

Abstract
Murine cytomegalovirus (MCMV) functions interfere with protein trafficking in the secretory pathway. In this report we used Δm138-MCMV, a recombinant virus with a deleted viral Fc receptor, to demonstrate that MCMV also perturbs endosomal trafficking in the early phase of infection. This perturbation had a striking impact on cell surface-resident major histocompatibility complex class I (MHC-I) molecules due to the complementary effect of MCMV immunoevasins, which block their egress from the secretory pathway. In infected cells, constitutively endocytosed cell surface-resident MHC-I molecules were arrested and retained in early endosomal antigen 1 (EEA1)-positive and lysobisphosphatidic acid (LBPA)-negative perinuclear endosomes together with clathrin-dependent cargo (transferrin receptor, Lamp1, and epidermal growth factor receptor). Their progression from these endosomes into recycling and degradative routes was inhibited. This arrest was associated with a reduction of the intracellular content of Rab7 and Rab11, small GTPases that are essential for the maturation of recycling and endolysosomal domains of early endosomes. The reduced recycling of MHC-I in Δm138-MCMV-infected cells was accompanied by their accelerated loss from the cell surface. The MCMV function that affects cell surface-resident MHC-I was activated in later stages of the early phase of viral replication, after the expression of known immunoevasins. MCMV without the three immunoevasins (the m04, m06, and m152 proteins) encoded a function that affects endosomal trafficking. This function, however, was not sufficient to reduce the cell surface expression of MHC-I in the absence of the transport block in the secretory pathway.
Herpesviruses are well known to interfere with major histocompatibility complex class I (MHC-I) molecules in order to ensure evasion from immune recognition. A majority of evidence so far indicates that they target MHC-I maturation events and MHC-I trafficking in the secretory pathway (33), although evidence exists suggesting that herpesviruses could also interfere with MHC-I functions in endosomal pathways (8). Murine cytomegalovirus (MCMV), a member of the herpesvirus family, dedicates a substantial part of its genome to encoding nonessential genes for the modulation of cellular functions (40), including MHC-I trafficking in the secretory pathway (24, 27, 45, 48, 49, 52). All known immune evasion functions encoded by MCMV are based on a direct interaction of viral gene products with MHC-I complexes in the secretory pathway. The egress of nascent MHC-I complexes to the cell surface of MCMV-infected cells is abolished as a consequence of their retention in the endoplasmic reticulum (ER)-cis-Golgi intermediate compartment (ERGIC) by the m152 gene product (10, 19, 24, 52, 56) as well as redirection of those that escape into the Golgi compartment toward late endosomes (LEs) for degradation by the m06 MCMV gene product (45). These effects are opposed by gp34, a product of the MCMV m04 gene, which associates with MHC-I complexes and reaches the cell surface (24, 27).
The loss of MHC-I from the cell surface is an expected consequence of the activity of m152 and m06, which act in the secretory pathway. The level of cell surface MHC-I is substantially reduced at later times of infection (10, 19, 24, 48, 52), and cells stably transfected with either the m152 or m06 gene do not display MHC-I at the cell surface (20, 24). If the loss of MHC-I from the cell surface is a consequence of the prevented egress from the secretory pathway, then the cell surface loss should follow the kinetics of the constitutive internalization of MHC-I complexes in the endosomal pathway. Given that the constitutive internalization is the net result of cell surface supply from the secretory pathway, endocytic uptake, and endocytic recycling, it is a slow process that occurs in normal fibroblasts at a rate of ∼6 to 8% per hour (36). Therefore, the effect of MCMV immunoevasins on cell surface MHC-I should be expected at later times of infection. However, several reports demonstrated that the level of MHC-I surface expression was already reduced in the early phase of infection (10, 45, 48, 52). Thus, it would be reasonable to expect that MCMV contributes with a function that causes the accelerated retrieval of cell surface-resident MHC-I complexes.
In this report we demonstrate that MCMV perturbs endosomal trafficking very early in infection by acting on distal parts of early endosome (EE) route and affecting the trafficking of both clathrin-dependent and clathrin-independent cargoes. Clathrin-dependent cargo does not share primary endocytic carriers with MHC-I proteins (12, 14), which enter the cell via the nonclathrin Arf6-associated endocytic carriers (12, 14, 41, 42, 53), but they meet in the proximal part of the common early endocytic route and redirect to distal endocytic carriers around the cell center (12, 14). The perturbation of the distal part of the EE route has dramatic consequences on MHC-I, since it supplements the viral mechanisms that act in the secretory pathway. The net result of this perturbation is a complete loss of MHC-I molecules from the cell surface.
MATERIALS AND METHODS
Cell lines, viruses, and infection conditions.
Primary murine embryonic fibroblasts (MEFs) were generated from BALB/c mice and bred in minimal essential medium (MEM) supplemented with 5% (vol/vol) fetal bovine serum (FBS), 2 mM l-glutamine, 100 mg/ml streptomycin, and 100 U/ml penicillin. All reagents were obtained from Gibco (Grand Island, NY). The cells were grown in petri dishes as adherent cell lines and used for infection after three in vitro passages when they were 90% confluent.
The following recombinant MCMVs were used: Δm138-MCMV (ΔMC95.15), with a deletion of the fcr1 (m138) gene (9); Δie1-MCMV, with a deletion of exon 4 of the ie1 gene (13); Δie2-MCMV, with a deletion of the ie2 (m128) gene (38); Δie3-MCMV, with a deletion of the ie3 gene (2); and Δm4m6m152-MCMV, with deletions of three known MCMV immunoevasins that affect MHC-I maturation (52). MCMV wild-type (wt) strain Smith (ATCC VR-194; American Type Culture Collection [ATCC]) and MCMV deletion mutants were propagated on third-passage BALB/c MEFs and purified by sucrose gradient centrifugation, as described previously (6). Δie3-MCMV was grown on the complementing cell line NIH 3T3 BAM25 (2).
MEFs were infected at a multiplicity of infection (MOI) of 10 with an enhancement of infectivity by centrifugation (5), and the efficiency of infection was monitored by the detection of the intracellular immediate-early 1 (ie1) protein. The selective expression of MCMV immediate-early (IE) proteins and controlled transition to the early (E) phase after infection with Δm138-MCMV were achieved with a protocol described previously (10, 48). Viral DNA replication was determined as described previously by Keil et al. (25).
Reagents and antibodies.
Monoclonal antibodies (MAbs) to Kd (clone MA-215) (23), Dd (clone 34-5-8s [ATCC HB-102]), transferrin (Tf) receptor (TfR) (clone R17 217.1.3 [ATCC TIB-219]), the MCMV ie1 protein (clone CRO101) (11, 30), and lysobisphosphatidic acid (LBPA) (clone 6C4) (28) were used as hybridoma culture supernatants for immunofluorescence or as semipurified ascites for immunoprecipitation. MAb to actin was obtained from Sigma-Aldrich Chemie GmbH (Germany), MAbs to GM130 and Lamp1 were obtained from Becton Dickinson & Co. (San Jose, CA), MAb to early endosomal antigen 1 (EEA1) was obtained from Zymed Laboratories (San Francisco, CA), and MAbs to Rab5, Rab7, and Rab11 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Alexa488- and Alexa555-conjugated anti-mouse IgG2a, anti-mouse IgG2b, anti-mouse IgG1, anti-rat IgG, anti-rabbit Ig, anti-chicken Ig, transferrin, dextran, and epidermal growth factor (EGF) were all obtained from Molecular Probes-Invitrogen (Eugene, OR). LysoTracker Red DND-99, anti-mouse fluorescein isothiocyanate (FITC)-Ig, and streptavidin-FITC were all obtained from Molecular Probes-Invitrogen (Eugene, OR). Transferrin-biotin, horseradish peroxidase (HRP)-conjugated anti-mouse IgG, brefeldin A (BFA), cycloheximide (CH), actinomycin D (Act D), and nocodazole were obtained from Sigma-Aldrich Chemie GmbH (Germany). Streptavidin-peroxidase conjugate was obtained from Roche Diagnostics GmbH (Mannheim, Germany), and HRP-conjugated anti-rabbit IgG was obtained from Santa Cruz Biotechnology (Santa Cruz, CA).
Cell surface expression and flow cytometry.
Uninfected or MCMV-infected fibroblasts were collected by short trypsin treatment and incubated at 4°C for 30 to 60 min with either fluorochrome-conjugated or unconjugated primary reagents in phosphate-buffered saline (PBS) containing 10 mM EDTA, HEPES (pH 7.2), 0.1% NaN3, and 2% fetal calf serum (FCS). Unbound reagents were removed by three washes with cold PBS, and when required, the cells were incubated with a fluorochrome-conjugated secondary reagent for 30 min at 4°C. After three washes with PBS, cells were analyzed by flow cytometry using a FACSCalibur flow cytometer (Becton Dickinson & Co., San Jose, CA). Dead cells were excluded by propidium iodide, and a total of 10,000 cells was acquired. The fluorescence signal was determined as the mean fluorescence intensity (MFI) after subtraction of the background fluorescence (ΔMFI).
