

Abstract
A selection of carbon–fluorine bond-forming reactions is presented with particular focus on transition metal-mediated fluorination. A brief summary of conventional fluorination reactions is followed by a discussion of fluorination reactions mediated by palladium and silver. Investigations into the mechanism as well as the conceptual difficulty associated with transition metal-mediated carbon–fluorine bond formation are presented.
Keywords: carbon–fluorine bond formation, transition metals, palladium, silver, catalysis
1 Introduction
Fluorinated molecules are valuable as pharmaceuticals,1 agrochemicals,2 tracers for positron emission tomography (PET),3 and new materials.4 The introduction of fluorine into organic molecules can affect the basicity and acidity of proximal functional groups, the dipole moment, and hydrogen bonding ability.5 In pharmaceuticals, fluorine is often introduced to increase lipophilicity, bioavailability, and metabolic stability.3a,6 The size of fluorine as a substituent is similar to a hydroxyl group (van der Waals radii: F: 1.47 Å; OH 1.40 Å; compare to H: 1.20 Å). The radioisotope 18F has a half-life of 110 minutes and is used in PET for the synthesis of 18F-PET tracers. Despite the utility of fluorine substituents, relatively few methods are available for selective carbon–fluorine bond formation,7 when compared to methods for other carbon–halogen bond formations.
Recently, much progress has been made in Csp3–F bond formation including the asymmertric α-fluorination of carbonyl compounds with organo-8 and metal9 catalysis. However, the general construction of Csp2–F bonds, especially aromatic carbon–fluorine bonds, in functionalized molecules remains an unsolved problem in organic chemistry. The transition metals copper, palladium, and silver have been used for aromatic carbon–fluorine bond formations.10 This review discusses state-of-the-art aromatic carbon–fluorine bond formation reactions with particular focus on recently emerged transition metal-mediated approaches. A brief outline of classical methods of aromatic carbon–fluorine bond formation reactions is presented, followed by a discussion of novel fluorination reactions mediated by palladium and silver.
2 Nucleophilic Fluorination
Nucleophilic aromatic substitution reactions can introduce fluorine atoms into electron-deficient arenes. A common method for the synthesis of fluorinated aromatics in industry is the Halex (halogen exchange) process, in which halogen atoms serve as leaving groups and inexpensive, inorganic fluoride sources such as spray-dried KF are used as nucleophiles.11 The rate-determining step in nucleophilic aromatic fluorination by substitution, including the Halex process, is the addition of fluoride to form a Meisenheimer complex. Therefore, aryl chlorides are more suitable substrates in the Halex process than the corresponding aryl bromides and iodides, because chlorine is more electronegative than bromine and iodine. High reaction temperatures and phase transfer catalysts can increase the efficiency of the Halex process due to increased solubility of fluoride (Scheme 1).
Scheme 1.

Example of the Halex process with high-boiling solvent and phase transfer catalyst.
A useful alternative to the Halex process is fluorodenitration, a process in which the nitro group functions as a leaving group.11a,12 The nitro group is often displaced preferentially in the presence of chloride substituents, presumably because the Meisenheimer complex derived from attack at the carbon bearing the nitro group is more stabilized (Scheme 2).13
Scheme 2.

Fluorodenitration. The nitro group is displaced selectively in the presence of chloride substituents.
Ammonium substituents can also be appropriate leaving groups for nucleophilic fluorination reactions and are often used for the introduction of radio labeled fluoride (18F−). Trimethylammonium groups are typically more electron-withdrawing than nitro groups and undergo nucleophilic aromatic fluorination more efficiently than nitroarenes (Table 1).14
Table 1.
Effect of leaving groups (Y) and activating groups (X) on the rate of nucleophilic aromatic fluorination.
 | |||
|---|---|---|---|
| X | Y | krel (120 °C) | krel (80 °C) |
| NO2 | NMe3ClO4 | 400 | 30000 |
| NO2 | NO2 | 40 | 420 |
| CN | NMe3ClO4 | 16 | 100 |
| COMe | NMe3ClO4 | 8 | 33 |
| CN | NO2 | 1 | 1 |
Fluorination of diaryliodonium salts15 was reported by Beringer in 1953.16 In 2007, Ross used aryl(2-thienyl)iodonium salts for nucleophilic introduction of fluorine-18 to control the regioselectivity of fluoride attack.17 Notably, arenes that do not possess electron-withdrawing groups were also successfully fluorinated (Scheme 3).
Scheme 3.

Fluorination of aryliodonium salts.
Tetrabutylammonium fluoride (TBAF) is a common fluorinating reagent that is available as a trihydrate. Tetraalkylammonium counterions for fluoride reduce the ionic bond strength and increases the solubility of fluoride salts in organic solvents.18 The presence of water reduces the nucleophilicity of fluoride by hydrogen bonding. Drying of most quaternary ammonium fluorides by heat is problematic due to competing E2-elimination (Hofmann elimination) with fluoride serving as a strong base under anhydrous conditions (Scheme 4, top).19 In 2005, DiMagno reported the preparation of anhydrous tetrabutylammonium fluoride (TBAFanh) from hexafluorobenzene and tetrabutylammonium cyanide (Scheme 4, bottom).20
Scheme 4.

Preparation of anhydrous TBAF. Drying at high temperature results in Hofmann elimination but the reaction between hexafluorobenzene and tetrabutylammonium cyanide affords anhydrous TBAF at low temperature.
The Halex processes and fluorodenitration reactions can proceed at lower temperatures with TBAFanh as the fluoride source. For example, a typical Halex fluorination of 2,6-dichloropyridine requires heating at 200 °C for 10 hr (Scheme 5, top).21 In comparison, the same substrate is fluorinated within 1.5 hr upon exposure to TBAFanh at 20 °C (Scheme 5, bottom).22
Scheme 5.

Efficient nucleophilic aromatic substitution with anhydrous TBAF in comparison to traditional conditions.
In 2008, the synthesis of fluoroarenes from unactivated haloarenes upon treatment with tetramethylammonium fluoride (TMAF) was reported by Grushin.23
Fluorobenzene could be prepared in 65% yield from bromobenzene (Scheme 6). The fluorination of 2-bromonaphthalene resulted in a 3:2 mixture of 2-fluoronaphthalene and 1-fluoronaphthalene. The observed regioselectivity is consistent with the formation of an aryne intermediate. Elimination to form an aryne demonstrates the high basicity of fluoride in an anhydrous solvent (“naked” fluoride24). It has been shown that “naked” fluoride can deprotonate DMSO to form HF,23 which can be problematic because the in situ generated HF quickly consumes another equivalent of fluoride to form bifluoride (FHF−), which is less nucleophilic than fluoride.
Scheme 6.

Fluorination of unactivated arenes via arynes using TMAF.
3 Electrophilic Fluorination
Electron-rich arenes react with electrophilic fluorinating reagents but the regioselectivity is typically low (Scheme 7).25 Common organometallic reagents such as organomagnesiums or organolithiums can afford regio-specific fluorination with electrophilic fluorinating reagents, though the fluorination yield is highly dependent on the substrate.26
Scheme 7.

Unselective fluorination of phenol with an electrophilic fluorinating reagent.
In 2010, Beller reported improved reaction conditions for the electrophilic fluorination of Grignard reagents (Table 2).27 The Beller conditions are especially useful for the fluorination of electron-rich aryl Grignard reagents.
Table 2.
Electrophilic fluorination of arylmagnesium reagents by Beller.
 | |||
|---|---|---|---|
| Product | Yield | Product | Yield |
 |
81% |  |
33% |
 |
73% |  |
40% |
In 2010, Knochel reported an improved procedure for the electrophilic fluorination of aryl- and heteroarylmagnesium reagents (Table 3).28 Knochel’s conditions are particularly convenient for the fluorination of a variety of heteroarenes including isoquinolines, pyrroles, pyridines, benzothiophenes, and furans.
Table 3.
Electrophilic fluorination of arylmagnesium reagents by Knochel.
 | |||
|---|---|---|---|
| Product | Yield | Product | Yield |
 |
90% |  |
63% |
 |
43% |  |
75% |
 |
60% |  |
49% |
Organometallics with lower basicity and therefore larger functional group tolerance than Grignard reagents such as arylzinc halides, arylsilanes, arylstannanes, arylgermaniums and arylboronic acids can be converted into fluorinated arenes. However, they usually require reactive electrophilic fluorinating reagents such as elemental fluorine, XeF2, or AcOF for successful fluorination,29 or are limited to electron-rich arenes (Table 4).30
Table 4.
Electrophilic fluorination of arylboronic acids and aryl-trifluoroborate.
 | |||
|---|---|---|---|
| Product | Yield | Product | Yield |
 |
75% |  |
67% |
 |
100% | 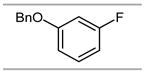 |
0% |
Electrophilic fluorination of arylsilanes with N-fluoro reagents gives aryl fluorides in low yields (Table 5).31 Therefore, the use of highly reactive electrophilic reagents is required. For example, XeF2 can be used to fluorodesilylate aryltrimethylsilanes in C6F6 at 18 °C in 61–86% yield.32
Table 5.
Fluorination of aryltrimethylsilanes.
 | |||
|---|---|---|---|
| R | a | b | c |
| t-Bu | 86 | 8 | 6 |
| Cl | 82 | 12 | 6 |
| OMe | 61 | 39 | 0 |
A successful example of fluorodestannylation is the preparation of 6-fluoro-L-DOPA.33 Treatment of the trimethylarylstannane with elemental fluorine allowed for the regioselective introduction of fluorine (Scheme 8). This strategy allows for the preparation of the [18F]-analogue (25% radiochemical yield), which is a PET-tracer for Parkinson’s disease34a and has been used in oncology.34b
Scheme 8.

