expressing A3F wt or A3F mt or transduced with empty vector DNA (Ctrl) as described in the legend to Fig. 4 were analyzed for A3F expression using an A3F-specific polyclonal rabbit antibody. To control for nonspecific background produced by the secondary antibody, vector-transduced cells were treated with the secondary antibody only (no 10). Labeled samples were viewed and photographed with a Zeiss Axio Imager Z1.
A3F from stable HeLa cells is packaged into viral cores but lacks antiviral activity.
To investigate the antiviral effect of A3F expressed in stable HeLa cells, cells were transfected with the pNL4-3 or pNL4-3Vif(−) proviral construct. HeLa cells stably expressing A3G-Myc were included for comparison. Cells and virus-containing supernatants were harvested 24 h after transfection. To directly compare the expression and packaging of A3G wt, A3F wt, and A3F mt into NL4-3 and NL4-3vif(−) particles by immunoblotting, we used a Myc-specific polyclonal antibody directed against the common C-terminal Myc tag present in these proteins (Fig. 6 A, parts I and IV). NL4-3 Vif only modestly reduced cellular levels of A3G but completely blocked viral packaging (Fig. 6A, lane 3), consistent with our previous observations (9). As expected, A3F wt and A3F mt were packaged into Vif-deficient HIV-1 virions. We estimate the efficiency of A3F packaging in the absence of Vif to be as good as or better than that of A3G packaging (Fig. 6A, compare lane 4 to lanes 6, 8, 10, and 12). NL4-3 Vif had variable effects on cellular levels of A3F wt and A3F mt in the stable HeLa clones, and the amounts of virus-associated A3F appeared to be directly proportional to the intracellular protein levels. It is interesting that A3F wt and A3F mt expressed by clones 1 each appear to be less sensitive to degradation by Vif than those expressed by the other two clones (Fig. 6A, part I). The reason for this is not clear. Differences in Vif expression among the samples are minor and are unlikely to account for the differential degradation of A3F. It is also interesting that while Vif efficiently blocks A3G encapsidation even when cellular degradation is modest, changes in A3F encapsidation appear to largely reflect intracellular Vif-induced protein levels.
FIG. 6.

A3F expressed in stable HeLa cell lines does not have antiviral activity. (A) Stable HeLa cell lines expressing A3G wt (34), A3F wt (clones 1 and 2), or A3F mt (clones 1 and 2) were each transfected with 5 μg of pNL4-3 or pNL4-3Vif(−) DNA. Transfected cells and virus-containing supernatants were harvested 24 h later and processed for immunoblot analysis. Blots were probed with a myc-specific antibody (parts I and IV), a Vif MAb (part II), and an HIV-positive patient serum (APS) for the detection of viral CA proteins (parts III and V). Proteins are identified on the right. (B) Virus samples from panel A were normalized for equivalent reverse transcriptase activity and used to infect TZM-bl cells. Tat-induced luciferase activity was quantified and used to calculate relative viral infectivity. The infectivity of WT virus was defined as 100% for each producer cell type (lanes 1, 3, 5, 7, 9, and 11), and the infectivity of Vif-deficient viruses was calculated relative to that of the corresponding WT viruses (lanes 2, 4, 6, 8, 10, and 12). Error bars reflect standard deviations calculated from three independent infections.
We next tested the infectivity of the individual virus preparations relative to that of virus produced in the absence of APOBEC. Viral supernatants were used to infect HeLa TZM-bl indicator cells as described in Materials and Methods. The infectivity of Vif-deficient viruses was calculated relative to that of WT NL4-3, which was defined as 100% for each cell type (Fig. 6B). As expected, APOBEC-deficient viruses were fully infectious (Fig. 6B, lanes 1 and 2) and expression of A3G was inhibited vif-defective HIV-1 (lane 4) but not WT NL4-3 (lane 3). Surprisingly, neither A3F wt nor deaminase-defective A3F mt inhibited viral infectivity. In fact, the relative infectivity of WT and vif-defective HIV-1 in the presence of A3F wt or A3F mt was identical in all samples despite significant differences in the amounts of virus-associated A3F (Fig. 6A, part IV; compare lane 7 to 8, 9 to 10, or 11 to 12). These results indicate that A3F from stable HeLa cells has little, if any, antiviral activity.