Immunofluorescence and confocal analysis.
Cells grown on coverslips were fixed with 4% formaldehyde (20 min at room temperature [RT]) and permeabilized with 0.5% Triton X-100 (7 min at RT) or 0.2% Tween (15 min at RT). For Rab protein expression, cells were fixed and permeabilized with ice-cold methanol (5 min). After permeabilization, cells were incubated with fluorochrome-conjugated or unconjugated primary immunofluorescence reagents for 60 min. Unbound reagents were washed with PBS, and cells were either embedded or incubated for 60 min with an appropriate fluorochrome-conjugated secondary reagent. After three washes in PBS, labeled cells were embedded in Mowiol and analyzed by confocal microscopy. Images were obtained by using an Olympus Fluoview FV300 confocal microscope (Olympus Optical Co., Tokyo, Japan) with 60× Plan Apo objectives. To visualize the level of colocalization, 8 to 10 cells per experimental condition were randomly selected on the same coverslip among those that were well spread and showed a well-resolved pattern. Images of single cells were acquired at the same magnification, exported in a TIFF format, and processed by Fluoview, version 4.3 FV 300 (Olympus Optical Co., Tokyo, Japan). Presentation of figures was accomplished with Adobe Photoshop (San Jose, CA).
Internalization assays.
The internalization of MHC-I, Lamp1, and TfR was analyzed by a modified internalization assay as described previously (8, 42). Internalization was performed on adherent cells grown either in six-well tissue culture plates for flow cytometry or on coverslips for immunofluorescence. Prior to their labeling, cells were washed three times with PBS. Cell surface proteins were labeled with MAb reagents at 4°C for 60 min, unbound MAbs were removed by the three washes with PBS, and internalization was performed at 37°C. Cells were detached by short trypsinization, a procedure which did not affect the association of MAbs with cell surface proteins (data not shown). Uninternalized MAbs bound to cell surface proteins were determined by flow cytometry using appropriate fluorochrome-conjugated secondary reagents. Internalized MAbs were visualized inside the adherent cells by immunofluorescence after a short low-pH (1 min at pH 2.2) treatment prior to fixation to remove MAbs from the cell surface.
The internalization of Tf on adherent fibroblasts was monitored by immunofluorescence and Western blot (WB) analysis. After 60 min of starvation in Tf-free medium, cells were exposed to Alexa555-Tf or Tf-biotin for 30 min at 37°C to gain continuous internalization and load Tf-containing intracellular compartments. The steady-state cell surface Tf-binding capacity was too low to ensure sufficient signal for quantitative flow cytometric analysis of internalization. Thus, Tf uptake was analyzed by the binding of Tf-biotin to the cell surface at 4°C, and internalization was achieved by incubation at 37°C in Tf-free medium. Uninternalized Tf-biotin was removed by a short low-pH treatment, and internalized Tf-biotin was quantitatively determined by WB analysis.
The internalization of EGF was monitored after 60 min of starvation in FCS-free medium followed by continuous exposure to Alexa555-EGF or Alexa488-EGF at 37°C. The internalization of Alexa488-EGF was terminated after 60 min by the replacement of the medium, and cells were chased for different periods of time prior to flow cytometric analysis. For confocal analysis, uninternalized Alexa555-EGF was removed by the low-pH treatment prior to processing.
The kinetic model for analysis of the internalization rate (IR) was based on parameters similar to those used for the transferrin receptor (7). From the half-life (t1/2) values, we calculated the first-order rate constant for internalization (ki), which is equal to ln(2)/t1/2.
Recycling assays.
The recycling of MHC-I molecules was determined by a modification of an assay described previously (53). MHC-I molecules were labeled with MAbs at 4°C for 60 min, and MHC-I-MAb complexes were internalized for 1 to 4 h at 37°C. After internalization, the remaining cell surface MAbs were removed by a short low-pH treatment, and cells were incubated at 37°C with Alexa488-conjugated secondary antibodies to capture recycled MHC-I-MAb complexes. The fluorescence signal was quantified by flow cytometry (ΔMFI). The percentage of recycled MHC-I was calculated by the division of ΔMFI intensities of recycled and internalized MHC-I proteins.
In order to distinguish between internalized and recycled MHC-I-MAb complexes, the recycling assay was performed at 16°C, a temperature at which the progression of early endosomes (EE) and recycling from EE are inhibited (49). Recycled MHC-I-MAb complexes were captured with Alexa488-anti-mouse IgG2a or IgG2b (green fluorescence), and internalized complexes that did not recycle were stained with Alexa555-anti-mouse IgG2a or IgG2b (red fluorescence) after fixation and permeabilization. Cells were analyzed by confocal microscopy.
Tf recycling was determined after Tf internalization and its loss from cells by recycling (50). Cells were starved in medium without Tf for 1 h and exposed to Tf-biotin for 60 min at 37°C. Uninternalized Tf-biotin was removed by washing (three times in medium with unlabeled Tf), and the recycling of Tf-biotin was determined by its loss from cells after incubation at 37°C in medium containing 200 μg/ml of unlabeled Tf (49, 50). Tf-biotin in cells was determined by WB analysis using streptavidin-peroxidase (POD).
Western blotting.
Cellular extracts for WB analysis were prepared in SDS sample buffer, separated by reducing SDS-PAGE, and blotted onto a polyvinylidene difluoride (PVDF) WB membrane (Roche Diagnostics GmbH, Mannheim, Germany) at 60 to 70 V for 1 h. Membranes were washed in Tris-HCl-buffered saline (TBS) (50 mM, pH 7.5) and incubated in 1% blocking reagent (Roche Diagnostics GmbH, Mannheim, Germany) for 2 h, followed by a 1-h incubation with MAbs, three cycles of washing (TBS with 0.1% Tween 20 [TBS-T buffer]), and a 45-min incubation with peroxidase-conjugated secondary reagent diluted in TBS buffer containing 0.5% blocking reagent. After being washed three times with TBS-T buffer (pH 7.5), membranes were enveloped into plastic wrap, incubated with Super Signal West Dura extended-duration substrate (Pierce Chemical Co., Rockford, IL) for 1 min, and exposed to BioMax film (Kodak). Signals of bands on WB membranes were quantified with a calibrated imaging densitometer (GS-710; Bio-Rad).
Cell surface biotinylation and immunoprecipitation.
Adherent cells were washed three times with PBS in tissue culture dishes and resuspended in biotinylation buffer (10 mM Na2B4O7·10H2O, 5 mM NaCl, 2 mM CaCl2, and 1 mM MgCl2 [pH 8.8]). The biotinylation reaction was initiated by the addition of 0.15 mg/ml d-biotinoyl-ɛ-amidicaproic acid-N-hydroxysuccinimide ester (Roche Diagnostics GmbH, Mannheim, Germany) at room temperature and stopped after 20 min with cold stop solution (50 mM NH4Cl in PBS) for 5 min on ice. After three washes with 10% tissue culture medium (supplemented with FBS), cells were incubated at 37°C in fresh medium. After the indicated times, cells were collected, washed in ice-cold PBS three times, and lysed in a buffer containing 50 mM Tris-Cl (pH 8.0), 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, 0.02% NaN3, and 2 mM phenylmethylsulfonyl fluoride (PMSF). Supernatants of cellular lysates were precleared with 30 μl of a protein A-Sepharose slurry (Amersham Pharmacia Biotech). Quantitative immunoprecipitation of MHC-I molecules was performed with the ascitic fluid of appropriate MAbs (3 μl). Immune complexes were retrieved with protein A-Sepharose (50 μl of a 50% slurry), and Sepharose beads were washed as described previously (56). Immune complexes were eluted at 96°C by incubation with sample buffer and analyzed by 13% SDS-PAGE under nonreducing conditions. After electrophoresis, polyacrylamide gels were blotted onto a PVDF membrane and probed with 100 mU/ml of streptavidin-POD as described above.
RESULTS
Cell surface MHC-I molecules are downregulated in the early phase of infection with MCMV.
Given that cell surface MHC-I molecules are downregulated at later times of MCMV infection (10, 19, 24, 48, 52), we first determined the kinetics of their loss from the cell surface. We used the recombinant virus Δm138-MCMV in order to avoid unspecific interactions of the m138 protein, which strongly binds the Fc portion of immunoglobulins (9), with monoclonal antibody (MAb) reagents. The cell surface level of MHC-I (Dd and Kd) was unchanged within the first 5 h of infection (Fig. 1 A), which is followed by their rapid loss, with a rate of approximately 0.33 ± 0.02 h−1, resulting in a reduction to ∼60% of the control level at 6 h postinfection (p.i.) and to ∼30% at 8 h p.i. (Fig. 1A). The cell surface loss of MHC-I molecules coincided with the appearance of their intracellular accumulation in perinuclear vesicles (Fig. 1B). Effects similar to those described above for Δm138-MCMV were also observed after infection with wild-type (wt) MCMV, including the degree of cell surface loss (Fig. 1C).