Fluorodestannylation in the preparation of 6-fluoro-L-DOPA.
4 Balz-Schiemann Reaction
A special class of nucleophilic aromatic fluorination reactions is the Balz-Schiemann reaction (Scheme 9).35 In the Balz-Schiemann reaction, aryl fluorides are formed by pyrolysis (typically 110–170 °C) of aromatic diazonium tetrafluoroborates (ArN2+BF4−).
Scheme 9.

The Balz-Schiemann reaction.
Diazotization of an aniline in the presence of hydrogen tetrafluoroborate (HBF4) followed by either thermal or photochemical decomposition of the resulting diazonium tetrafluoroborate affords the aryl fluoride. Yields can often be improved when hexafluorophosphates (PF6−) or hexafluoroantimonates (SbF6−) are used instead of tetra-fluoroborates (Table 6).36
Table 6.
Substrate scope of the Balz-Schiemann reaction.
 | |||
|---|---|---|---|
| R | HBF4 (%) | HPF6 (%) | HSbF6 (%) |
| p-CH3 | 70 | 71 | 47 |
| p-COOH | 40 | 49 | 76 |
| p-NO2 | 40–58 | 63 | 60 |
| p-OH | –a | 10–20 | trace |
| o-OCH3 | 54–67 | 60 | 37 |
| o-COOH | 9 | 60 | 62 |
Preparation of the diazonium salt failed.
Alternative approaches include diazotization with NOBF4 followed by in situ fluorodediazoniation, which provided improved yields and broader substrate scope (Table 7).37
Table 7.
One-pot procedure for the Balz-Schiemann reaction.
 | ||
|---|---|---|
| R | NOBF4 reaction (%) | Traditional method |
| H | 72 | 60 |
| 2-Cl | 90 | 40 |
| 2-COMe | 56 | 47 |
| 4-CH2COOH | 73 | failed |
| 2-OH | 58 | failed |
Other variations involve fluorodediazoniation in HF/pyridine and in BF3·Et2O,38 the use of triazenes in HF/pyridine as precursors to aryldiazonium salts,39 and fluorodediazoniation in ionic liquids.40
5 Palladium-Mediated C–X Bond Formation
Transition metals, especially palladium, have been widely used for carbon–heteroatom bond formation reactions including C–O, C–N, C–S, and C–P bonds.41 Despite efforts in the last decades, only recently have successful transition metal-mediated C–F bond formations been achieved.42 In this chapter, the general considerations for carbon–heteroatom bond formations are discussed. Chapter 5 introduces concepts regarding general features of C–X bond-forming reactions, with a focus on hypotheses on why C–F bond formation is difficult. We subsequently discuss transition metal-mediated nucleophilic and electrophilic fluorination reactions in chapters 6–9.
A series of studies from the Hartwig group, the Buchwald group, and others have shown that reductive elimination to form carbon–heteroatom bonds occurs more rapidly from complexes with more nucleophilic heteroatoms.43 The rate of reductive eliminations to form C–X bonds from arylpalladium(II) amido, alkoxo, thiolato, and phosphido complexes that contain similar substituents on the heteroatoms occur in the order C–P > C–S > C–N > C–O as shown in Scheme 10.
Scheme 10.

Rate of C–X reductive elimination as a function of the heteroatom.
Reductive elimination from the alkylamido complex shown is faster than reductive elimination from an analogous arylamido complex, which is faster than reductive elimination from the corresponding diarylamido complex.44 Recently, it has been shown that arylpalladium amidate complexes undergo reductive elimination more slowly than the diarylamido complexes (Scheme 11).45
Scheme 11.

Electronic effect on the rate of C–N reductive elimination.
With increasing nucleophilicity of the heteroatom, reductive elimination becomes faster, presumably due to a more favorable attack of the electron pair onto the ipso carbon of the aryl group in the transition state (π effect).46 Alternatively, faster reductive elimination may be attributed to a decrease in the polarity, and thereby bond strength, of the metal–heteroatom bond. In the absence of steric or mesomeric effects, increased polarity of a metal–heteroatom bond typically leads to increased bond strength due to stronger ionic contribution to the bond energy (σ effect). A comparison between the electronic effects on the rate of reductive elimination from arylpalladium amido and amidate complexes (Scheme 11) with those on the rate of reductive elimination from arylpalladium alkyl complexes (Scheme 12)47 helps to elucidate the potential contributions of the σ and π effects.
Scheme 12.

Electronic effect on the rate of C–C reductive elimination.
Studies on the rates of reductive elimination from arylpalladium alkyl complexes containing varying functional groups on the carbon (R) are shown in Scheme 12. Reductive elimination from the arylpalladium alkyl complexes occurs more slowly from the alkyl complexes containing an electron-withdrawing group on the R carbon. Because the arylpalladium alkyl complexes lack lone pair electrons on the alkyl carbon bound to the metal, only the σ effect need be considered.
Fluorine is the most electronegative element, which should result in metal–fluorine bond energies with a large ionic contribution that lead to slower reductive elimination. Likewise, for the anions in the series C, N, O, F, fluorine is the least nucleophilic atom. Both properties of fluorine may explain why C–F reductive elimination is so challenging when compared to other carbon–heteroatom bond formations. In fact, despite significant research, a well-defined C–F reductive elimination reaction to form an aryl fluoride was reported only in 2008.48 In 2009, Buchwald reported C–F reductive elimination from a palladium(II) complex utilizing a bulky monodentate phosphine ligand, BrettPhos.49
6 Palladium-Mediated Nucleophilic Fluorination
A general scheme of palladium-catalyzed nucleophilic fluorination is shown in Scheme 13. Oxidative addition of an aryl halide or triflate to palladium(0) followed by ligand exchange with fluoride (F−) provides an arylpalladium(II) fluoride complex, which undergoes C–F bond formation via reductive elimination.
Scheme 13.

General scheme of Pd-mediated nucleophilic fluorination.
Oxidative addition of aryl halides and pseudohalides to palladium(0) is well established.50 Grushin showed that halide to fluoride exchange is possible to make arylpalladium(II) fluoride complexes.51 Spectroscopic evidence has been reported52 for the formation of [(Et3P)3PdIIF]+ in solution, although the complex was never isolated, likely due to its rapid decomposition by an intramolecular redox process to produce [(Et3P)nPd0] and Et3PF2.52b,53 The redox reaction Pd(II)/P(III)→ Pd(0)/P(V)53 may be the reason that complexes of the type [(R3P)2PdIIF2] have not yet been prepared, while several corresponding chloro, bromo, and iodo analogues are stable and well-known.
Grushin has developed two methods for the preparation of palladium(II) fluoride complexes. The reaction of organopalladium hydroxides with Et3N·(HF)3 (TREAT HF) in the presence of a free phosphine results in the formation of mononuclear fluoride complexes (Scheme 14, top).54 Alternatively, palladium(II) fluoride complexes can be prepared by I/F ligand exchange with AgF in benzene54 or toluene55 in the presence of ArI (5–10 mol%) (Scheme 14, bottom).
Scheme 14.

Preparation of PdII fluoride complexes by Grushin.
The palladium fluoride [(Ph3P)2PdII(Ph)(F)] is stable in the solid state and in anhydrous solution.51 NMR analysis of wet solutions, however, suggested that water may facilitate cleavage of the Pd–F bond as shown in Scheme 15.56 While reversible, ionization could be problematic for synchronous C–F reductive elimination.
Scheme 15.

Water-induced ionization of a Pd–F bond.
The thermal decomposition of [(Ph3P)2PdII(Ph)(F)] in anhydrous toluene at 110 °C was slow, and no C–F reductive elimination products were observed. Instead, P–F, P–P, and C–C bond formation products were formed (Scheme 16, top).57 In addition, mechanistic studies of the thermal decomposition of [(Ph3P)2PdII(Ph)(F)] and [(Ph3P)2PdII(C6D5)(F)] established reversible C–P oxidative addition/reductive elimination that results in scrambling of the phenyl groups on the phosphine ligand and the aryl group on the palladium (Scheme 16, bottom).
Scheme 16.