A3F is packaged into the viral nucleoprotein complex.
Previous studies of A3A demonstrated that the antiviral activity of APOBEC correlates with targeting of the proteins to the viral core (2, 9). We therefore considered the possibility that the lack of A3F antiviral activity was due to mislocalization of the deaminase in the viral particles. To address this question, we performed detergent stripping of HIV-1 virions to assess the core association of A3F. This method was previously used to demonstrate the core association of A3G (9, 22). Clones 2 of A3F wt and A3F mt cell lines were randomly chosen for this analysis. Cells were transfected with pNL43Vif(−) in the presence of vesicular stomatitis virus G protein (VSV-G). VSV-G was included to enable reinfection of the cells by viruses produced early on. This strategy allowed us to strongly boost virus production and obtain sufficient material for our analysis. Viral supernatants were processed for sucrose step gradient separation in the presence or absence of Triton X-100 as described in Materials and Methods. Individual fractions of the gradient were then subjected to immunoblot analysis to identify the A3F, CA, and MA proteins (Fig. 7). As expected, in the absence of detergent, all proteins were found in fraction 3 of the step gradient (Fig. 7, untreated), consistent with the accumulation of virions at the interphase of 20% and 60% sucrose (cartoon in Fig. 7) and attesting to the absence of soluble contaminants in the virus preparations. Particles subjected to step gradient analysis in the presence of Triton X-100 were quantitatively stripped of their MA proteins, which accumulated in fraction 1 at the top of the gradient (Fig. 7, X-100, MA). As noted before (9, 22), CA was partially sensitive to detergent treatment and partitioned between fractions 1 and 3. A smaller fraction of CA protein was found at the bottom of the gradient (fraction 4), presumably due to sampling error (samples were collected from the top). Importantly, however, A3F was completely resistant to detergent treatment and accumulated in fraction 3 of the gradient. We conclude that A3F wt and A3F mt were both properly targeted to the viral core.
FIG. 7.

A3F wt and A3F mt expressed in stable HeLa cell lines is packaged into the core of HIV-1 virions. Stable HeLa cell lines (clones 2 of A3F wt and A3F mt) were transfected with 4.5 μg of pNL4-3Vif(−) together with 0.5 μg of pCMV-VSV-G. Virus-containing supernatants were harvested at 48 h after transfection, and viruses were purified and concentrated by ultracentrifugation through 20% sucrose. Pelleted viruses were suspended in DMEM and divided into two equal fractions. One fraction was left untreated, and the other fraction was treated with 0.5% Triton X-100. Untreated and detergent-treated samples were subjected to step gradient centrifugation. Four equal fractions (1 to 4 in the cartoon on the left) were collected from the top of the gradients. Aliquots from each fraction were analyzed by immunoblotting using a myc-specific antibody (A3F). The same blot was then reprobed with an HIV-positive human serum to identify viral CA and MA proteins.
DISCUSSION
The question of which cytidine deaminases contribute to the accumulation of G-to-A mutations in viral genomes has occupied the field for a number of years (for a review, see reference 10). Previous studies demonstrated that cytidine deaminases have preferential target specificities based on their dinucleotide context. A3G preferentially targets CC, while the preferred target site for A3F is TC (underlining denotes the target for deamination) (3, 4, 13, 18, 52, 56). Interestingly, analysis of viral sequences derived from patient peripheral blood mononuclear cells revealed a trend for preferential mutation of G in the GA context (i.e., deamination occurs in the TC context) (17). This observation, however, does not provide conclusive evidence for the in vivo importance of A3F, since A3B and A3C, and to a lesser extent A3G as well, can catalyze cytidine deamination in the TC context (3-5, 25, 55).