FIG. 1.

Loss of cell surface MHC-I in the early phase of MCMV infection. (A) Cell surface expression of Kd molecules determined on murine embryonic fibroblasts (MEFs) by flow cytometry after infection with Δm138-MCMV and after treatment with cycloheximide (CH) or brefeldin A (BFA). Results are presented as percentages of initial expression. Data are averages of data from four independent experiments ± standard deviations (SD). (B) Detection of Kd by indirect immunofluorescence on PFA-fixed (cell surface) and PFA-fixed and Triton X-100-permeabilized (intracellular) cells. Bars, 10 μm. (C) Cell surface level of Kd after 8 h of infection with wt MCMV, Δm138-MCMV, Δie1-MCMV, Δie2-MCMV, and Δie3-MCMV. (D) Cell surface levels of Dd and Kd after infection with Δm138-MCMV under conditions of selective and enhanced expression of immediate-early (IE) and early (E) genes. The protocol used for the definition of the period of E gene transcription (10) required for the synthesis of the protein(s) that downregulates cell surface MHC-I is presented above the data. (Top) Infection in the presence of CH results in enhanced transcription of IE genes, which is followed by selective IE protein synthesis after the replacement of CH with actinomycin D (Act D). (Bottom) After the removal of CH, the synthesized IE proteins activate the transcription of E genes until Act D is added. (E) Effect of ganciclovir (GC) (8 μM) on cell surface levels of Kd (left axis and circles) and DNA replication (right axis and lines) in Δm138-MCMV-infected fibroblasts. The incorporation of [3H]thymidine was determined in 2-h intervals.
MCMV immunoevasins that arrest MHC-I transport in the secretory pathway and abolish the supply of the cell surface (10, 19, 24, 45, 52, 56) may lead to the constitutive endocytic clearance of preformed MHC-I from the plasma membrane. However, the rate of loss of MHC-I from the cell surface was much lower (t1/2 of >24 h) when cells were treated with either cycloheximide (CH) or brefeldin A (Fig. 1A), drugs that prevent export from the secretory pathway. This finding suggests that the viral interference mechanism is distinct from the transport block of newly synthesized MHC-I proteins.
The cell surface MHC-I proteins were lost to a similar degree after infection with mutant viruses lacking either the immediate-early 1 (ie1) or ie2 gene (Fig. 1C). However, after infection with Δie3-MCMV, the cell surface level of MHC-I even increased (Fig. 1C), indicating that the function that rapidly downregulates cell surface MHC-I is related to the effect of either the ie3 protein or viral early functions that are under ie3 regulation. The loss of cell surface MHC-I in cells treated for 3 h with CH followed by 8 h of treatment with actinomycin D (Act D), conditions for an enhanced expression of IE proteins (10, 25), indicates that neither IE gene products nor transcripts that occur in the IE phase affect cell surface-resident MHC-I. However, when CH was replaced with inhibitor-free medium prior to the addition of Act D, a procedure that allows limited and enhanced early (E) gene expression (10, 48), cell surface-resident MHC-I proteins were reduced under conditions of at least 3 h of E gene expression (Fig. 1D). Given that viral E genes that block transport in the secretory pathway require only 30 min of E gene expression (10, 48), these data suggest that the viral interference mechanism is not related to the function of these genes but rather to viral functions that are expressed later. Cell surface MHC-I was downregulated at a similar rate in infected cells in which viral DNA replication was inhibited with ganciclovir (Fig. 1E) or foscarnet (data not shown), indicating that the viral interference mechanism does not require the function of viral DNA replication machinery and expression of late genes.
MHC-I molecules were cleared from the cell surface with kinetics that differ from those of clathrin-dependent mechanisms.
In order to display MHC-I complexes that are relocated from the cell surface, we labeled cell surface MHC-I molecules of Δm138-MCMV-infected fibroblasts with MAbs at 4 h p.i., when their cell surface level was still unchanged, and monitored their endosomal trafficking (Fig. 2 A). This assay is widely used in internalization studies (3, 6, 12, 41, 42, 53). Labeled MHC-I complexes, both Kd (Fig. 2A) and Dd (data not shown), internalized with similar kinetics in Δm138-MCMV-infected cells, with average internalization rates (IRs) of 0.33 ± 0.018 h−1 for Kd and 0.315 ± 0.012 h−1 for Dd (t1/2 of ∼2 h). This rate was 4- to 6-fold higher than that in uninfected fibroblast (Fig. 2A), in which the average IRs were 0.077 ± 0.012 h−1 for Kd (t1/2 of ∼9 h) and 0.058 ± 0.01 h−1 for Dd (t1/2 of ∼12 h). After 4 h of internalization, approximately 20% of MHC-I remained at the surface of Δm138-MCMV-infected cells (Fig. 2A).
FIG. 2.

Internalization of cell surface MHC-I and clathrin-dependent cargo in uninfected and Δm138-MCMV-infected fibroblasts. (A) The loss of Kd from the cell surface was determined by an internalization assay. The experimental design is presented on the right. Cell surface Kd of uninfected and Δm138-MCMV-infected (at 4 h p.i.) MEFs was labeled with MAbs for 60 min at 4°C, and the remaining MAbs at the cell surface were determined by flow cytometry after the indicated periods of incubation at 37°C (internalization). Results are presented as a percentage of initial expression ± SD from six independent experiments. (B) Intracellular distribution of cell surface-labeled Kd after 4 and 8 h of internalization in Δm138-MCMV-infected MEFs. Internalized Kd-MAb complexes were determined on permeabilized cells with Alexa555-anti-mouse IgG2b. (C) Internalization rate (IR) and recycling rate (RR) of TfR in L-Ld fibroblasts, determined by anti-TfR MAb (R17) and by Tf-biotin or Alexa488-Tf. The rates were calculated from t1/2 values. Results represent the means ± SD of data from six independent experiments. (D) Tf uptake in uninfected and Δm138-MCMV-infected MEFs determined by binding of Tf-biotin to cell surface TfRs at 4°C and internalization at 37°C for the indicated times. Uninternalized Tf-biotin was removed by low-pH treatment, and the level of intracellular Tf-biotin was determined by Western blotting and quantified by densitometry; black bars represent uninfected cells, and gray bars represent infected cells. (E) Confocal images of internalized TfR in uninfected and Δm138-MCMV-infected MEFs after labeling of cell surface TfR with MAbs at 4°C and internalization at 37°C. Bars, 10 μm.
Internalized MHC-I-MAb complexes were first found dispersed in cytoplasmic vesicles, and after 2 h, they concentrated in the perinuclear area (data not shown). After 4 h of internalization (Fig. 2B), a majority of internalized MHC-I-MAb was found accumulated in enlarged perinuclear vesicles. Although many of these vesicles were concentrated in the juxtanuclear area, internalized MHC-I complexes could be found accumulated in the juxtanuclear tubulovesicular endosomes only after prolonged internalization (8 h of internalization) (Fig. 2B).
In order to rule out the possibility that the binding of MAbs changes the rate or the route of endosomal trafficking, we performed a control experiment by using two indicators of transferrin (Tf) receptor (TfR) trafficking. Our data (Fig. 2C) demonstrate that the IR and recycling rate (RR) of TfR are indistinguishable when TfR was labeled either with MAbs or with biotinylated and fluorochrome-conjugated Tf, respectively.
Although the IR of MHC-I was 4- to 6-fold higher in Δm138-MCMV-infected cells than in uninfected fibroblasts, it was still much lower than the IR of the clathrin-dependent cargo. Given that the cell surface level of TfR was low on MEFs, we compared the uptakes of Tf-biotin in uninfected and infected cells. Our data demonstrate that the rates of uptake of Tf-biotin were similar in infected and uninfected cells (Fig. 2D), indicating that the clathrin-dependent process is functional in the infected cell. However, we observed a delay of TfR accumulation in the juxtanuclear recycling compartment, suggesting a disturbance of TfR trafficking (Fig. 2E). Taken together, our data indicate that MHC-I complexes are removed from the surface of infected cells with kinetics that differ from those of clathrin-dependent mechanisms.
Internalized MHC-I molecules are retained in a perinuclear early endosomal compartment that is insensitive to brefeldin A.
