

Abstract

A series of π-extended phosphorescent palladium(II) and platinum(II) porphyrin complexes were synthesized, in which additional benzene rings are fused radially onto at least one of the four peripheral benzo groups. The photophysical properties of the metalloporphyrins palladium(II)-meso-tetra-(4-fluorophenyl)mononaphthotribenzoporphyrin (Pd1NF), cis-palladium(II)-meso-tetra-(4-fluorophenyl)dibenzodinaphthoporphyrin (Pd2NF), and palladium(II)-meso-tetra-(4-fluorophenyl)monobenzotrinaphthoporphyrin (Pd3NF) and the corresponding platinum(II) compounds (Pt1NF, cis-Pt2NF, Pt3NF) were investigated. The compounds under investigation absorb intensively in the near-infrared region (628−691 nm) and emit at room temperature at 815−882 nm. Phosphorescence quantum yields of the platinum(II) porphyrins range from 25 to 53% with luminescence decay times of 21 to 44 μs in deoxygenated toluene solutions at room temperature. The corresponding palladium(II) complexes exhibit quantum yields in the range of 7 to 18% with lifetimes of 106 to 206 μs. Density functional theory (DFT) calculations revealed nonplanar geometries for all complexes and corroborate the absorption characteristics. The subsequent π extension of the porphyrin system leads to near-infrared absorbing oxygen indicators with tailor-made luminescence properties as well as tunable oxygen sensitivity.
Short abstract
The synthesis and characterization of novel “hybrid” porphyrin complexes is presented. Photophysical properties, such as absorption in the near-infrared, emission maxima, quantum yields, and lifetimes, are determined by the conjugation length of the porphyrin system, which leads to “tailor-made” NIR indicators for optical oxygen detection.
Introduction
The class of π-extended porphyrins,1 in which the pyrrole rings are fused with external aromatic fragments (e.g., tetrabenzoporphyrins (TBP) and tetranaptho[2,3]porphyrins (TNP)), continues to be intensively investigated in the fields of materials research,2 biomedical imaging and sensing,3 and photodynamic therapy.4 Platinum(II) and palladium(II) complexes are of particular interest since they possess strong room temperature near-infrared (NIR) phosphorescence.5,6 They were successfully applied in OLEDs,2b,7 photovoltaics,2a,8 and optical oxygen sensors.9 Particularly, NIR oxygen indicators overcome many drawbacks of the UV−vis indicators, such as the adverse effects of autofluorescence and light scattering in biological media.10 Additionally, NIR indicators are particularly promising for subcutaneous glucose sensing (implantable sensors or “smart tattoos”)11 which rely on oxygen transducers.
One way for shifting the absorption bands of transition metal porphyrins into the lower energy region is the extension of the porphyrin core by fusing it with external aromatic fragments.1,12 This method results in the so-called “π-extended porphyrins”. Tetra-annulated porphyrins like tetrabenzoporphyrins and tetranaphthoporphyrins and their metal complexes have already been intensively studied due to their attractive spectral features.13 However, the complexes of tetranaphthoporphyrins with palladium(II) or platinum(II) show relatively low luminescence quantum yields12,14 as well as extremely low photostability.15 In contrast to that, the corresponding tetrabenzoporphyrin complexes exhibit moderate photostability as well as high brightness.10
This work highlights the hybridization of both benzo- and naphthoporphyrins resulting in novel functional chromophores with interesting photophysical properties. The systematic condensation of additional external aromatic fragments leads to “tailor-made” NIR absorbing dyes, which cover the complete region from 630 to 690 nm and, therefore, perfectly match the commercially available laser diodes with excitation wavelengths of 635, 650, and 670 nm, respectively. We demonstrate that the extension of the aromatic system has a deep impact on the luminescence decay times as well as quantum yield of the corresponding palladium(II) and platinum(II) complexes. In this context, our method opens up a powerful tool to control the sensitivity to oxygen and at the same time to provide optimal dynamics in most sensor materials.10
Results and Discussion
Synthesis
The synthesis of π-extended porphyrins is based on the direct oxidative aromatization of porphyrins annealed with nonaromatic saturated hydrocarbon rings.1,12,13 The basic molecules such as the 4,5,6,7-tetrahydroisoindole as well as 4,9-dihydro-2H-benz[f]isoindole were prepared according to literature methods (cf. Supporting Information).12,13 Porphyrin synthesis was accomplished under optimized Lindsey conditions (cf. Scheme 1),16 introducing the “benzo” component 4,5,6,7-tetrahydroisoindole and the “naphtho” component 4,9-dihydro-2H-benz[f]isoindole in a molar ratio of 1.1/1 (10−2 M) in dry and deoxygenated CH2Cl2. The reaction mixture was protected from light, and 4-fluorobenzaldehyde was added in an equimolar amount (10−2 M). After stirring the reaction mixture at room temperature for 10 min, BF3·Et2O (10−3 M, 0.2 equiv) was added as a catalyst. After 1 h of reaction time, the water scavenger triethylorthoacetate (10−2 M) was added, which significantly improved the yield (approximately 10%). Subsequent addition of the catalyst leads to complete conversion after 4 h. The reaction mixture was allowed to stir overnight at room temperature in the presence of an excess of 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ), which then was removed by extraction with aqueous Na2SO3 and water. After purification by column chromatography on neutral alumina, the three main fractions, 1NF, 2NF, and 3NF, were isolated (overall yield, 42%; thereof, 3NF approximately 37%; 2NF approximately 34%; 1NF approximately 29%) and characterized by 1H NMR, UV−vis, and MALDI-TOF analysis (cf. Supporting Information). The 1H NMR spectra of the corresponding ligands display the correct number of proton signals, and the mass spectra confirm the molecular mass of the corresponding ligand (cf. Experimental Section). In this context, it is noteworthy that a complete oxidation of the naphtho moieties had taken place already during oxidative aromatization with DDQ. According to UV−vis and 1H NMR spectroscopy, the fraction of 2NF was isolated as a single isomer. Further evidence for the nature of the isomer could be retrieved from additional NMR studies. The 19F NMR spectrum of 2NF exhibits two distinct signals at −109.5 and −113.6 ppm (cf. Supporting Information Figure S4), whereas the latter signal represents two fluorophenyl moieties. In our hands, these peaks altogether indicate three chemically nonequivalent fluorophenyl groups, which evidence the presence of the cis isomer. For the corresponding trans isomer, only one signal would be expected due to its higher symmetry compared to the cis isomer. The 2D 1H/13C NMR−HMBC spectrum (cf. Supporting Information Figure S5) at peak positions 8.24/164.5 ppm and 8.24/162.5 ppm, respectively, reveals the characteristic one-bond carbon fluorine coupling constant JC-1,F = 249 Hz, which is comparable to the corresponding coupling constant of 4-fluorobenzaldehyde (JC-1,F = 255 Hz).17 The 2D 1H/13C NMR−HSQC spectrum (cf. Supporting Information Figure S6) gives additional information about the nature of the fluorophenyl protons. The signals at around 8.23/135.0 ppm represent the meta protons bound to the Ph3 carbons. The ortho protons bound to the Ph2 carbons reside at around 7.54/114.9 ppm. According to the 13C NMR spectrum (cf. Supporting Information Figure S3), the signals ranging from 116.6 to 115.0 ppm further indicate the presence of the three chemically nonequivalent fluorophenyl groups of the cis isomer. In fact, for the trans isomer, only two signals would be expected in this region due to symmetry reasons, which correspond to the two Ph3 carbon signals of 4-fluorobenzaldehyde at around 118 ppm.18 In this context, the signals for 2NF at 116.4 and 116.2 ppm reveal a coupling constant JC-2,F = 21.9 Hz, consistent with the corresponding coupling constant of 4-fluorobenzaldehyde (JC-2,F = 22.4 Hz).17
Scheme 1.