In vitro, A3F has potent antiviral activity (52, 58), although at least one study reported that human A3F is 10 to 50 times less potent than human A3G (57). Surprisingly, however, the antiviral activity of A3F appears to be less dependent on catalytic deaminase activity than A3G. In fact, WT and deaminase-defective A3F inhibited viral infectivity equally well in a transient-expression system (14). The results of our current study are consistent with these observations and confirm that A3F has strong antiviral activity when transiently expressed and this effect does not depend on catalytic activity (Fig. 1). We also noted that deaminase-deficient A3F, like its deaminase-defective A3G counterpart, did not have antiviral activity when expressed from stable cell lines at close-to-endogenous levels (Fig. 6) (34). Importantly, however, A3F wt also lost its antiviral activity when expressed in stable HeLa cells. In that respect, A3F is clearly distinct from A3G, which maintained high antiviral activity under similar conditions (Fig. 6).
Why does stably expressed catalytically active A3F lack antiviral effects? The enzyme retains catalytic activity, as evidenced by the appearance of low-level G-to-A mutations in the GA context in viruses produced from stable A3F cells (data not shown). However, this does not affect the overall viral infectivity, possibly because of purifying selection, as reported for A3G (41). The low antiviral potential of A3F could explain why G-to-A mutations in the context of GA dinucleotides can be identified in HIV-infected individuals. One could argue that sublethal mutagenesis of retroviral genomes may benefit the virus by increasing its potential to adapt to environmental pressure.
The difference between the strong antiviral effects of A3F and A3F transiently expressed in stable HeLa cells is most likely explained by a simple dose effect since transient expression of A3F can lead to severe overexpression of the protein (Fig. 4). It is important to keep in mind that the rate of A3F synthesis in transiently transfected cells is much greater than in stable cell lines in which protein synthesis and degradation have reached a steady state that is tolerated by the cell (34). Since A3F packaging is dependent on intracellular protein levels (Fig. 1), we conclude that viruses produced in cells transiently expressing A3F contain significantly more A3F than viruses derived from stable A3F-producing cells. A second factor potentially contributing to the poor antiviral effect of stably expressed A3F is that both A3G and A3F gradually transition from a low-molecular-mass configuration to a high-molecular-mass complex and eventually accumulate in P bodies and stress granules (24, 46, 49, 51). Since APOBEC appears to be packaged primarily from the pool of newly synthesized protein (46), transiently expressed A3F may be more packaging competent, which would explain its stronger antiviral effect than that of the stable expressed protein.
Differences in the relative expression of transiently and stably expressed A3F do not, however, explain why stably expressed A3G retains antiviral activity and A3F does not. We did not detect any drastic differences in the relative packaging efficiencies of A3G and A3F (Fig. 6A), and A3F was properly targeted to the viral core, a criterion we previously determined to be critical for APOBEC antiviral activity (9). It is therefore possible that A3F is catalytically less active or that deamination is generally more efficient in the CC context than in the TC context. The fact that neither WT nor deaminase-deficient A3F inhibits viral infectivity in stable HeLa cells explains why transient overexpression of the protein did not reveal any difference in relative antiviral potency between the catalytically active and inactive forms of the enzyme. A recent study proposed that transiently overexpressed A3F inhibits viral DNA integration (33). This effect was largely independent of A3F catalytic activity and is therefore consistent with our data. A number of other mechanisms have been proposed to explain deamination-independent inhibition of viral infectivity by A3G (for a review, see reference 10). It remains to be investigated if any of these mechanisms apply to viral inhibition by A3F.
On a final note, we observed that Vif more potently inhibited the encapsidation of A3G than that of A3F (Fig. 6A). In fact, packaging of A3F appeared to strictly parallel intracellular A3F levels both in the presence and in the absence of Vif. In contrast, inhibition of A3G packaging by Vif was much more effective than intracellular depletion, consistent with previous observations (19, 30). This suggests that Vif has the ability to inhibit A3G packaging via degradation-dependent and degradation-independent mechanisms while A3F packaging can only be inhibited through a degradation-dependent process. Details of this interesting phenomenon will have to be investigated in future studies.