In order to characterize endosomes with retained MHC-I, we performed a colocalization analysis of internalized MHC-I in Δm138-MCMV-infected cells with a panel of well-known endosomal markers (Fig. 3). A majority of internalized MHC-I localized in the enlarged perinuclear vesicles that were positive for early endosomal antigen 1 (EEA1) (Fig. 3Aa) and TfR (Fig. 3Ab), indicating that these vesicles represent a transitional stage of “classical” EEs (12, 14, 49). A majority of the MHC-I-loaded vesicles were found around the cell center in the area that does not contain membranes positive for the late endosomal (LE) markers lysobisphosphatidic acid (LBPA) (Fig. 3Ad) and ganglioside M1 (GM1) (Fig. 3Ae) (28, 39). Very little colocalization with these markers was observed, suggesting that MHC-I molecules either do not reach or do not stay long in the predegradative LEs. In contrast, a colocalization of internalized MHC-I with Lamp1, a marker of limiting LE membranes, was found in the perinuclear localization typical for LEs but also in the juxtanuclear area (Fig. 3Ac). Given that Lamp1 recirculates between the LEs and plasma membrane, through EEs (28, 47), these data suggest that Lamp1 also arrests with MHC-I. Taken together, the colocalization analysis indicates that internalized MHC-I molecules are retained in the juxtanuclear early endosomal vesicles that are in a transitional stage before their fusion or maturation into LEs. This is also supported by the lack of their colocalization with LysoTracker Red DND-99 (Fig. 3Af), a marker of acidic endosomes (lysosomes).
FIG. 3.

Characterization of the MHC-I-loaded perinuclear endosomal compartment in Δm138-MCMV-infected cells. (A) Colocalization of internalized MHC-I with early and late endosomal markers. At 4 h p.i., MHC-I molecules were labeled with MAbs and internalized for an additional 4 h. Internalized MHC-I-MAb complexes were stained with Alexa555-anti-mouse IgG2b secondary reagent (red fluorescence [a to d]) and colocalized with the early endosomal markers EEA1 (Alexa488-anti-chicken IgG [green fluorescence]) (a) and TfR (Alexa488-anti-rat IgG [green fluorescence]) (b) and the late endosomal markers Lamp1 (Alexa488-conjugated anti-rat IgG [green fluorescence]) (c) and LBPA (Alexa488-conjugated anti-mouse IgG1 [green fluorescence]) (d). Also shown is the colocalization of internalized MHC-I complexes (stained with Alexa488-anti-mouse IgG2b [green fluorescence]) with GM1 (stained with Alexa555-CTxB [red fluorescence]) (e) and fluid-phase internalized LysoTracker Red DND-99-labeled endosomes (red fluorescence) (f). (B) Internalized MHC-I-MAb complexes (red fluorescence) were colocalized with the Golgi marker GM130 (green fluorescence) in untreated cells or in cells treated for an additional 60 min with nocodazole (4 μM) and BFA (10 μg/ml). Bars, 10 μm.
MHC-I-loaded endosomes partially intertwined with the Golgi cisternae in the pericentriolar area and displayed a pattern of colocalization with the Golgi marker GM130 (Fig. 3B). The disruption of the microtubular network by nocodazole dispersed both MHC-I-loaded vesicles and the Golgi cisternae throughout the cytosol but did not lead to their complete separation (Fig. 3B). Therefore, to exclude the fusion of these compartments, we treated Δm138-MCMV-infected cells with brefeldin A (BFA) after the internalization of MHC-I complexes. BFA treatment (34), at a concentration that dispersed the Golgi compartment (Fig. 3B), did not disperse juxtanuclear vesicles loaded with internalized MHC-I (Fig. 3B), indicating that MHC-I retention endosomes did not fuse with the Golgi compartment. In addition, BFA treatment caused the pericentriolar concentration and tubulation of MHC-I-loaded endosomes (Fig. 3B), suggesting that the BFA-sensitive machinery contributes to the maintenance of the MHC-I-loaded endosomes in the vesicular form.
MCMV perturbs recycling from early endosomal compartments.
The increased internalization rate and retention of MHC-I in Δm138-MCMV-infected cells may be associated with the reduced capacity for recycling from the retention endosomes. Thus, we next determined the efficiency of recycling from MHC-I-loaded endosomes (53). In uninfected fibroblasts, 30% ± 8% of internalized MHC-I was returned to the cell surface with a t1/2 of 16 ± 3 min (Fig. 4 A). However, in Δm138-MCMV-infected cells, the recycling efficiency was reduced to 12% ± 3%, and the half-value was reached after 45 ± 12 min (Fig. 4A). These data demonstrate that MHC-I molecules could recycle from the retention vesicles but that their recycling was reduced and delayed. Although more MHC-I was internalized in Δm138-MCMV-infected cells than uninfected cells, these data suggest that the formation of the recycling domain in the retention endosomes was limited. Prolonged internalization did not increase the recycling efficiency in either uninfected or infected cells (data not shown), indicating that the capacity of the recycling domain is determined by components of the recycling machinery and not by the amount of internalized cargo.
FIG. 4.
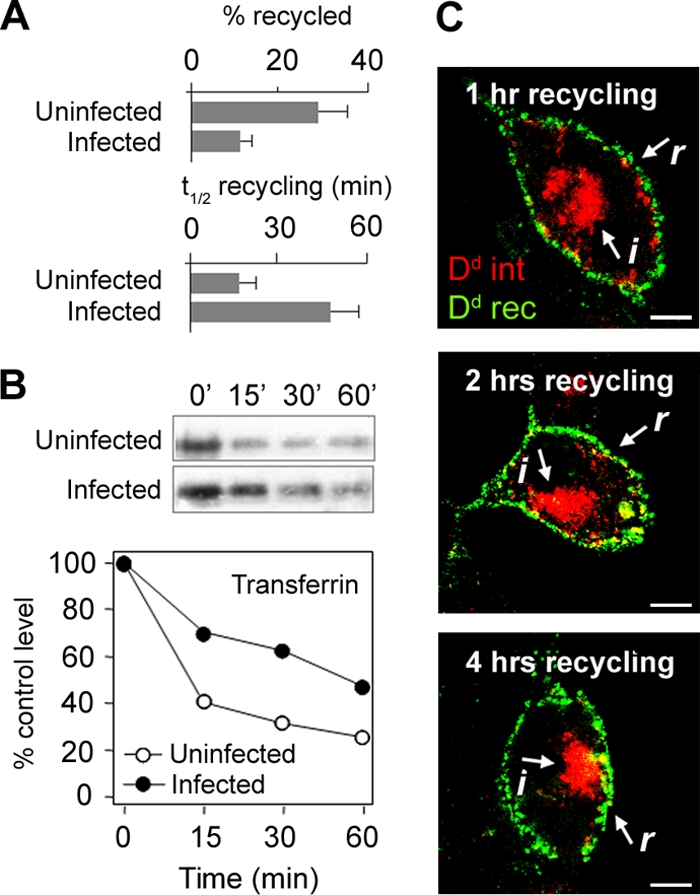
Recycling of MHC-I molecules in MCMV-infected cells. (A) Recycling efficiencies, expressed as the percentage recycled, and recycling kinetics, expressed as the t1/2 required to reach the maximal signal, were determined for uninfected and Δm138-MCMV-infected (6 h p.i.) fibroblasts after 1 h of internalization of Kd-MAb complexes. Recycled complexes were captured by Alexa488-conjugated anti-mouse IgG2b, and fluorescence intensity was determined by flow cytometry. (B) Recycling of internalized Tf determined by Western blot detection of intracellular Tf-biotin. The loss of Tf-biotin from cells occurs by recycling. Quantification was made by densitometry of blots, and results are expressed as a percentage of Tf-biotin after internalization (control value). Shown are data from one representative experiment out of four experiments. (C) Recycling of internalized (i) Dd-MAb (red fluorescence) from the juxtanuclear retention compartment at 16°C. Recycled (r) Dd-MAb was captured by Alexa488-conjugated anti-mouse IgG2a (green fluorescence) and stacked in subplasmalemmal endosomes at 16°C. Bars, 10 μm.
Given that internalized MHC-I colocalized with steady-state (Fig. 3A) and internalized (data not shown) TfR in perinuclear endosomes, we next examined whether Tf recycling is also altered in infected cells. Internalized Tf rapidly recycled from uninfected cells, resulting in the loss of internalized Tf-biotin from cells at a t1/2 of ∼10 min (Fig. 4B). In contrast, in Δm138-MCMV-infected cells, the loss of internalized Tf-biotin was delayed (t1/2 of ∼45 min), indicating that TfR recycling is also reduced in infected fibroblasts. This supports our previous conclusion that MCMV infection affects the formation of recycling domains in early endosomes.