Unsuccessful C–F reductive elimination due to the competing P–F, P–P, C–C, and C–P bond formation.
Arylpalladium(II) fluoride complex 1 features a Xantphos ligand,58 a bidentate phosphine ligand with a large bite angle that is known to facilitate reductive elimination.59 The trifluoromethyl complex 2 successfully afforded C–CF3 bond formation in quantitative yield upon heating in anhydrous benzene at 80 °C for 3 hr. However, thermolysis of the fluoride complex 1 in anhydrous benzene at 60 °C led only to P–F bond formation and no C–F reductive elimination (Scheme 17).
Scheme 17.

Unsuccessful C–F reductive elimination and successful C–CF3 reductive elimination.
In an attempt to avoid competing P–F bond formation, arylpalladium(II) complex 3, which lacks phosphine ligands, was synthesized.60 Thermal decomposition of 3 and 4 in anhydrous benzene at 80 °C did not result in C–F bond formation. Palladium black and the C–C bond formation product biphenyl were produced instead, along with palladium(II) difluoride complex 4 (Scheme 18).
Scheme 18.

C–C bond formation from bis(pyridyl)phenylpalladium(II) fluoride complex (3).
In 2007, Yandulov reported a computational analysis on C–F reductive elimination from arylpalladium(II) fluorides.61 Yandulov explored N-heterocyclic carbenes (NHC)62,63 and phosphines as auxiliary ligands.
Potential C–F reductive elimination from T-shaped (PMe3)PdII(Ph)F (5) was computed. The activation enthalpy for Ph–F bond formation was predicted to be ΔH‡ = 25.1 kcal/mol; however, reversible migration of the phenyl group to the phosphine ligand, as observed by Grushin, had a lower activation enthalpy of ΔH‡ = 21.1 kcal/mol. Moreover, dimerization of 5 to form 6 was computed to be exothermic by ΔH° = 21.8 kcal/mol (Scheme 19).
Scheme 19.

Computed C–F reductive elimination from the T-shaped three-coordinate PMe3 palladium(II) fluoride complex 5.
A four-coordinate palladium fluoride complex would avoid dimer formation; however, the enthalpy of activation for C–F reductive elimination from four-coordinate complex [(Me3P)2PdII(Ph)(F)] (7) was predicted to be 38.8 kcal/mol, which is too high for practical synthetic applications (Scheme 20).
Scheme 20.

Computed C–F reductive elimination from four-coordinate PMe3 palladium(II) fluoride complex.
Yandulov’s calculations predicted that in the related tricoordinate NHC complex 8 reductive elimination of Ar–F is more facile for electron-withdrawing substituents, similar to related carbon–heteroatom reductive elimination reactions64 and nucleophilic aromatic substitution reactions (Scheme 21).65
Scheme 21.

Computed effect of substituents on the aryl group on C–F reductive elimination.
Yandulov further performed calculations on the effect of the presence of a hydrogen bond donor and found that hydrogen bonding stabilized the starting material more than the transition state (Scheme 22).
Scheme 22.

Computed effect of hydrogen bond donor on C–F reductive elimination.
Yandulov’s calculations predicted that 1) NHC ligands would undergo unproductive C–C reductive elimination preferentially over C–F reductive elimination, 2) palladium complexes with phosphines would have lower activation barriers than those with NHC ligands, 3) C–F reductive elimination from a four-coordinate bisphosphine complex would have a high activation barrier, 4) electron-withdrawing groups on the aryl substituent would decrease the TS energy, and 5) presence of hydrogen bond donors would increase the activation barrier.
Yandulov proposed that destabilization of a palladium fluoride dimer such as 6 by bulky ligands should increase the concentration of the three-coordinate arylpalladium(II) fluoride complex that could undergo C–F reductive elimination. Arylpalladium(II) fluoride dimer 9 features an electron-withdrawing aryl group and a bulky phosphine ligand P(t-Bu)3. Complex 9 did not afford any C–F bond formation product upon thermolysis at 60 °C for 160 hr; however, upon addition of the bulky phosphine ligand t-BuXPhos, developed by Buchwald and shown to mediated various carbon–heteroatom bond formations including C–O bonds,66 circa 10% of C–F bond formation was observed after 22 hr at 60 °C (Scheme 23).
Scheme 23.

C–F bond formation from arylpalladium complex 9.
The first observation of Ar–F bond formation from an arylpalladium(II) fluoride complex was a significant and promising result. Conclusive evidence for concerted C–F reductive elimination was not obtained. In 2007, Grushin proposed the possibility of C–P reductive elimination with subsequent nucleophilic aromatic fluorination to account for Yandulov’s observation of C–F bond formation in 10% yield (Scheme 24).67 As of yet, the mechanism of Yandulov’s C–F bond formation remains unknown.
Scheme 24.

C–P reductive elimination/nucleophilic aromatic substitution sequence for C–F bond formation proposed by Grushin.
In 2009, Buchwald communicated the first palladium-catalyzed nucleophilic aromatic fluorination as well as C–F reductive elimination from a palladium(II) complex.49 Key to the successful reaction was the use of the recently developed bulky monodentate phosphine ligand BrettPhos68 and its t-butyl derivative that afford mononuclear, tricoordinate Pd(II) complexes (Figure 1).
Figure 1.

Structure of BrettPhos and its t–butyl derivative.
ArPdII(X)(L) (L = Buchwald’s 2-biphenylphosphine ligand) are known to exist in a T-shaped three-coordinate structure where the fourth coordination site is occupied by the biphenyl moiety of the phosphine ligand (Scheme 25, left).69 When monomeric three-coordinate palladium(II) bromide complex 10 was treated with AgF, the corresponding three-coordinate palladium(II) fluoride complex 11 was obtained and its solid state structure was confirmed by X-ray crystallography. Upon thermolysis of 11 in toluene at 100 °C for 2 hr, C–F bond formation occurred in 15 to 25% yield (Scheme 25, right).
Scheme 25.

C–F bond formation from T-shape three-coordinate arylpalladium complex 11.
The palladium byproduct of the reaction shown in Scheme 25 was not identified, presumably due to its instability and high reactivity. However, when complex 11 was thermally decomposed in the presence of excess aryl bromide, formation of complex 10 was confirmed by 31P NMR, which suggests the intermediacy of a Pd(0) complex upon C–F bond formation (Scheme 26). In the solid state structure of 11, the phosphine ligand and the fluoride ligand occupy mutually trans coordination sites, likely due to the strong trans-influence of the σ-aryl ligand.70 This trans relationship between the phosphine ligand and the fluoride might be responsible for the successful C–F reductive elimination over the competing P–F reductive elimination, which was observed for other palladium(II) fluoride phosphine complexes.71
Scheme 26.

Proposed C–F reductive elimination. The presumed Pd(0) intermediate could not be observed but the Pd(II) complex after oxidative addition was characterized.
Buchwald successfully extended the stoichiometric reaction to catalysis and used the stable palladium(0) precursor [(cinnamyl)PdIICl]2, the sterically demanding t-BuBrettPhos, and CsF. The substrate scope of the catalytic fluorination is shown in Scheme 27. Electron-rich, electron-poor, ortho, ortho-disubstituted arenes, as well as heterocycles were compatible with the reaction conditions. Substrates with protic functional groups were not demonstrated to undergo fluorination, presumably due to the high basicity of fluoride under anhydrous conditions. The reactions are carried out with rigorous exclusion of moisture.
Scheme 27.

Substrate scope of palladium-catalyzed nucleophilic fluorination by Buchwald.
For most of the substrates only small amounts (up to 4%) of protodetriflated products (Ar–H) were obtained, which is advantageous for purification because the protodetriflated products typically have similar physical properties (Rf value, boiling point) to the corresponding fluorinated products. The intriguing formation of regioisomers was observed when para electron-donating or meta electron-withdrawing groups were present (Scheme 28).
Scheme 28.

Formation of regioisomers.
The mechanism for the formation of the regioisomers is not yet elucidated; however, it was rationalized that attack of a fluoride anion to the in situ formed benzyne is unlikely because the observed selectivities of regioisomers were distinct from those reported for the benzyne process reported by Grushin (Scheme 29).23
Scheme 29.

Comparison of the ratio of regioisomers with benzyne fluorination reported by Grushin.
7 Palladium-Mediated Directed Electrophilic Fluorination
A general pathway for palladium-mediated directed electrophilic fluorination is shown in Scheme 30. A palladium(II) complex undergoes cyclometallation followed by electrophilic fluorination resulting in regiospecific fluorination of the arene.
Scheme 30.