Acknowledgments
We are grateful to Amy Andrew, Takeshi Yoshida, and Hiroaki Takeuchi for valuable suggestions and for critical comments on the manuscript. We thank Melissa Gilden for constructing deaminase-defective A3F mt.
Antibodies to Vif (MAb 319) and A3F were obtained from Michael Malim through the NIH AIDS Research and Reference Reagent Program. The vector for expression of A3F was obtained from Matija Peterlin and Yong-Hui Zheng through the NIH AIDS Research and Reference Reagent Program. This work was supported in part by a grant from the NIH Intramural AIDS Targeted Antiviral Program to K.S. and by the Intramural Research Program of the NIH, NIAID.
Footnotes
Published ahead of print on 11 August 2010.
REFERENCES
- 1.Adachi, A., H. E. Gendelman, S. Koenig, T. Folks, R. Willey, A. Rabson, and M. A. Martin. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59:284-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aguiar, R. S., N. Lovsin, A. Tanuri, and B. M. Peterlin. 2008. Vpr.A3A chimera inhibits HIV replication. J. Biol. Chem. 283:2518-2525. [DOI] [PubMed] [Google Scholar]
- 3.Bishop, K. N., R. K. Holmes, and M. H. Malim. 2006. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J. Virol. 80:8450-8458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bishop, K. N., R. K. Holmes, A. M. Sheehy, N. O. Davidson, S. J. Cho, and M. H. Malim. 2004. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr. Biol. 14:1392-1396. [DOI] [PubMed] [Google Scholar]
- 5.Bourara, K., T. J. Liegler, and R. M. Grant. 2007. Target cell APOBEC3C can induce limited G-to-A mutation in HIV-1. PLoS Pathog. 3:1477-1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Browne, E. P., C. Allers, and N. R. Landau. 2009. Restriction of HIV-1 by APOBEC3G is cytidine deaminase-dependent. Virology 387:313-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiu, Y. L., V. B. Soros, J. F. Kreisberg, K. Stopak, W. Yonemoto, and W. C. Greene. 2005. Cellular APOBEC3G restricts HIV-1 infection in resting CD4+ T cells. Nature 435:108-114. (Retracted 8 July 2010.) [DOI] [PubMed] [Google Scholar]
- 8.Dang, Y., X. Wang, W. J. Esselman, and Y. H. Zheng. 2006. Identification of APOBEC3DE as another antiretroviral factor from the human APOBEC family. J. Virol. 80:10522-10533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goila-Gaur, R., M. A. Khan, E. Miyagi, S. Kao, and K. Strebel. 2007. Targeting APOBEC3A to the viral nucleoprotein complex confers antiviral activity. Retrovirology 4:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goila-Gaur, R., and K. Strebel. 2008. HIV-1 Vif, APOBEC, and intrinsic immunity. Retrovirology 5:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo, F., S. Cen, M. Niu, J. Saadatmand, and L. Kleiman. 2006. Inhibition of tRNA3Lys-primed reverse transcription by human APOBEC3G during human immunodeficiency virus type 1 replication. J. Virol. 80:11710-11722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo, F., S. Cen, M. Niu, Y. Yang, R. J. Gorelick, and L. Kleiman. 2007. The interaction of APOBEC3G with human immunodeficiency virus type 1 nucleocapsid inhibits tRNA3Lys annealing to viral RNA. J. Virol. 81:11322-11331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris, R. S., K. N. Bishop, A. M. Sheehy, H. M. Craig, S. K. Petersen-Mahrt, I. N. Watt, M. S. Neuberger, and M. H. Malim. 2003. DNA deamination mediates innate immunity to retroviral infection. Cell 113:803-809. [DOI] [PubMed] [Google Scholar]
- 14.Holmes, R. K., F. A. Koning, K. N. Bishop, and M. H. Malim. 2007. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J. Biol. Chem. 282:2587-2595. [DOI] [PubMed] [Google Scholar]