In order to distinguish whether internalized MHC-I molecules are able to reach juxtanuclear recycling endosomes in Δm138-MCMV-infected cells, we performed recycling at 16°C (Fig. 4C). This temperature is permissive for recycling from juxtanuclear recycling endosomes but not from EEs (49). In addition, at this temperature, EE trafficking was inhibited, and endocytosed cargo was stacked in subplasmalemmal vesicles, also including recycled MHC-I complexes, which enabled a distinction between recycled (r) and internalized (i) cargoes (Fig. 4C). At 16°C, approximately half (data not shown) of the internalized MHC-I was recycled within the first hour (detected as subplasmalemmal cargo), and the rest remained inside the cells, even after 4 h of recycling (Fig. 4C). These data indicate that internalized MHC-I molecules are able to reach the recycling domain, which has characteristics of the juxtanuclear recycling endosomes.
MCMV retards progression of MHC-I and clathrin-dependent cargo toward the degradative late endosomal compartments.
The retention of internalized MHC-I in the perinuclear endosomes also suggests the reduced maturation of the endolysosomal domain and its progression toward LEs. A majority of internalized MHC-I did not localize in Lamp1- and LBPA-positive (Fig. 3Ac and d) LE compartments, which argues against a massive membrane flow toward the endolysosomal route. In order to estimate the rate of progression toward LEs, we first determined the degradation rate of cell surface MHC-I in infected cells. Cell surface proteins were biotinylated at 4 h p.i., and the intracellular level of biotin-labeled MHC-I molecules was determined by immunoprecipitation. In uninfected fibroblasts (Fig. 5 A), biotinylated Kd (and Dd [data not shown]) remained in the cells for a long period of time (t1/2 of ∼12 h), which gives a degradation rate of ∼0.06 h−1. This is in a range similar to the internalization rate determined by MAb labeling (Fig. 2A) and indicates that MAb labeling did not facilitate degradation. Biotinylated MHC-I degraded faster in Δm138-MCMV-infected fibroblasts than in uninfected cells (Fig. 5A), and very little biotinylated Dd and Kd were found at 20 h postinfection (Fig. 5B). However, their degradation did not correlate with internalization. The degradation rate of Kd was determined to be ∼0.17 h−1 (Fig. 5A), which is significantly higher than that in uninfected cells but still half of the internalization rate (average of 0.315 h−1). This indicates that after fast removal from the cell surface and retention in the early endosomal compartment, MHC-I complexes have restricted entry into the degradative route.
FIG. 5.

Degradation kinetics of cell surface MHC-I and progression of clathrin-dependent cargo molecules into late endosomes. (A) Degradation kinetics of cell surface Kd molecules were determined by cell surface biotinylation of uninfected and Δm138-MCMV-infected (at 4 h p.i.) cells and immunoprecipitation of Kd from cytoplasmic extracts. Immunoprecipitates were quantified by a densitometric analysis of blots. Shown are data representative of data from four experiments. HC, heavy chain. (B) Immunoprecipitates of Dd and Kd from uninfected and Δm138-MCMV-infected cells 20 h after cell surface labeling. (C) Colocalization of internalized of Alexa555 (red fluorescence)- or Alexa488 (green fluorescence)-EGF (60 min) with steady-state TfR, LBPA, and Lamp1 and with cointernalized LysoTracker Red DND-99 (DND) in uninfected and Δm138-MCMV-infected (7 h p.i.) cells. (D) Cointernalized Kd-MAb and Alexa555-EGF or Lamp1 in Δm138-MCMV-infected cells (4 to 8 h p.i.). (E) Degradation kinetics of internalized EGF. Uninfected and Δm138-MCMV-infected (7 h p.i.) cells were loaded for 60 min with Alexa488-EGF, and the fluorescence intensity inside the cells was determined by flow cytometry after the indicated periods of chase. Results are presented as percentages of initial expression.
To examine the degradative routes in Δm138-MCMV-infected cells, we cointernalized MHC-I with two clathrin-dependent cargo molecules that travel into LEs via either dynamic (EGF) or static (Lamp1) early endosomes (4, 31). In infected cells, internalized EGF dispersed into TfR-negative, Lamp1-positive, and DND-99-positive perinuclear vesicles, as in uninfected cells (Fig. 5C), and did not colocalize with internalized MHC-I (Fig. 5D). This finding suggests that MCMV does not alter the formation of dynamic EEs and that the MHC-I retention compartments are not generated from dynamic EEs. Although EGF was able to reach the LBPA-negative predegradative compartment (Fig. 5C), its loss from Δm138-MCMV-infected cells was much slower (t1/2 of >2 h) than that from the uninfected cells (t1/2 of 30 min) (Fig. 5E), indicating that the degradation of clathrin-dependent cargo was also delayed in the infected cells.
A smaller fraction of the internalized Alexa555-EGF that reached the juxtanuclear area in Δm138-MCMV-infected cells colocalized with internalized TfR and MHC-I (Fig. 5C and D), which is consistent with their retention in the static subpopulation of EEs (31). This conclusion was supported by the cointernalization of MHC-I and Lamp1, which travels via static early endosomes (22). Both MHC-I and Lamp1 were found in the same perinuclear vesicles (Fig. 5D), indicating that MHC-I molecules are retained in static early endosomes with a reduced capacity for progression toward LEs.
MCMV infection decreases the cellular level of Rab GTPases.
The maturation of the early endosomal sorting domain requires the replacement of the Rab5 small GTPase either with Rab7, which leads to the formation of a late endosomal domain, or with Rab11, which is essential for the tubulation of the recycling domain (17, 46). Therefore, defective early endosomal maturation in Δm138-MCMV-infected cells could be associated with the reduced level or defective functioning of Rab proteins. In order to test this, we determined the levels of expression of Rab proteins that act at proximal (Rab5) and distal (Rab7 and Rab11) stages of endosomal maturation. At 6 h p.i. and later, a significant reduction of the Rab7 (data not shown) and Rab11 (Fig. 6 A) associations with endosomal membranes was observed by immunofluorescence. This reduction was not the result of cytoplasmic dispersion but rather was the consequence of a decreased cellular level (Fig. 6B). With the progression of infection, the cellular levels of all three Rab proteins declined, leading to a reduction of the level of Rab5 to the 35% control value and an almost-complete loss of Rab7 and Rab11 at 24 h p.i. (Fig. 6B). Given that a balance of Rab proteins is essential for the modeling of early endosomes, we calculated the levels of expression of Rab7 and Rab11 in relation to that of Rab5 (Fig. 6C). Our data demonstrate that the cellular levels of these two Rab proteins is altered more than that of Rab5, suggesting that the distal part of the early endosomal route is more affected by MCMV infection.
FIG. 6.
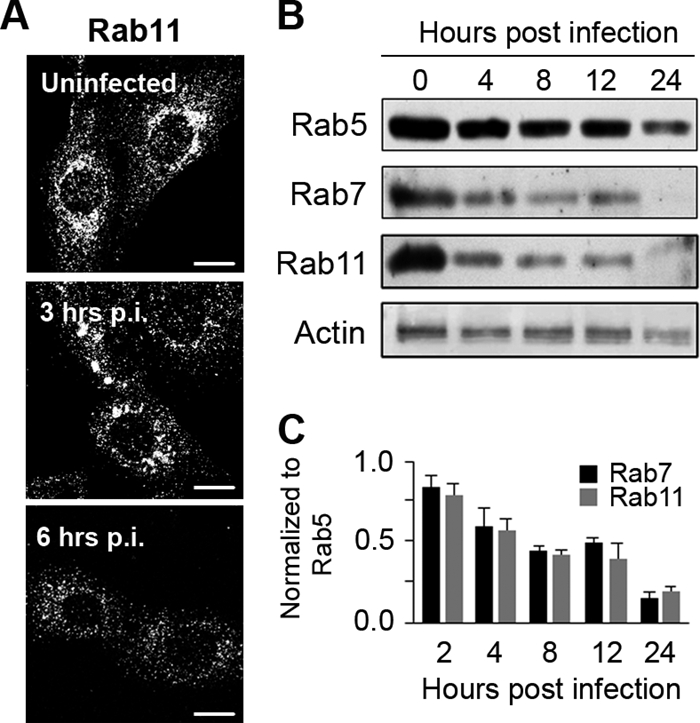
Expression of Rab GTPases in MCMV-infected cells. (A) Intracellular distribution of Rab11 in uninfected and Δm138-MCMV-infected cells determined by immunofluorescence. Bars, 10 μm. (B) Western blot analysis of Rab5, Rab7, and Rab11 expression levels in uninfected (0) and Δm138-MCMV-infected cells (4 to 24 h p.i.). (C) Relative expression levels of Rab7 and Rab11 normalized to that of Rab5. The blots were analyzed by densitometry, and the relative expression level of Rab5b at the corresponding time was set up as the 100% value. Data represent average data for four blots ± standard errors of the means (SEM).
The known MCMV immunoevasins are not required for endosomal MHC-I retention, but without them, the disruption of endosomal MHC-I trafficking is not sufficient to reduce cell surface expression of MHC-I.