General scheme of Pd-mediated directed electrophilic fluorination.
In 2006, the first palladium-catalyzed fluorination of phenylpyridine derivatives was reported by Sanford.72 Sanford pioneered palladium-catalyzed oxidative C–H functionalization of arenes that bear ortho-directing groups73 and extended the methodology to electrophilic fluorination. Phenylpyridines with electron-donating and electron-withdrawing groups were fluorinated in the presence of 10 mol% of Pd(OAc)2 and N-fluoropyridinium tetrafluoroborate in 33–75% yield under microwave irradiation (Table 8).
Table 8.
Substrate scope of the palladium-catalyzed directed electrophilic fluorination by Sanford.
 | |||
|---|---|---|---|
| Product | Yield | Product | Yield |
 |
75% |  |
52% |
 |
67% |  |
33% |
The development of the first palladium-catalyzed aromatic fluorination reaction was a significant advance in this research area. Challenges that remain are the harsh reaction conditions (microwave, reaction temperature of 150 °C), the necessity for ortho-directing groups, and the need for blocking groups in the ortho’ or meta’ position to avoid difluorination (Scheme 31).
Scheme 31.

Problematic second fluorination event.
In 2009, Yu reported a similar palladium-catalyzed directed electrophilic fluorination of C–H bonds of N-benzyltriflamide derivatives (Table 9).74
Table 9.
Substrate scope of the palladium-catalyzed directed electrophilic fluorination by Yu.
 | |||
|---|---|---|---|
| Product | Yield | Product | Yield |
 |
82% |  |
81% |
 |
60% |  |
70% |
The use of N-fluoro-2,4,6-trimethylpyridinium triflate, Pd(OTf)2·2H2O, and NMP (N-methylpyrrolidinone) was important for obtaining the aryl fluorides in high yields. The reaction reported by Yu employed milder reaction conditions than those reported by Sanford, but the necessity of an ortho-directing group and ortho’ or meta’ blocking groups remained. A major advantage of Yu’s fluorination reaction is the identity of the directing group; the triflamide can be readily converted into other functional groups (Scheme 32).75 The mechanism of the directed electrophilic fluorination has not been established and fluorination from Pd(II), Pd(III),76 and Pd(IV)77 intermediates are conceivable.
Scheme 32.

Interconversion of the triflamide directing group after fluorination.
8 Pd-Mediated Electrophilic Fluorination
A general scheme of palladium-mediated electrophilic fluorination without directing groups is described in Scheme 33. The carbon–palladium bond is introduced by transmetalation rather than cyclopalladation as discussed above. The potential substrate scope is therefore significantly larger than for directed fluorination reactions because directing groups are not required. However, substrates need to be pre-functionalized to introduce appropriate functionality for transmetalation. Oxidation of an arylpalladium(II) complex with an electrophilic fluorinating reagent (F+) can provide a high-valent arylpalladium complex that subsequently can afford C–F bond formation by reductive elimination.
Scheme 33.

General scheme of Pd-mediated electrophilic fluorination.
In 2008, Vigalok reported a study on reactivity of arylpalladium(II) complexes toward electrophilic fluorinating reagents.78 Vigalok observed a C–F bond forming reaction from a palladium(II) complex that possesses a monodentate aryl group. Upon treatment with the electrophilic fluorinating reagent N-fluoro-2,4,6-trimethylpyridinium tetrafluoroborate, aryl fluoride was obtained in circa. 10% yield (Scheme 34). Both electrophilic cleavage of the Pd–C bond with the electrophilic fluorinating reagent and a C–F reductive elimination from a Pd(IV) complex are potential mechanisms for this transformation.
Scheme 34.

C–F bond formation from an arylpalladium(II) complex upon treatment with an electrophilic fluorinating reagent.
In 2009, Sanford reported fluorination of arylpalladium(II) complex 12 upon treatment with the strong electrophilic fluorinating agent XeF2 (Scheme 35).79 When the reaction was stopped after 2.5 min, the intermediate arylpalladium(IV) trifluoride complex 13 was isolated. Complex 13 did not undergo C–F bond formation by reductive elimination, but gave C–F bond formation upon addition of oxidants such as XeF2, (PhSO2)2NF, and N-bromosuccinimide in high yield. The mechanism of C–F bond formation has not yet been established.
Scheme 35.

Oxidant-promoted C–F bond formation from palladium(IV) difluoride complex 13.
In 2008, fluorination of arylboronic acids via stoichiometric palladium complexes was reported by our group.80 An electron-poor sulfonamide ligand was introduced via nitrene insertion to benzo[h]quinoline palladium chloride dimer complex 18 (Scheme 36).81 Chloro palladium complex 19 did not undergo transmetalation with arylboronic acids. The acetate analogue 20, however, smoothly afforded the corresponding arylpalladium complex 21 in 65–91% yield when treated with a variety of functionalized arylboronic acids. The arylpalladium complexes derived from 21 are stable to moisture and air, and readily purified by column chromatography on silica gel.
Scheme 36.

Synthesis of arylpalladium(II) pyridyl-sulfonamide complexes via transmetalation.
Fluorination of arylpalladium complexes 21 using the electrophilic reagent F-TEDA-BF4 (22) afforded the corresponding aryl fluorides 23a–h regioselectively in 31–82% yield (Table 10). The scope of the reaction is large and a variety of functional groups can be tolerated; however, palladium is used in stoichiometric quantities.
Table 10.
Substrate scope of the palladium-mediated electrophilic fluorination by our group
 | |||
|---|---|---|---|
| Product | Yield | Product | Yield |
 |
79% |
 |
61% |
 |
46% |
 |
73% |
 |
70% |
 |
82% |
 |
74% |
 |
60% |
Under the reaction conditions that afforded fluorination products 23a–h, a high-valent palladium fluoride was not observed by NMR analysis. We sought to design an analogue of 23 that upon oxidation and formation of a putative high-valent palladium fluoride would exhibit higher stability than the presumed intermediate generated by fluorination of 21. Rigid ligands have been shown to stabilize high-valent metal centers including palladium(IV) intermediates.82 We therefore targeted the benzo[h]quinoline derivative 24 (Scheme 37). Treatment of benzo[h]quinoline palladium acetate dimer 25 with one equivalent of pyridine-sulfonamide ligand 26 in CH2Cl2 at 23 °C afforded arylpalladium complex 24 in 95% yield.48
Scheme 37.

Synthesis of more rigid pyridyl-sulfonamido palladium(II) complex 24.
Fluorination of 24 in acetonitrile at 50 °C afforded 10-fluorobenzo[h]quinoline (30) in 94% yield (Scheme 38). Moreover, a deep purple, well-defined intermediate 27 was spectroscopically observed at 23 °C. The pyridine complex 28 was significantly more stable than 27 and afforded C–F bond formation product in 90% yield. When 27 was treated with tetramethylammonium fluoride tetrahydrate (TMAF·4H2O) at 23°C, palladium(IV) difluoride 29 was obtained. The neutral palladium di-fluoride 29 was more stable than monofluorides 27 and 28, and the solid state structure of 29 was identified by X-ray crystallography. Complex 29 afforded 30 by reductive elimination in 97% yield when heated in DMSO at 150 °C for 10 min. Reductive elimination from compounds 27–29 were the first established examples of C–F reductive elimination to form aryl fluorides from transition metal complexes.
Scheme 38.

First synthesis of Pd(IV) fluoride complexes and establishment of Ar–F reductive elimination.
In 2010, the first mechanism study of C–F reductive elimination reactions from arylpalladium(IV) fluoride complexes was reported with particular focus on the C–F reductive elimination from 27.83 The C–F reductive elimination from 27 showed first-order kinetics. The proposed mechanism of C–F reductive elimination is shown in Scheme 39 and is based on activation parameters, rate dependence on the polarity of the reaction medium, Hammett analysis, and DFT calculations (Figure 2).
Scheme 39.

Proposed dissociative mechanism for C–F reductive elimination.
Figure 2.

Calculated structure of the transition state of C–F reductive elimination from 27.
The mechanism study confirmed that C–F reductive elimination proceeds efficiently from arylpalladium(IV) fluoride complexes, supported by pyridyl-sulfonamide ancillary ligands. It was proposed that the pyridyl-sulfonamide ligand plays a crucial role for facile and efficient C–F bond formation. The ability of the pyridyl-sulfonamide ligand to function as a bidentate-tridentate-bidentate coordinating ligand during oxidation and reductive elimination, combined with the appropriate electronic requirements of the sulfonamide to position the aryl substituent trans to the sulfonamide ligand in the Pd(II) complex and the fluoride ligand trans to the sulfonamide ligand in the Pd(IV) complex, may be the reasons for facile C–F bond formation (Scheme 40).
Scheme 40.