- 15.Hou, W., X. Wang, L. Ye, L. Zhou, Z. Q. Yang, E. Riedel, and W. Z. Ho. 2009. Lambda interferon inhibits human immunodeficiency virus type 1 infection of macrophages. J. Virol. 83:3834-3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iwatani, Y., H. Takeuchi, K. Strebel, and J. G. Levin. 2006. Biochemical activities of highly purified, catalytically active human APOBEC3G: correlation with antiviral effect. J. Virol. 80:5992-6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janini, M., M. Rogers, D. R. Birx, and F. E. McCutchan. 2001. Human immunodeficiency virus type 1 DNA sequences genetically damaged by hypermutation are often abundant in patient peripheral blood mononuclear cells and may be generated during near-simultaneous infection and activation of CD4(+) T cells. J. Virol. 75:7973-7986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jonsson, S. R., G. Hache, M. D. Stenglein, S. C. Fahrenkrug, V. Andresdottir, and R. S. Harris. 2006. Evolutionarily conserved and non-conserved retrovirus restriction activities of artiodactyl APOBEC3F proteins. Nucleic Acids Res. 34:5683-5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kao, S., M. A. Khan, E. Miyagi, R. Plishka, A. Buckler-White, and K. Strebel. 2003. The human immunodeficiency virus type 1 Vif protein reduces intracellular expression and inhibits packaging of APOBEC3G (CEM15), a cellular inhibitor of virus infectivity. J. Virol. 77:11398-11407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kao, S., E. Miyagi, M. A. Khan, H. Takeuchi, S. Opi, R. Goila-Gaur, and K. Strebel. 2004. Production of infectious human immunodeficiency virus type 1 does not require depletion of APOBEC3G from virus-producing cells. Retrovirology 1:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karczewski, M. K., and K. Strebel. 1996. Cytoskeleton association and virion incorporation of the human immunodeficiency virus type 1 Vif protein. J. Virol. 70:494-507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khan, M. A., S. Kao, E. Miyagi, H. Takeuchi, R. Goila-Gaur, S. Opi, C. L. Gipson, T. G. Parslow, H. Ly, and K. Strebel. 2005. Viral RNA is required for the association of APOBEC3G with human immunodeficiency virus type 1 nucleoprotein complexes. J. Virol. 79:5870-5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koning, F. A., E. N. Newman, E. Y. Kim, K. J. Kunstman, S. M. Wolinsky, and M. H. Malim. 2009. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J. Virol. 83:9474-9485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kozak, S. L., M. Marin, K. M. Rose, C. Bystrom, and D. Kabat. 2006. The anti-HIV-1 editing enzyme APOBEC3G binds HIV-1 RNA and messenger RNAs that shuttle between polysomes and stress granules. J. Biol. Chem. 281:29105-29119. [DOI] [PubMed] [Google Scholar]
- 25.Langlois, M. A., R. C. Beale, S. G. Conticello, and M. S. Neuberger. 2005. Mutational comparison of the single-domained APOBEC3C and double-domained APOBEC3F/G anti-retroviral cytidine deaminases provides insight into their DNA target site specificities. Nucleic Acids Res. 33:1913-1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lei, Y. C., Y. J. Tian, H. H. Ding, B. J. Wang, Y. Yang, Y. H. Hao, X. P. Zhao, M. J. Lu, F. L. Gong, and D. L. Yang. 2006. N-terminal and C-terminal cytosine deaminase domain of APOBEC3G inhibit hepatitis B virus replication. World J. Gastroenterol. 12:7488-7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li, X. Y., F. Guo, L. Zhang, L. Kleiman, and S. Cen. 2007. APOBEC3G inhibits DNA strand transfer during HIV-1 reverse transcription. J. Biol. Chem. 282:32065-32074. [DOI] [PubMed] [Google Scholar]
- 28.Liddament, M. T., W. L. Brown, A. J. Schumacher, and R. S. Harris. 2004. APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr. Biol. 14:1385-1391. [DOI] [PubMed] [Google Scholar]