MCMV encodes three proteins (m04, m06, and m152) that interfere with MHC-I trafficking in the secretory pathway (24, 27, 45, 56), resulting in the downregulation of MHC-I cell surface expression (52). In order to examine their role in the formation of perinuclear retention endosomes, we infected cells with the MCMV deletion mutant Δm4m6m152-MCMV, which is known not to abolish cell surface MHC-I expression (52). The cell surface expression levels of Dd and Kd were reduced to the ∼80% values in cells infected with the mutant virus at 12 h p.i. (Fig. 7 A), similar to values described previously for B12 cells (52). Although cell surface MHC-I was not downregulated after infection with Δm4m6m152-MCMV (Fig. 7A), MAb-labeled MHC-I internalized with an increased rate similar to those of cells infected with wt MCMV (data not shown) and Δm138-MCMV (Fig. 7B). Internalized Dd (data not shown) and Kd (Fig. 7C) were retained in the perinuclear endosomes. Thus, our data indicate that MCMVs without the three known MHC-I immunoevasins encode functions that affect endosomal trafficking, which are not sufficient to reduce the cell surface expression of MHC-I in the absence of a transport block in the secretory pathway.
FIG. 7.

The known MCMV immunoevasins are not required for endosomal MHC-I retention. (A) Cell surface expression of Dd and Kd on MEFs infected for 12 h with either Δm138-MCMV or Δm4m6m152-MCMV. Results are presented as percentages of initial expression. Data are averages of data from four independent experiments ± SD. (B) IRs of Dd and Kd in uninfected cells and cells infected with either Δm138-MCMV or Δm4m6m152-MCMV. (C) Confocal image of juxtanuclear accumulation of internalized Kd (red fluorescence, 4 h of internalization, as described in the legend Fig. 2) and nuclear MCMV ie1 (green fluorescence) in cells infected with the triple immunoevasin deletion mutant Δm4m6m152-MCMV. Bar, 10 μm.
DISCUSSION
In this report we demonstrate that MCMV perturbs endosomal trafficking in the early phase of infection. This perturbation has a striking impact on MHC-I trafficking because of the simultaneous effects of MCMV immunoevasins that act in the secretory pathway (24, 27, 45, 52, 56). Two of them (m152 and m06) are expressed in the early phase of infection (45, 56) and prevent the exit of the nascent MHC-I complexes to the cell surface (19, 40, 45, 56). Without them, MHC-I cell surface downregulation cannot occur (52). However, our data demonstrated that the loss of MHC-I from the cell surface is rapid and occurs early in the infection with a rate of ∼33% per hour. This cannot be explained by their constitutive internalization, which is five to six times slower in normal fibroblasts (36), indicating that MCMV encodes an additional function that is expressed in the early phase of infection (Fig. 1), after the expression of the three known MCMV immunoevasins (48). This function arrests the endosomal trafficking of cell surface-resident MHC-I in perinuclear early endosomes by inhibiting their recycling and LE progression. Together with the reduced egress of newly synthesized MHC-I complexes from the secretory pathway, this results in their accelerated loss from the cell surface. However, the effect is not MHC-I specific, since clathrin-dependent cargo molecules that travel via static early endosomes (31) are also collected in the perinuclear endosomes, and their progression toward either the recycling or degradative route is inhibited. Given that MHC-I and clathrin-dependent cargo meet in the proximal part of the common endosomal route (12, 14, 42), the effect of MCMV is focused on the distal stage of endosomal maturation to EEA1-positive EEs prior to the segregation of recycling and endolysosomal domains. This is associated with a reduction in levels of Rab7 and Rab11, small GTPases that bring the domain identity to the endosomal membrane (17, 46), indicating that MCMV thoroughly remodels the endosomal system in the early phase of infection.
MHC-I molecules are constitutively internalized via Arf6-associated clathrin-independent endocytic carriers that fuse with EEA1-positive transferrin-containing endosomes generated by clathrin-dependent endocytosis (6, 12, 14, 41, 42). These endosomes, by heterotypic fusion, generate “classical” early endosomes (14), which on microtubular trails partition their membranes and thereby sort their cargo into either recycling or endolysosomal domains (31). Recycling domains mature into recycling tubular carriers in a process that involves Rab11 GTPase (46), and endolysosomal domains fuse with or mature into LEs with the help of Rab7 (46). Given that the level of constitutive endocytosis of the plasma membrane is very high and occurs several times per hour (12, 18), constitutively endocytosed cargo, also including MHC-I molecules, should be returned either by rapid recycling from the primary acceptor compartment or by slow recycling from EEs (14, 53). The net result of constitutive internalization is a removal of approximately 5 to 10% of MHC-I molecules from the cell surface each hour, depending on the cell type and MHC-I allele (36). In uninfected fibroblasts, less than one-third of endocytosed MHC-I molecules are recycled back to the cell surface from EEs and the juxtanuclear tubulovesicular recycling compartment (41, 53). Another two-thirds were directed into degradation each hour, and the cell surface loss was reconstituted by newly synthesized MHC-I from the secretory pathway. Recycling is thereby critical for the maintenance of the cell surface level and cell surface retention of MHC-I. In MCMV-infected fibroblasts, the recycling of MHC-I complexes is delayed and reduced, which cannot keep the balance with the constitutive endocytosis, and one-third of cell surface MHC-I molecules were redistributed to the intracellular localization each hour, resulting in their retention in the perinuclear EEA1-positive EEs. This retention suggests a defective maturation of endosomal domains, which can be associated with the insufficient amount of corresponding Rab proteins. This is supported by the observation that the recycling of Tf was reduced and the degradation of EGF was delayed in Δm138-MCMV-infected cells. Similar effects on TfR recycling after interference with Rab11 functions (54) and on EGF trafficking after the depletion of Rab7 by RNA interference (51) were observed. In the absence of Rab7, the trafficking of EGF through the EEs to the LEs does not change, but their exit from the LEs into lysosomes is blocked (51).
Our data indicate that the MCMV-generated perinuclear retention endosomes represent an intermediate in EE maturation at the stage of remodeling of their EEA1-positive (also Rab5-positive) sorting domain into either recycling or endolysosomal domains, a process known to require Rab conversion (46). It has the characteristics of sorting endosomes at the terminal stage of their maturation, such as localization, vesicular nature, attachment of EEA1, accessibility to the incoming cargo from the plasma membrane known to cross the sorting endosome (TfR, EGF receptor [EGFR], and Lamp1), attachment to microtubules, and propensity for tubulation after BFA treatment (55). In addition, the retention endosomes are concentrated in the juxtanuclear area devoid of LE membranes, the site of early endosomal transition or conversion into recycling endosomes or LEs (37). The retention vesicles tend to fuse in the pericentriolar area and demonstrate tubulation, which is a characteristic of endosomal recycling domains (34, 55). From this compartment, recycling occurs at 16°C, conditions that do not allow recycling from EEs (49), but also after BFA treatment (M. Ilić Tomaš and P. Lučin, unpublished data), which inhibits recycling from juxtanuclear recycling endosomes (49, 50). Thus, the perinuclear retention compartments share characteristics of EEs and the juxtanuclear recycling compartment. It is attractive to speculate that MCMVs conserve the process of endosomal conversion displaying various stages of converting endosomes, a process which is rapid in untreated cells (46) and very slow in MCMV-infected cells.
It was recently reported that a sustained activation of protein kinase C (PKC) drives the formation of an endosomal compartment in the juxtanuclear localization, named the pericentrion, which sequesters membrane recycling components (1, 21) but not components and markers of the endolysosomal route (21). Although the MCMV-generated perinuclear endosomal compartment has many similarities with the pericentrion, it also sequesters cargo molecules that travel toward LEs (EGF, Lamp1, and dextran). In addition, PKC inhibitors did not cause a dispersal of MCMV retention compartments (Tomaš and Lučin, unpublished). Thus, although PKCs are essential for the early phase of MCMV replication (30) and may be activated during herpesvirus infection (43), it is unlikely that MCMV remodeling of early endosomal trafficking is the consequence of sustained PKC activation.
A similar effect on the perturbation of MHC-I trafficking was described previously for the HIV early gene product Nef, which reduces MHC-I surface expression by the internalization of MHC-I molecules (15). For this effect, Nef does not use classical clathrin-mediated endocytosis, as in the case of CD4 downregulation, but internalizes cell surface MHC-I molecules via a nonclathrin Arf6-dependent pathway (4, 44) and redistributes them into the perinuclear area that overlaps with the trans-Golgi network (TGN) membranes (15). However, it appears that Nef does not specifically interact with Arf6 but rather nonspecifically perturbs the trafficking route used by MHC-I (32), including alterations in the morphology and functions of the endosomal recycling compartment (35).