Oxidation and reductive elimination supported by the pyridyl-sulfonamide ligand.
9 Silver-Mediated Fluorination
Ag-catalyzed reactions have emerged as important synthetic methods for a variety of organic transformations84 including halogenations of terminal alkynes, additions of OH or NH groups85 across allenes, alkynes or alkenes, group transfer reactions86 of carbenes, nitrenes, or silylenes, C–H insertion reactions,87 enantioselective carbonyl/imine addition reactions such as Mukaiyama-Aldol reaction,88 Mannich reaction,89 or allylation reactions,90 and decarboxylation reactions.91
In 2009, we reported silver-mediated C–F bond formation.92 Encouraged by a literature report of facile transmetalation from arylstannanes to silver(I) nitrate, known for almost 100 years,93 silver-mediated fluorination was developed. Arylstannanes afford the corresponding aryl fluorides when reacted with F-TEDA-PF6 in the presence of 2.0 equiv of AgOTf in acetone within 20 min (Scheme 41). The silver-mediated fluorination tolerates electron-rich, electron-poor, ortho, ortho-disubstituted, and heterocyclic aromatics, as well as protic functional groups. The fluorination was applicable to functionalized biologically active molecules such as camptothecin and quinine, enabling access to complex aryl fluorides.
Scheme 41.

Substrate scope of AgOTf-mediated fluorination of aryl-stannanes by our group.
Nucleophilic functional groups such as amines and sulfides were not compatible with the fluorination reaction conditions because they react with F-TEDA-PF6 to form N-fluoro or S-fluoro compounds which, if appropriately positioned β-hydrogen atoms are present, eliminate hydrogen fluoride (Scheme 42). Although the yields for this reaction are uniformly high, 10–20% of the corresponding protodestannylated products were observed. Recently, a catalytic version of the silver-mediated fluorination reaction has been developed by our group.94
Scheme 42.

Decomposition of a tertiary amine with F-TEDA-PF6.
In 2009, the silver-mediated fluorination reaction was extended to arylboronic acids and their derivatives.95 Boronic acid derivatives are versatile, virtually non-toxic, commercially available in great diversity, and readily prepared.96 Upon addition of NaOH in methanol, arylboronic acids underwent transmetalation to Ag(I) to afford the corresponding arylsilver complexes. After methanol evaporation and dissolving the reaction mixture in acetone, fluorination was achieved (Scheme 43).
Scheme 43.

One-pot fluorination of arylboronic acids.
The substrate scope of the silver-mediated one-pot fluorination of arylboronic acids is shown in Table 11. Electron-rich (28a, 28d), electron-poor (28c, 28e), protic (28a, 28b), halogenated (28e), ortho, ortho-disubstituted (28d) arenes, as well as heterocycles (28f–28h) were fluorinated successfully. The stoichiometric accumulation of the thermally unstable arylsilver complex proved increasingly problematic for complex molecules, limiting the utility of the silver-mediated fluorination of boronic acids. Importantly, only trace amounts of proto-deborylation were observed during this process.
Table 11.
Substrate scope of the silver-mediated fluorination of aryl-boronic acids reported by our group.
 | |||
|---|---|---|---|
| Product | Yield | Product | Yield |
 |
70% |
 |
73% |
 |
77% |
 |
75% |
 |
76% |
 |
71% |
 |
73% |
 |
75% |
The postulated mechanism for the silver-mediate fluorination involves a high-valent silver species from which C–F reductive elimination can occur.
10 Conclusion
Carbon–fluorine bond formation for the synthesis of aryl fluorides is challenging by conventional synthesis and transition metal-mediated transformations. The properties of fluorine, such as high electronegativity, high reactivity of F2, and the high basicity of naked fluoride are responsible for the difficulties associated with C–F bond formation. Carbon–fluorine reductive elimination from transition metal complexes is more challenging than C–C, C–N, and C–O reductive eliminations, presumably due to the strong ionic contribution of transition metal–fluorine bonds, which increase metal–fluorine bond strength and result in basic fluoride ligands that can form bifluorides.
Significant advances in transition metal-mediated fluorination reactions have been reported in the past five years. Palladium-catalyzed cross coupling chemistry was extended to C–F bond formation by the Buchwald group. Pd(0)/(II)-catalyzed nucleophilic fluorination has been a sought after reaction and can access fluoroarenes from aryl triflates. Current challenges include anhydrous reaction conditions that render the naked fluoride ion basic, which so far has prevented the fluorination of substrates with protic functional groups. The directed electrophilic palladium-catalyzed fluorination reactions developed by Sanford and Yu can fluorinate C–H bonds directly if appropriate directing groups are present. The palladium and silver-mediated fluorination reactions developed by our group have a broader substrate scope, likely due to the milder reaction conditions, but the palladium-mediated fluorination reaction currently requires stoichiometric amounts of transition metal.
Acknowledgments
We acknowledge NIH-NIGMS (GM088237) for financial support. We thank Adam S. Kamlet and Eunsung Lee for critical proofreading of this review.
Biographies

Takeru Furuya was born in 1983 in Tokyo, Japan. He received his undergraduate chemistry degree in 2006 at Tokyo University where he conducted research on a total synthesis of (−)-Tetrodotoxin under Prof. Tohru Fukuyama. He then moved to Harvard University in 2006 for graduate study with Prof. Tobias Ritter, where he is currently pursuing graduate work on transition-metal-mediated aromatic C–F bond formation. He is expected to obtain a Ph.D. degree in 2010 and will join the Du Bois group at Stanford University.

Johannes Klein was born in 1985 in Wuppertal, Germany. He received his undergraduate degree in 2007 from the Dortmund University of Technology. He then moved to University College Dublin where he worked on heterocyclic chemistry under the guidance of Dr. P. Evans and obtained his M.Sc. in 2009. He then joined the Ritter lab as a visiting scholar and worked on silver-mediated fluorination reactions and mechanistic studies on bimetallic palladium catalysis for 6 months. He has now moved to the University of York where he started working towards his Ph.D. under the supervision of Prof. Taylor.