- 29.Luo, K., T. Wang, B. Liu, C. Tian, Z. Xiao, J. Kappes, and X. F. Yu. 2007. Cytidine deaminases APOBEC3G and APOBEC3F interact with human immunodeficiency virus type 1 integrase and inhibit proviral DNA formation. J. Virol. 81:7238-7248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mariani, R., D. Chen, B. Schrofelbauer, F. Navarro, R. Konig, B. Bollman, C. Munk, H. Nymark-McMahon, and N. R. Landau. 2003. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell 114:21-31. [DOI] [PubMed] [Google Scholar]
- 31.Martinez, M. A., M. Sala, J. P. Vartanian, and S. Wain-Hobson. 1995. Reverse transcriptase and substrate dependence of the RNA hypermutagenesis reaction. Nucleic Acids Res. 23:2573-2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mbisa, J. L., R. Barr, J. A. Thomas, N. Vandegraaff, I. J. Dorweiler, E. S. Svarovskaia, W. L. Brown, L. M. Mansky, R. J. Gorelick, R. S. Harris, A. Engelman, and V. K. Pathak. 2007. Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J. Virol. 81:7099-7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mbisa, J. L., W. Bu, and V. K. Pathak. 2010. APOBEC3F and APOBEC3G inhibit HIV-1 DNA integration by different mechanisms. J. Virol. 84:5250-5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miyagi, E., S. Opi, H. Takeuchi, M. Khan, R. Goila-Gaur, S. Kao, and K. Strebel. 2007. Enzymatically active APOBEC3G is required for efficient inhibition of human immunodeficiency virus type 1. J. Virol. 81:13346-13353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miyagi, E., F. Schwartzkopff, R. Plishka, A. Buckler-White, K. A. Clouse, and K. Strebel. 2008. APOBEC3G-independent reduction in virion infectivity during long-term HIV-1 replication in terminally differentiated macrophages. Virology 379:266-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Newman, E. N., R. K. Holmes, H. M. Craig, K. C. Klein, J. R. Lingappa, M. H. Malim, and A. M. Sheehy. 2005. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr. Biol. 15:166-170. [DOI] [PubMed] [Google Scholar]
- 37.Nguyen, D. H., S. Gummuluru, and J. Hu. 2007. Deamination-independent inhibition of hepatitis B virus reverse transcription by APOBEC3G. J. Virol. 81:4465-4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Opi, S., H. Takeuchi, S. Kao, M. A. Khan, E. Miyagi, R. Goila-Gaur, Y. Iwatani, J. G. Levin, and K. Strebel. 2006. Monomeric APOBEC3G is catalytically active and has antiviral activity. J. Virol. 80:4673-4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peng, G., K. J. Lei, W. Jin, T. Greenwell-Wild, and S. M. Wahl. 2006. Induction of APOBEC3 family proteins, a defensive maneuver underlying interferon-induced anti-HIV-1 activity. J. Exp. Med. 203:41-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosler, C., J. Kock, M. Kann, M. H. Malim, H. E. Blum, T. F. Baumert, and F. von Weizsacker. 2005. APOBEC-mediated interference with hepadnavirus production. Hepatology 42:301-309. [DOI] [PubMed] [Google Scholar]
- 41.Russell, R. A., M. D. Moore, W. S. Hu, and V. K. Pathak. 2009. APOBEC3G induces a hypermutation gradient: purifying selection at multiple steps during HIV-1 replication results in levels of G-to-A mutations that are high in DNA, intermediate in cellular viral RNA, and low in virion RNA. Retrovirology 6:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sasada, A., A. Takaori-Kondo, K. Shirakawa, M. Kobayashi, A. Abudu, M. Hishizawa, K. Imada, Y. Tanaka, and T. Uchiyama. 2005. APOBEC3G targets human T-cell leukemia virus type 1. Retrovirology 2:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmitt, K., M. S. Hill, A. Ruiz, N. Culley, D. M. Pinson, S. W. Wong, and E. B. Stephens. 