Interference with endocytic trafficking can be exploited by MCMV as a way for surveillance of the LE peptide-loading compartments. By retaining cell surface MHC-I at the endosomal crossroads, MCMV may prevent not only the exit of nascent viral peptide-loaded MHC-I complexes to the cell surface but also peptide loading in the exogenous pathway. The exchange of peptides can occur in acidic LEs (16), and viral peptide-loaded MHC-I can be presented at the cell surface by exocytosis (26). Therefore, it is important for MCMV to prevent the progression of MHC-I from EEs toward an LE peptide-loading compartment. This may be achieved by their retention at the sorting site without a direct interaction of MCMV-encoded proteins with MHC-I. Together with the prevented egress from the secretory pathway, this can be sufficient for a complete disarming of the antigen presentation machinery.
Taken together, the striking effect of MCMV on the MHC-I cell surface level in the early phase of infection uncovers a mechanism of endosomal remodeling very early in infection. It is attractive to speculate that this remodeling evolved for the purpose of MCMV morphogenesis and that the remodeling of cellular functions occurs much before viral DNA replication. This is supported by a recent observation that the endosomal recycling compartment contributes during the envelopment of human cytomegalovirus (29). Thus, an understanding of the morphogenesis of the retention compartment may give insight into the earliest events in the formation of the assembly compartment. In addition, an understanding of the mechanism of formation of the perinuclear endosomal retention compartment that retains clathrin-dependent and clathrin-independent cargoes may contribute to an understanding of molecular and mechanistic events in endosomal maturation and the formation of recycling endosomes and LEs. These mechanisms are not yet resolved, and, as for many cellular functions, MCMV may point to main principles that drive endosomal maturation in normal cells.
Acknowledgments
This work was supported by the Ministry of Science, Education, and Sport of the Republic of Croatia (grants 062006, 062-0620238-0223, and 062-0000000-3540) and EMBIC project European FP6, NoE (grants 512040 and LSHM-CT-2004-512040).
We are grateful to Jean Gruenberg (University of Geneva, Geneva, Switzerland) for providing anti-LBPA antibody. We thank Peter Ghazal and Paul Lacaze (University of Edinburgh, Edinburgh, Scotland) for providing Δie3-MCMV stock. We thank Ulrich H. Koszinowski, Martin Messerle, and Stipan Jonjić for providing us with mutant viruses. We also thank Stipan Jonjić, Biserka Mulac-Jeričević, and Nikola Petković for critical reading of the manuscript. We thank Jelena Đirlić and Ksenija Tulić for technical assistance.
Footnotes
Published ahead of print on 18 August 2010.
REFERENCES
- 1.Alvi, F., J. Idkowiak-Baldys, A. Baldys, J. R. Raymond, and Y. A. Hannun. 2007. Regulation of membrane trafficking and endocytosis by protein kinase C: emerging role of the pericentrion, a novel protein kinase C-dependent subset of recycling endosomes. Cell. Mol. Life Sci. 64:263-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Angulo, A., P. Ghazal, and M. Messerle. 2000. The major immediate-early gene ie3 of mouse cytomegalovirus is essential for viral growth. J. Virol. 74:11129-11236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Basha, G., G. Lizée, A. T. Reinicke, R. P. Seipp, K. D. Omilusik, and W. A. Jefferies. 2008. MHC class I endosomal and lysosomal trafficking coincides with exogenous antigen loading in dendritic cells. PLoS One 3:e3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blagoveshchenskaya, A. D., L. Thomas, S. F. Feliciangeli, C. H. Hung, and G. Thomas. 2002. HIV-1 Nef downregulates MHC-I by a PACS-1- and PI3K-regulated ARF6 endocytic pathway. Cell 111:853-866. [DOI] [PubMed] [Google Scholar]
- 5.Brune, W., H. Hengel, and U. H. Koszinowski. 2001. A mouse model for cytomegalovirus infection. Curr. Protoc. Immunol. 19:19.7. [DOI] [PubMed] [Google Scholar]
- 6.Caplan, S., N. Naslavsky, L. M. Hartnell, R. Lodge, R. S. Polishchuk, J. G. Donaldson, and J. S. Bonifacino. 2002. A tubular EHD1-containing compartment involved in the recycling of major histocompatibility complex class I molecules to the plasma membrane. EMBO J. 21:2557-2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ciechanover, A., A. L. Schwartz, A. Dautry-Varsat, and H. F. Lodish. 1983. Kinetics of internalization and recycling of transferrin and the transferrin receptor in a human hepatoma cell line. Effect of lysosomotropic agents. J. Biol. Chem. 258:9681-9689. [PubMed] [Google Scholar]
- 8.Coscoy, L., and D. Ganem. 2000. Kaposi's sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc. Natl. Acad. Sci. U. S. A. 97:8051-8056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crnković-Mertens, I., M. Messerle, I. Milotić, U. Szepan, N. Kučić, A. Krmpotić, S. Jonjić, and U. H. Koszinowski. 1998. Virus attenuation after deletion of the cytomegalovirus Fc receptor gene is not due to antibody control. J. Virol. 72:1377-1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Del Val, M., H. Hengel, H. Häcker, U. Hartlaub, T. Ruppert, P. Lučin, and U. H. Koszinowski. 1992. Cytomegalovirus prevents antigen presentation by blocking the transport of peptide-loaded major histocompatibility complex class I molecules into the medial-Golgi compartment. J. Exp. Med. 176:729-738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dobonici, M., J. Podlech, H. P. Steffens, S. Maiberger, and M. J. Reddehase. 1998. Evidence against a key role for transforming growth factor-β1 in cytomegalovirus-induced bone marrow aplasia. J. Gen. Virol. 79:867-876. [DOI] [PubMed] [Google Scholar]
- 12.Eyster, C. A., J. D. Higginson, R. Huebner, N. Porat-Shliom, R. Weigert, W. W. Wu, R. F. Shen, and J. G. Donaldson. 2009. Discovery of new cargo proteins that enter cells through clathrin-independent endocytosis. Traffic 10:590-599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghazal, P., A. E. Visser, M. Gustems, R. García, E. M. Borst, K. Sullivan, M. Messerle, and A. Angulo. 2005. Elimination of ie1 significantly attenuates murine cytomegalovirus virulence but does not alter replicative capacity in cell culture. J. Virol. 79:7182-7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grant, B. D., and J. G. Donaldson. 2009. Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 10:597-608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greenberg, M. E., A. J. Iafrate, and J. Skowronski. 1998. The SH3 domain-binding surface and an acidic motif in HIV-1 Nef regulate trafficking of class I MHC complexes. EMBO J. 17:2777-2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grommé, M., F. G. Uytdehaag, H. Janssen, J. Calafat, R. S. van Binnendijk, M. J. Kenter, A. Tulp, D. Verwoerd, and J. Neefjes. 1999. Recycling MHC class I molecules and endosomal peptide loading. Proc. Natl. Acad. Sci. U. S. A. 96:10323-10331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grosshans, B. L., D. Ortiz, and P. Novick. 2006. Rabs and their effectors: achieving specificity in membrane traffic. Proc. Natl. Acad. Sci. U. S. A. 103:11821-11827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hao, M., and F. R. Maxfield. 2000. Characterization of rapid membrane internalization and recycling. J. Biol. Chem. 275:15279-15288. [DOI] [PubMed] [Google Scholar]
- 19.Hengel, H., U. Reusch, A. Gutermann, H. Ziegler, S. Jonjić, P. Lučin, and U. H. Koszinowski. 1999. Cytomegaloviral control of MHC class I function in the mouse. Immunol. Rev. 168:167-176. [DOI] [PubMed] [Google Scholar]
- 20.Holtappels, R., D. Gillert-Marien, D. Thomas, J. Podlech, P. Deegen, S. Herter, S. A. Oehrlein-Karpi, D. Strand, M. Wagner, and M. J. Reddehase. 2006. Cytomegalovirus encodes a positive regulator of antigen presentation. J. Virol. 80:7613-7624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Idkowiak-Baldys, J., K. P. Becker, K. Kitatani, and Y. A. Hannun. 2006. Dynamic sequestration of the recycling compartment by classical protein kinase C. J. Biol. Chem. 281:22321-22331. [DOI] [PubMed] [Google Scholar]