Tobias Ritter was born in 1975 in Lübeck, Germany. He received his undergraduate education in Braunschweig, Germany, Bordeaux, France, Lausanne, Switzerland, and Stanford, US, and received a master of science from Braunschweig University in 1999. He conducted undergraduate research with Prof. Barry M. Trost at Stanford, obtained his PhD with Prof. Erick M. Carreira at ETH Zurich in 2004, and was a postdoc with Prof. Robert H. Grubbs at Caltech. In 2006, Tobias was appointed as Assistant Professor in the Department of Chemistry and Chemical Biology at Harvard. His research program is based on synthetic organic and organometallic chemistry. The Ritter lab currently focuses on fluorination chemistry for late-stage functionalization of complex natural and unnatural products and bimetallic transition metal redox catalysis.
References
- 1.(a) Müller K, Faeh C, Diederich F. Science. 2007;317:1881. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]; (b) Purser S, Moore PR, Swallow S, Gouverneur V. Chem Soc Rev. 2008;37:320. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]
- 2.Jeschke P. ChemBioChem. 2004;5:570. doi: 10.1002/cbic.200300833. [DOI] [PubMed] [Google Scholar]
- 3.(a) Phelps ME. Proc Natl Acad Sci USA. 2000;97:9226. doi: 10.1073/pnas.97.16.9226. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lasne M-C, Perrio C, Rouden J, Barré L, Roeda D, Dolle F, Crouzel C. Chemistry of β+-Emitting Compounds Based on Fluorine-18. In: Krause W, editor. Topics in Current Chemistry. Vol. 222. Springer; Berlin: 2002. pp. 201–258. [Google Scholar]; (c) Ametamey SM, Honer M, Schubiger PA. Chem Rev. 2008;108:1501. doi: 10.1021/cr0782426. [DOI] [PubMed] [Google Scholar]
- 4.(a) Böhm HJ, Banner D, Bendels S, Kansy M, Kuhn B, Müller K, Obst-Sander U, Stahl M. ChemBioChem. 2004;5:637. doi: 10.1002/cbic.200301023. [DOI] [PubMed] [Google Scholar]; (b) Shimizu M, Hiyama T. Angew Chem, Int Ed. 2005;44:214. doi: 10.1002/anie.200460441. [DOI] [PubMed] [Google Scholar]; (c) Hung MH, Farnham WB, Feiring AE, Rozen S. Fluoropolymers. Plenum Publishing Co; New York, NY, USA: 1999. Fluoromonomers and Fluoropolymers. [Google Scholar]
- 5.O’Hagan D. Chem Soc Rev. 2008;37:308. [Google Scholar]
- 6.(a) Smart BE. J Fluorine Chem. 2001;109:3. [Google Scholar]; (b) Bondi A. J Phys Chem. 1964;68:441. [Google Scholar]; (c) Hudlicky M, Pavlath AE, editors. Chemistry of Organic Fluorine Compounds II. American Chemical Society; Washington, DC, USA: 1995. [Google Scholar]; (d) Rowley M, Hallett DJ, Goodacre S, Moyes C, Crawforth J, Sparey TJ, Patel S, Marwood R, Patel S, Thomas S, Hitzel L, O’Connor D, Szeto N, Castro JL, Hutson PH, MacLeod AM. J Med Chem. 2001;44:1603. doi: 10.1021/jm0004998. [DOI] [PubMed] [Google Scholar]; (e) Berkowitz DB, Bose M. J Fluorine Chem. 2001;112:13. [Google Scholar]; (f) Tanake F, Fukuse H, Wada H, Fukushima M. Curr Pharm Biotechnol. 2000;1:137. doi: 10.2174/1389201003378979. [DOI] [PubMed] [Google Scholar]; (g) Couturier O, Luxen A, Chatal JF, Vuillez JP, Rigo P, Hustinx R. Eur J Nucl Med Mol Imaging. 2004;31:1182. doi: 10.1007/s00259-004-1607-9. [DOI] [PubMed] [Google Scholar]
- 7.(a) Kirk KL. Org Process Res Dev. 2008;12:305. [Google Scholar]; (b) Baudoux J, Cahard D. Org React. 2007;69:347. [Google Scholar]; (c) Furuya T, Kuttruff CA, Ritter T. Curr Opin Drug Discovery Dev. 2008;11:803. [PubMed] [Google Scholar]
- 8.(a) Beeson TD, MacMillan DWC. J Am Chem Soc. 2005;127:8826. doi: 10.1021/ja051805f. [DOI] [PubMed] [Google Scholar]; (b) Marigo M, Fielenbach D, Braunton A, Kjærsgaard A, Jørgensen KA. Angew Chem, Int Ed. 2005;44:3703. doi: 10.1002/anie.200500395. [DOI] [PubMed] [Google Scholar]; (c) Steiner DD, Mase N, Barbas CF., III Angew Chem, Int Ed. 2005;44:3706. doi: 10.1002/anie.200500571. [DOI] [PubMed] [Google Scholar]
- 9.(a) Hintermann L, Togni A. Angew Chem, Int Ed. 2000;39:4359. doi: 10.1002/1521-3773(20001201)39:23<4359::AID-ANIE4359>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]; (b) Pihko PM. Angew Chem, Int Ed. 2006;45:544. doi: 10.1002/anie.200502425. [DOI] [PubMed] [Google Scholar]; (c) Hamashima Y, Yagi K, Takano H, Tamás L, Sodeoka M. J Am Chem Soc. 2002;124:14530. doi: 10.1021/ja028464f. [DOI] [PubMed] [Google Scholar]; (d) Ma JA, Cahard D. Tetrahedron: Asymmetry. 2004;15:1007. [Google Scholar]; (e) Shibata N, Ishimaru T, Nagai T, Kohno J, Toru T. Synlett. 2004:1703. [Google Scholar]
- 10.For copper-mediated aromatic fluorinations that are not discussed in this review, see: EI DuPont de Nemours & Co. (Grushin V): Processes for preparing fluoroarenes from haloarenes. 07202388. US. 2007Subramanian MA, Manzer LE. Science. 2002;297:1665. doi: 10.1126/science.1076397.Janmanchi KM, Dolbier WR., Jr Org Process Res Dev. 2008;12:349.
- 11.(a) Adams DJ, Clark JH. Chem Soc Rev. 1999;28:225. [Google Scholar]; (b) Horwitz JP, Tomson AJ. J Org Chem. 1961;26:3392. [Google Scholar]; (c) Barlin GB, Young AC. J Chem Soc, Perkin Trans 1. 1972:1269. [Google Scholar]; (d) Pike VW, Aigbirhio FI. J Chem Soc, Chem Commun. 1995:2215. [Google Scholar]; (e) Shah A, Pike VW, Widdowson DA. J Chem Soc, Perkin Trans 1. 1998:2043. [Google Scholar]; (f) Ermert J, Hocke C, Ludwig T, Gail R, Coenen HH. J Labelled Compd Radiopharm. 2004;47:429. [Google Scholar]
- 12.Adams DJ, Clark JH, McFarland H. J Fluorine Chem. 1998;92:127. [Google Scholar]
- 13.(a) Artamkina GA, Egorov MP, Beletskaya IP. Chem Rev. 1982;82:427. [Google Scholar]; (b) Terrier F. Chem Rev. 1982;82:77. [Google Scholar]
- 14.Angelini G, Speranza M, Wolf AP, Shiue CY. J Fluorine Chem. 1985;27:177. [Google Scholar]
- 15.Zhdankin VV, Stang PJ. Chem Rev. 2008;108:5299. doi: 10.1021/cr800332c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beringer FM, Brierley A, Drexler M, Gindler EM, Lumpkin CC. J Am Chem Soc. 1953;75:2708. [Google Scholar]
- 17.Ross TL, Ermert J, Hocke C, Coenen HH. J Am Chem Soc. 2007;129:8018. doi: 10.1021/ja066850h. [DOI] [PubMed] [Google Scholar]
- 18.Yoshida Y, Kimura Y. Chem Lett. 1988;17:1355. [Google Scholar]
- 19.Sharma RK, Fry JL. J Org Chem. 1983;48:2112. [Google Scholar]
- 20.Sun H, DiMagno SG. J Am Chem Soc. 2005;127:2050. doi: 10.1021/ja0440497. [DOI] [PubMed] [Google Scholar]
- 21.Asahi Glass Co. Kumai S, Seki T, Wada A. 04164068. JP. 1992
- 22.Sun H, DiMagno SG. Angew Chem, Int Ed. 2006;45:2720. doi: 10.1002/anie.200504555. [DOI] [PubMed] [Google Scholar]
- 23.Grushin VV, Marshall WJ. Organometallics. 2008;27:4825. [Google Scholar]
- 24.(a) Christe KO, Wilson WW. J Fluorine Chem. 1990;47:117. [Google Scholar]; (b) Christe KO, Wilson WW, Wilson RD, Bau R, Feng JA. J Am Chem Soc. 1990;112:7619. [Google Scholar]; (c) Schwesinger R, Link R, Thiele G, Rotter H, Honert D, Limbach HH, Männle F. Angew Chem, Int Ed Engl. 1991;30:1372. [Google Scholar]; (d) Schwesinger R, Link R, Wenzl P, Kossek S. Chem — Eur J. 2006;12:438. doi: 10.1002/chem.200500838. [DOI] [PubMed] [Google Scholar]; (e) Grushin VV. Angew Chem, Int Ed. 1998;37:994. doi: 10.1002/(SICI)1521-3773(19980420)37:7<994::AID-ANIE994>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 25.Adachi K, Ohira Y, Tomizawa G, Ishihara S, Oishi S. J Fluorine Chem. 2003;120:173. [Google Scholar]