2009. Mutations in the highly conserved SLQYLA motif of Vif in a simian-human immunodeficiency virus result in a less pathogenic virus and are associated with G-to-A mutations in the viral genome. Virology 383:362-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schumacher, A. J., G. Hache, D. A. Macduff, W. L. Brown, and R. S. Harris. 2008. The DNA deaminase activity of human APOBEC3G is required for Ty1, MusD, and human immunodeficiency virus type 1 restriction. J. Virol. 82:2652-2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Simon, J. H., T. E. Southerling, J. C. Peterson, B. E. Meyer, and M. H. Malim. 1995. Complementation of vif-defective human immunodeficiency virus type 1 by primate, but not nonprimate, lentivirus vif genes. J. Virol. 69:4166-4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Soros, V. B., W. Yonemoto, and W. C. Greene. 2007. Newly synthesized APOBEC3G is incorporated into HIV virions, inhibited by HIV RNA, and subsequently activated by RNase H. PLoS Pathog. 3:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vartanian, J.-P., A. Meyerhans, B. Asjo, and S. Wain-Hobson. 1991. Selection, recombination, and G—-A hypermutation of human immunodeficiency virus type 1 genomes. J. Virol. 65:1779-1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang, T., C. Tian, W. Zhang, P. T. Sarkis, and X. F. Yu. 2008. Interaction with 7SL RNA but not with HIV-1 genomic RNA or P bodies is required for APOBEC3F virion packaging. J. Mol. Biol. 375:1098-1112. [DOI] [PubMed] [Google Scholar]
- 49.Wang, X., P. T. Dolan, Y. Dang, and Y. H. Zheng. 2007. Biochemical differentiation of APOBEC3F and APOBEC3G proteins associated with HIV-1 life cycle. J. Biol. Chem. 282:1585-1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wei, X., J. M. Decker, H. Liu, Z. Zhang, R. B. Arani, J. M. Kilby, M. S. Saag, X. Wu, G. M. Shaw, and J. C. Kappes. 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 46:1896-1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wichroski, M. J., G. B. Robb, and T. M. Rana. 2006. Human retroviral host restriction factors APOBEC3G and APOBEC3F localize to mRNA processing bodies. PLoS Pathog. 2:e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wiegand, H. L., B. P. Doehle, H. P. Bogerd, and B. R. Cullen. 2004. A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. EMBO J. 23:2451-2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang, Y., F. Guo, S. Cen, and L. Kleiman. 2007. Inhibition of initiation of reverse transcription in HIV-1 by human APOBEC3F. Virology 365:92-100. [DOI] [PubMed] [Google Scholar]
- 54.Ying, S., X. Zhang, P. T. Sarkis, R. Xu, and X. Yu. 2007. Cell-specific regulation of APOBEC3F by interferons. Acta Biochim. Biophys. Sin. (Shanghai) 39:297-304. [DOI] [PubMed] [Google Scholar]
- 55.Yu, Q., D. Chen, R. Konig, R. Mariani, D. Unutmaz, and N. R. Landau. 2004. APOBEC3B and APOBEC3C are potent inhibitors of simian immunodeficiency virus replication. J. Biol. Chem. 279:53379-53386. [DOI] [PubMed] [Google Scholar]
- 56.Yu, Q., R. Konig, S. Pillai, K. Chiles, M. Kearney, S. Palmer, D. Richman, J. M. Coffin, and N. R. Landau. 2004. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat. Struct. Mol. Biol. 11:435-442. [DOI] [PubMed] [Google Scholar]
- 57.Zennou, V., and P. D. Bieniasz. 2006. Comparative analysis of the antiretroviral activity of APOBEC3G and APOBEC3F from primates. Virology 349:31-40. [DOI] [PubMed] [Google Scholar]
- 58.Zheng, Y. H., D. Irwin, T. Kurosu, K. Tokunaga, T. Sata, and B. M. Peterlin. 2004. Human APOBEC3F is another host factor that blocks human immunodeficiency virus type 1 replication. J. Virol. 78:6073-6076. [DOI] [PMC free article] [PubMed] [Google Scholar]