- 22.Janvier, K., and J. S. Bonifacino. 2005. Role of the endocytic machinery in the sorting of lysosome-associated membrane proteins. Mol. Biol. Cell 16:4231-4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jefferies, W. A., and H. G. Burgert. 1990. E3/19K from adenovirus 2 is an immunosubversive protein that binds to a structural motif regulating the intracellular transport of major histocompatibility complex class I proteins. J. Exp. Med. 172:1653-1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kavanagh, D. G., M. C. Gold, M. Wagner, U. H. Koszinowski, and A. B. Hill. 2001. The multiple immune-evasion genes of murine cytomegalovirus are not redundant: m4 and m152 inhibit antigen presentation in a complementary and cooperative fashion, J. Exp. Med. 194:967-978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keil, G. M., A. Ebeling-Keil, and U. H. Koszinowski. 1984. Temporal regulation of murine cytomegalovirus transcription and mapping of viral RNA synthesized at immediately early times after infection. J. Virol. 50:784-795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kleijmeer, M. J., J. M. Escola, F. G. UytdeHaag, E. Jakobson, J. M. Griffith, A. D. Osterhaus, W. Stoorvogel, C. J. Melief, C. Rabouille, and H. J. Geuze. 2001. Antigen loading of MHC class I molecules in the endocytic tract. Traffic 2:124-137. [DOI] [PubMed] [Google Scholar]
- 27.Kleijnen, M. F., J. B. Huppa, P. Lučin, S. Mukherjee, H. Farrell, A. E. Campbell, U. H. Koszinowski, A. B. Hill, and H. L. Ploegh. 1997. A mouse cytomegalovirus glycoprotein, gp34, forms a complex with folded class I MHC molecules in the ER which is not retained but is transported to the cell surface. EMBO J. 16:685-694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kobayashi, T., M. H. Beuchat, M. Lindsay, S. Frias, R. D. Palmiter, H. Sakuraba, R. G. Parton, and J. Gruenberg. 1999. Late endosomal membranes rich in lysobisphosphatidic acid regulate cholesterol transport. Nat. Cell Biol. 1:113-118. [DOI] [PubMed] [Google Scholar]
- 29.Krzyzaniak, M. A., M. Mach, and W. J. Britt. 2009. HCMV-encoded glycoprotein M (UL100) interacts with Rab11 effector protein FIP4. Traffic 10:1439-1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kučić, N., H. Mahmutefendić, and P. Lučin. 2005. Inhibition of protein kinases C prevents murine cytomegalovirus replication. J. Gen. Virol. 86:2153-2161. [DOI] [PubMed] [Google Scholar]
- 31.Lakadamyali, M., M. J. Rust, and X. Zhuang. 2006. Ligands for clathrin-mediated endocytosis are differentially sorted into distinct populations of early endosomes. Cell 124:997-1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Larsen, J. E., R. H. Massol, T. J. Nieland, and T. Kirchhausen. 2004. HIV Nef-mediated major histocompatibility complex class I down-modulation is independent of Arf6 activity. Mol. Biol. Cell 15:323-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lilley, B. N., and H. L. Ploegh. 2005. Viral modulation of antigen presentation: manipulation of cellular targets in the ER and beyond. Immunol. Rev. 207:126-144. [DOI] [PubMed] [Google Scholar]
- 34.Lippincott-Schwartz, J., J. G. Donaldson, A. Schweizer, E. G. Berger, H. P. Hauri, L. C. Yuan, and R. D. Klausner. 1990. Microtubule-dependent retrograde transport of proteins into the ER in the presence of brefeldin A suggests an ER recycling pathway. Cell 60:821-836. [DOI] [PubMed] [Google Scholar]
- 35.Madrid, R., K. Janvier, D. Hitchin, J. Day, S. Coleman, C. Noviello, J. Bouchet, A. Benmerah, J. Guatelli, and S. Benichou. 2005. Nef-induced alteration of the early/recycling endosomal compartment correlates with enhancement of HIV-1 infectivity. J. Biol. Chem. 280:5032-5044. [DOI] [PubMed] [Google Scholar]
- 36.Mahmutefendić, H., G. Blagojević, N. Kučić, and P. Lučin. 2007. Constitutive internalization of murine MHC class I molecules. J. Cell. Physiol. 210:445-455. [DOI] [PubMed] [Google Scholar]
- 37.Maxfield, F. R., and T. E. McGraw. 2004. Endocytic recycling. Nat. Rev. Mol. Cell Biol. 5:121-132. [DOI] [PubMed] [Google Scholar]
- 38.Ménard, C., M. Wagner, Z. Ruzsics, K. Holak, W. Brune, A. E. Campbell, and U. H. Koszinowski. 2003. Role of murine cytomegalovirus US22 gene family members in replication in macrophages. J. Virol. 77:5557-5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Möbius, W., V. Herzog, K. Sandhoff, and G. Schwarzmann. 1999. Intracellular distribution of a biotin-labeled ganglioside, GM1, by immunoelectron microscopy after endocytosis in fibroblasts. J. Histochem. Cytochem. 47:1005-1014. [DOI] [PubMed] [Google Scholar]
- 40.Mohr, C. A., L. Cicin-Sain, M. Wagner, T. Sacher, M. Schnee, Z. Ruzsics, and U. H. Koszinowski. 2008. Engineering of cytomegalovirus genomes for recombinant live herpesvirus vaccines. Int. J. Med. Microbiol. 298:115-125. [DOI] [PubMed] [Google Scholar]
- 41.Naslavsky, N., R. Weigert, and J. G. Donaldson. 2004. Characterization of a nonclathrin endocytic pathway: membrane cargo and lipid requirements. Mol. Biol. Cell 15:3542-3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Naslavsky, N., R. Weigert, and J. G. Donaldson. 2003. Convergence of non-clathrin- and clathrin-derived endosomes involves Arf6 inactivation and changes in phosphoinositides. Mol. Biol. Cell 14:417-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park, R., and J. D. Baines. 2006. Herpes simplex virus type 1 infection induces activation and recruitment of protein kinase C to the nuclear membrane and increased phosphorylation of lamin B. J. Virol. 80:494-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Piguet, V., L. Wan, C. Borel, A. Mangasarian, N. Demaurex, G. Thomas, and D. Trono. 2000. HIV-1 Nef protein binds to the cellular protein PACS-1 to downregulate class I major histocompatibility complexes. Nat. Cell Biol. 2:163-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reusch, U., W. Muranyi, P. Lučin, H. G. Burgert, H. Hengel, and U. H. Koszinowski. 1999. A cytomegalovirus glycoprotein re-routes MHC class I complexes to lysosomes for degradation. EMBO J. 18:1081-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rink, J., E. Ghigo, Y. Kalaidzidis, and M. Zerial. 2005. Rab conversion as a mechanism of progression from early to late endosomes. Cell 122:735-749. [DOI] [PubMed] [Google Scholar]
- 47.Rohrer, J., A. Schweizer, D. Russell, and S. Kornfeld. 1996. The targeting of Lamp1 to lysosomes is dependent on the spacing of its cytoplasmic tail tyrosine sorting motif relative to the membrane. J. Cell Biol. 132:565-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thäle, R., U. Szepan, H. Hengel, G. Geginat, P. Lučin, and U. H. Koszinowski. 1995. Identification of the mouse cytomegalovirus genomic region affecting MHC class I molecule transport. J. Virol. 69:6098-6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van Dam, E. M., T. Ten Broeke, K. Jansen, P. Spijkers, and W. Stoorvogel. 2002. Endocytosed transferrin receptors recycle via distinct dynamin and phosphatidylinositol 3-kinase-dependent pathways. J. Biol. Chem. 277:48876-48883. [DOI] [PubMed] [Google Scholar]
- 50.van Dam, E. M., and W. Stoorvogel. 2002. Dynamin-dependent transferrin receptor recycling by endosome-derived clathrin-coated vesicles. Mol. Biol. Cell 13:169-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vanlandingham, P. A., and B. P. Ceresa. 2009. Rab7 regulates late endocytic trafficking downstream of multivesicular body biogenesis and cargo sequestration. J. Biol. Chem. 284:12110-12124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wagner, M., A. Gutermann, J. Podlech, M. J. Reddehase, and U. H. Koszinowski. 2002. Major histocompatibility complex class I allele-specific cooperative and competitive interactions between immune evasion proteins of cytomegalovirus. J. Exp. Med. 16:805-816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weigert, R., A. C. Yeung, J. Li, and J. G. Donaldson. 2004. Rab22a regulates the recycling of membrane proteins internalized independently of clathrin. Mol. Biol. Cell 15:3758-3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wilcke, M., L. Johannes, T. Galli, V. Mayau, B. Goud, and J. Salamero. 2000. Rab11 regulates the compartmentalization of early endosomes required for efficient transport from early endosomes to the trans-Golgi network. J. Cell Biol. 151:1207-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wilson, J. M., M. de Hoop, N. Zorzi, B. H. Toh, C. G. Dotti, and R. G. Parton. 2000. EEA1, a tethering protein of the early sorting endosome, shows a polarized distribution in hippocampal neurons, epithelial cells, and fibroblasts. Mol. Biol. Cell 11:2657-2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ziegler, H., R. Thäle, P. Lučin, W. Muranyi, T. Flohr, H. Hengel, H. Farrell, W. Rawlinson, and U. H. Koszinowski. 1997. A mouse cytomegalovirus glycoprotein retains MHC class I complexes in the ERGIC/cis-Golgi compartments. Immunity 6:57-66. [DOI] [PubMed] [Google Scholar]