- 26.For examples, see Davis FA, Han W, Murphy CK. J Org Chem. 1995;60:4730.Lal GS. J Org Chem. 1993;58:2791.Differding E, Wehrli M. Tetrahedron Lett. 1991;32:3819.Barnette WE. J Am Chem Soc. 1984;106:452.
- 27.Anbarasan P, Neumann H, Beller M. Angew Chem, Int Ed. 2010 doi: 10.1002/anie.200905855. Early View. [DOI] [PubMed] [Google Scholar]
- 28.Yamada S, Gavryushin A, Knochel P. Angew Chem, Int Ed. 2010 doi: 10.1002/anie.200905052. Early View. [DOI] [PubMed] [Google Scholar]
- 29.(a) Bryce MR, Chambers RD, Mullins ST, Parkin A. J Chem Soc, Chem Commun. 1986:1623. [Google Scholar]; (b) Tius MA, Kawakami JK. Synth Commun. 1992;22:1461. [Google Scholar]
- 30.Cazorla C, Métay E, Andrioletti B, Lemaire M. Tetrahedron Lett. 2009;50:3936. [Google Scholar]
- 31.Tredwell M, Gouverneur V. Org Biomol Chem. 2006;4:26. doi: 10.1039/b513399h. [DOI] [PubMed] [Google Scholar]
- 32.Lothian AP, Ramsden CA. Synlett. 1993:753. [Google Scholar]
- 33.Namavari M, Bishop A, Satyamurthy N, Bida G, Barrio JR. Appl Radiat Isot. 1992;43:989. doi: 10.1016/0883-2889(92)90217-3. [DOI] [PubMed] [Google Scholar]
- 34.(a) Barrio JR, Huang SC, Phelps ME. Biochem Pharmacol. 1997;54:341. doi: 10.1016/s0006-2952(97)00031-2. [DOI] [PubMed] [Google Scholar]; (b) Hoegerle S, Altehoefer C, Ghanem N, Brink I, Moser E, Nitzsche E. Eur J Nucl Med. 2001;28:64. doi: 10.1007/s002590000404. [DOI] [PubMed] [Google Scholar]
- 35.Balz G, Schiemann G. Ber Deut Chem Ges. 1927;60:1186. [Google Scholar]
- 36.(a) Rutherford KG, Redmond W, Rigamonti J. J Org Chem. 1961;26:5149. [Google Scholar]; (b) Sellers C, Suschitzky H. J Chem Soc, C. 1968:2317. [Google Scholar]
- 37.Milner DJ. Synth Commun. 1992;22:73. [Google Scholar]
- 38.(a) Olah GA, Welch J. J Am Chem Soc. 1975;97:208. [Google Scholar]; (b) Shinhama K, Aki S, Furuta T, Minamikawa JI. Synth Commun. 1993;23:1577. [Google Scholar]
- 39.Rosenfeld MN, Widdowson DA. J Chem Soc, Chem, Commun. 1979:914. [Google Scholar]
- 40.Laali KK, Gettwert VJ. J Fluorine Chem. 2001;107:31. [Google Scholar]
- 41.Hartwig JF. Acc Chem Res. 2008;41:1534. doi: 10.1021/ar800098p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.For a recent review, see: Brown MJ, Gouverneur V. Angew Chem, Int Ed. 2009;48:8610. doi: 10.1002/anie.200902121.
- 43.(a) Hartwig JF. Inorg Chem. 2007;46:1936. doi: 10.1021/ic061926w. [DOI] [PubMed] [Google Scholar]; (b) Muci AR, Buchwald SL. Top Curr Chem. 2002;219:131. [Google Scholar]
- 44.Driver MS, Hartwig JF. J Am Chem Soc. 1997;119:8232. [Google Scholar]
- 45.Fujita KI, Yamashita M, Puschmann F, Alvarez-Falcon MM, Incarvito CD, Hartwig JF. J Am Chem Soc. 2006;128:9044. doi: 10.1021/ja062333n. [DOI] [PubMed] [Google Scholar]
- 46.Widenhoefer RA, Buchwald SL. J Am Chem Soc. 1998;120:6504. [Google Scholar]
- 47.Culkin DA, Hartwig JF. Organometallics. 2004;23:3398. [Google Scholar]
- 48.Furuya T, Ritter T. J Am Chem Soc. 2008;130:10060. doi: 10.1021/ja803187x. [DOI] [PubMed] [Google Scholar]
- 49.Watson DA, Su M, Teverovskiy G, Zhang Y, García-Fortanet J, Kinzel T, Buchwald SL. Science. 2009;325:1661. doi: 10.1126/science.1178239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stille JK, Lau KSY. Acc Chem Res. 1977;10:434. [Google Scholar]
- 51.Grushin VV. Chem —Eur J. 2002;8:1006. doi: 10.1002/1521-3765(20020301)8:5<1006::aid-chem1006>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 52.(a) Dixon KR, McFarland JJ. J Chem Soc, Chem Commun. 1972:1274. [Google Scholar]; (b) Cairns MA, Dixon KR, McFarland JJ. J Chem Soc, Dalton Trans. 1975:1159. [Google Scholar]
- 53.Mason MR, Verkade JG. Organometallics. 1992;11:2212. [Google Scholar]
- 54.(a) Fraser SL, Antipin MYu, Khroustalyov VN, Grushin VV. J Am Chem Soc. 1997;119:4769. [Google Scholar]; (b) Pilon MC, Grushin VV. Organometallics. 1998;17:1774. [Google Scholar]
- 55.Marshall WJ, Thorn DL, Grushin VV. Organometallics. 1998;17:5427. [Google Scholar]
- 56.Roe DC, Marshall WJ, Davidson F, Soper PD, Grushin VV. Organometallics. 2000;19:4575. [Google Scholar]
- 57.Grushin VV. Organometallics. 2000;19:1888. [Google Scholar]
- 58.Grushin VV, Marshall WJ. J Am Chem Soc. 2006;128:12644. doi: 10.1021/ja064935c. [DOI] [PubMed] [Google Scholar]
- 59.(a) Kamer PCJ, van Leeuwen PWNM, Reek JNH. Acc Chem Res. 2001;34:895. doi: 10.1021/ar000060+. [DOI] [PubMed] [Google Scholar]; (b) Yin J, Buchwald SL. J Am Chem Soc. 2002;124:6043. doi: 10.1021/ja012610k. [DOI] [PubMed] [Google Scholar]; (c) Zuideveld MA, Swennenhuis BHG, Boele MDK, Guari Y, van Strijdonck GPF, Reek JNH, Kamer PCJ, Goubitz K, Fraanje J, Lutz M, Spek AL, van Leeuwen PWNM. J Chem Soc, Dalton Trans. 2002:2308. [Google Scholar]
- 60.Grushin VV, Marshall WJ. J Am Chem Soc. 2009;131:918. doi: 10.1021/ja808975a. [DOI] [PubMed] [Google Scholar]
- 61.Yandulov DV, Tran NT. J Am Chem Soc. 2007;129:1342. doi: 10.1021/ja066930l. [DOI] [PubMed] [Google Scholar]
- 62.(a) Beller M, Bolm C, editors. Transition Metals for Organic Synthesis: Building Blocks and Fine Chemicals. Wiley-VCH; Weinheim: 2004. [Google Scholar]; (b) Cornils B, Herrmann WA, editors. Applied Homogeneous Catalysis with Organometallic Compounds: A Comprehensive Handbook in Three Volumes. Wiley-VCH; Weinheim: 2002. [Google Scholar]
- 63.(a) McGuinness DS, Green MJ, Cavell KJ, Skelton BW, White AH. J Organomet Chem. 1998;565:165. [Google Scholar]; (b) McGuinness DS, Saendig N, Yates BF, Cavell KJ. J Am Chem Soc. 2001;123:4029. doi: 10.1021/ja003861g. [DOI] [PubMed] [Google Scholar]; (c) Lewis AKdeK, Caddick S, Cloke FGN, Billingham NC, Hitchcock PB, Leonard J. J Am Chem Soc. 2003;125:10066. doi: 10.1021/ja035565k. [DOI] [PubMed] [Google Scholar]; (d) Crudden CM, Allen DP. Coord Chem Rev. 2004;248:2247. [Google Scholar]; (e) Cavell KJ, McGuinness DS. Coord Chem Rev. 2004;248:671. [Google Scholar]
- 64.(a) Hartwig JF. Acc Chem Res. 1998;31:852. [Google Scholar]; (b) Hartwig JF. Synlett. 2006:1283. [Google Scholar]
- 65.Smith MB, March J. March’s AdVanced Organic Chemistry: Reactions, Mechanisms and Structure. Wiley-Interscience; New York: 2001. [Google Scholar]
- 66.(a) Burgos CH, Barder TE, Huang X, Buchwald SL. Angew Chem, Int Ed. 2006;45:4321. doi: 10.1002/anie.200601253. [DOI] [PubMed] [Google Scholar]; (b) Vorogushin AV, Huang X, Buchwald SL. J Am Chem Soc. 2005;127:8146. doi: 10.1021/ja050471r. [DOI] [PubMed] [Google Scholar]; (c) Billingsley KL, Anderson KW, Buchwald SL. Angew Chem, Int Ed. 2006;45:3484. doi: 10.1002/anie.200600493. [DOI] [PubMed] [Google Scholar]
- 67.Grushin VV, Marshall WJ. Organometallics. 2007;26:4997. [Google Scholar]
- 68.Fors BP, Watson DA, Biscoe MR, Buchwald SL. J Am Chem Soc. 2008;130:13552. doi: 10.1021/ja8055358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Barder TE, Walker SD, Martinelli JR, Buchwald SL. J Am Chem Soc. 2005;127:4685. doi: 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]
- 70.Landis CR, Firman TK, Root DM, Cleveland T. J Am Chem Soc. 1998;120:1842. [Google Scholar]
- 71.Crabtree RH. The organometallic chemistry of the transition metals. John Wiley & Sons; Hoboken, New Jersey: 2005. [Google Scholar]
- 72.Hull KL, Anani WQ, Sanford MS. J Am Chem Soc. 2006;128:7134. doi: 10.1021/ja061943k. [DOI] [PubMed] [Google Scholar]
- 73.(a) Dick AR, Hull KL, Sanford MS. J Am Chem Soc. 2004;126:2300. doi: 10.1021/ja031543m. [DOI] [PubMed] [Google Scholar]; (b) Desai LV, Hull KL, Sanford MS. J Am Chem Soc. 2004;126:9542. doi: 10.1021/ja046831c. [DOI] [PubMed] [Google Scholar]; (c) Kalyani D, Deprez NR, Desai LV, Sanford MS. J Am Chem Soc. 2005;127:7330. doi: 10.1021/ja051402f. [DOI] [PubMed] [Google Scholar]; (d) Kalyani D, Sanford MS. Org Lett. 2005;7:4149. doi: 10.1021/ol051486x. [DOI] [PubMed] [Google Scholar]; (e) Desai LV, Malik HA, Sanford MS. Org Lett. 2006;8:1141. doi: 10.1021/ol0530272. [DOI] [PubMed] [Google Scholar]; (f) Lyons TW, Sanford MS. Chem Rev. 2010;110:1147. doi: 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang X, Mei TS, Yu JQ. J Am Chem Soc. 2009;131:7520. doi: 10.1021/ja901352k. [DOI] [PubMed] [Google Scholar]
- 75.(a) Glass RS. J Chem Soc, Chem Commun. 1971:1546. [Google Scholar]; (b) Glass RS, Hoy RC. Tetrahedron Lett. 1976;17:1777. [Google Scholar]
- 76.(a) Powers DC, Ritter T. Nat Chem. 2009;1:302. doi: 10.1038/nchem.246. [DOI] [PubMed] [Google Scholar]; (b) Powers DC, Geibel MAL, Klein JEMN, Ritter T. J Am Chem Soc. 2009;131:17050. doi: 10.1021/ja906935c. [DOI] [PubMed] [Google Scholar]
- 77.(a) Byers PK, Canty AJ, Crespo M, Puddephatt RJ, Scott JD. Organometallics. 1988;7:1363. [Google Scholar]; (b) Whitfield SR, Sanford MS. J Am Chem Soc. 2007;129:15142. doi: 10.1021/ja077866q. [DOI] [PubMed] [Google Scholar]; (c) Fu Y, Li Z, Liang S, Guo QX, Liu L. Organometallics. 2008;27:3736. [Google Scholar]; (d) Racowski JM, Dick AR, Sanford MS. J Am Chem Soc. 2009;131:10974. doi: 10.1021/ja9014474. [DOI] [PubMed] [Google Scholar]
- 78.(a) Kaspi AW, Yahav-Levi A, Goldberg I, Vigalok A. Inorg Chem. 2008;47:5. doi: 10.1021/ic701722f. [DOI] [PubMed] [Google Scholar]; (b) Vigalok A. Chem–Eur J. 2008;14:5102. doi: 10.1002/chem.200701738. [DOI] [PubMed] [Google Scholar]
- 79.Ball ND, Sanford MS. J Am Chem Soc. 2009;131:3796. doi: 10.1021/ja8054595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Furuya T, Kaiser HM, Ritter T. Angew Chem, Int Ed. 2008;47:5993. doi: 10.1002/anie.200802164. [DOI] [PubMed] [Google Scholar]
- 81.Dick AR, Remy MS, Kampf JW, Sanford MS. Organometallics. 2007;26:1365. [Google Scholar]
- 82.(a) Canty AJ. Acc Chem Res. 1992;25:83. [Google Scholar]; (b) Canty AJ, Jin H, Roberts AS, Skelton BW, Traill PR, White AH. Organometallics. 1995;14:199. [Google Scholar]; (c) Canty AJ, Denney MC, van Koten G, Skelton BW, White AH. Organometallics. 2004;23:5432. [Google Scholar]; (d) Cámpora J, Palma P, del Río D, López JA, Álvarez E, Connelly NG. Organometallics. 2005;24:3624. [Google Scholar]; (e) Dick AR, Kampf JW, Sanford MS. J Am Chem Soc. 2005;127:12790. doi: 10.1021/ja0541940. [DOI] [PubMed] [Google Scholar]
- 83.Furuya T, Benitez D, Tkatchouk E, Strom AE, Tang P, Goddard WA, Ritter T. J Am Chem Soc. 2010 doi: 10.1021/ja909371t. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Naodovic M, Yamamoto H. Chem Rev. 2008;108:3132. doi: 10.1021/cr068413r. [DOI] [PubMed] [Google Scholar]
- 85.(a) Yang CG, Reich NW, Shi Z, He C. Org Lett. 2005;7:4553. doi: 10.1021/ol051065f. [DOI] [PubMed] [Google Scholar]; (b) Yoshida S, Fukui K, Kikuchi S, Yamada T. Chem Lett. 2009;38:786. [Google Scholar]; (c) Ye D, Zhang X, Zhou Y, Zhang D, Zhang L, Wang H, Jiang H, Liu H. Adv Synth Catal. 2009;351:2770. [Google Scholar]; (d) Weibel JM, Blanc A, Pale P. Chem Rev. 2008;108:3149. doi: 10.1021/cr078365q. [DOI] [PubMed] [Google Scholar]
- 86.(a) Li Z, He C. Eur J Org Chem. 2006:4313. [Google Scholar]; (b) Cui Y, He C. J Am Chem Soc. 2003;125:16202. doi: 10.1021/ja038668b. [DOI] [PubMed] [Google Scholar]; (c) Dias HVR, Lovely CJ. Chem Rev. 2008;108:3223. doi: 10.1021/cr078362d. [DOI] [PubMed] [Google Scholar]
- 87.(a) Cui Y, He C. Angew Chem, Int Ed. 2004;43:4210. doi: 10.1002/anie.200454243. [DOI] [PubMed] [Google Scholar]; (b) Lovely CJ, Flores JA, Meng X, Dias HVR. Synlett. 2009:129. [Google Scholar]
- 88.(a) Yanagisawa A, Matsumoto Y, Nakashima H, Asakawa K, Yamamoto H. J Am Chem Soc. 1997;119:9319. [Google Scholar]; (b) Yanagisawa A, Matsumoto Y, Asakawa K, Yamamoto H. J Am Chem Soc. 1999;121:892. [Google Scholar]; (c) Yanagisawa A, Nakatsuka Y, Asakawa K, Wadamoto M, Kageyama H, Yamamoto H. Bull Chem Soc Jpn. 2001;74:1477. [Google Scholar]; (d) Yanagisawa A, Nakatsuka Y, Asakawa K, Kageyama H, Yamamoto H. Synlett. 2001:69. doi: 10.1002/(sici)1521-3773(19991216)38:24<3701::aid-anie3701>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]; (e) Yanagisawa A, Ichikawa T, Arai T. J Organomet Chem. 2007;692:550. [Google Scholar]; (f) Ohkouchi M, Masui D, Yamaguchi M, Yamagishi T. J Mol Catal A. 2001;170:1. [Google Scholar]; (g) Akullian LC, Snapper ML, Hoveyda AH. J Am Chem Soc. 2006;128:6532. doi: 10.1021/ja061166o. [DOI] [PubMed] [Google Scholar]
- 89.(a) Mandai H, Mandai K, Snapper ML, Hoveyda AH. J Am Chem Soc. 2008;130:17961. doi: 10.1021/ja807243t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Josephsohn NS, Snapper ML, Hoveyda AH. J Am Chem Soc. 2004;126:3734. doi: 10.1021/ja049388e. [DOI] [PubMed] [Google Scholar]; (c) Wieland LC, Vieira EM, Snapper ML, Hoveyda AH. J Am Chem Soc. 2009;131:570. doi: 10.1021/ja8062315. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Josephsohn NS, Carswell EL, Snapper ML, Hoveyda AH. Org Lett. 2005;7:2711. doi: 10.1021/ol050910r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Carswell EL, Snapper ML, Hoveyda AH. Angew Chem, Int Ed. 2006;45:7230. doi: 10.1002/anie.200603496. [DOI] [PubMed] [Google Scholar]
- 90.(a) Yanagisawa A, Kageyama H, Nakatsuka Y, Asakawa K, Matsumoto Y, Yamamoto H. Angew Chem, Int Ed. 1999;38:3701. doi: 10.1002/(sici)1521-3773(19991216)38:24<3701::aid-anie3701>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]; (b) Yanagisawa A, Nakashima H, Nakatsuka Y, Ishiba A, Yamamoto H. Bull Chem Soc Jpn. 2001;74:1129. [Google Scholar]; (c) Wadamoto M, Ozasa N, Yanagisawa A, Yamamoto H. J Org Chem. 2003;68:5593. doi: 10.1021/jo020691c. [DOI] [PubMed] [Google Scholar]; (d) Wadamoto M, Yamamoto H. J Am Chem Soc. 2005;127:14556. doi: 10.1021/ja0553351. [DOI] [PubMed] [Google Scholar]; (e) Wadamoto M, Yamamoto H. J Am Chem Soc. 2005;127:14556. doi: 10.1021/ja0553351. [DOI] [PubMed] [Google Scholar]
- 91.Lu P, Sanchez C, Cornella J, Larrosa I. Org Lett. 2009;11:5710. doi: 10.1021/ol902482p. [DOI] [PubMed] [Google Scholar]
- 92.Furuya T, Strom AE, Ritter T. J Am Chem Soc. 2009;131:1662. doi: 10.1021/ja8086664. [DOI] [PubMed] [Google Scholar]
- 93.(a) Krause E, Schmitz M. Ber Deutsch Chem Ges. 1919;52:2150. [Google Scholar]; (b) Beverwijk CDM, van der Kerk GJM. J Organomet Chem. 1972;43:C7. [Google Scholar]
- 94.Tang P, Furuya T, Ritter T. submitted. [Google Scholar]
- 95.Furuya T, Ritter T. Org Lett. 2009;11:2860. doi: 10.1021/ol901113t. [DOI] [PubMed] [Google Scholar]
- 96.Hall DG. Preparation and applications in organic synthesis and medicine. Wiley-VCH; Weinheim: 2005. Boronic acids. [Google Scholar]