meso-Tetra(4-fluorophenyl)tetracyclohexenoporphyrin (B) was isolated as a side product and not further characterized. The formation of meso-tetra(4-fluorophenyl)tetranaphthoporphyrin (4NF) could not be observed for these optimized reaction conditions with a “benzo” to “naphtho” ratio of 1.1 to 1. In this context, it should be mentioned that the product distribution can potentially be modified by applying a different initial ratio of the “benzo” and “naphtho” starting materials. Moreover, 4-fluorobenzaldehyde was used in the synthesis because fluorine substitution of the phenyl groups in the meso position results in metalloporphyrins with improved photophysical properties, such as increased luminescence quantum yield as well as higher photostability.10 The title compounds Pd1NF, Pd2NF, and Pd3NF and Pt1NF, Pt2NF, and Pt3NF were prepared by the reaction of the corresponding ligand and the palladium and platinum precursors (MCl2(PhCN)2, M = Pd, Pt), respectively, and subsequent oxidation with DDQ (cf. Scheme 2). Palladation reactions proceeded smoothly in refluxing THF with 2 equiv of the Pd precursor. For platination reactions, harsher conditions were applied, using a Pt precursor to ligand ratio of 3:1 in 1,2,4-trimethylbenzene under reflux conditions (180 °C). Purification was accomplished by column chromatography using neutral Al2O3 (eluent: toluene for all complexes) as well as by recrystallization from CH2Cl2/MeOH after the removal of Al2O3 by filtration. Yields of analytically pure compounds ranged from 48 to 53% for the palladium(II) complexes and from 21 to 32% for the platinum(II) porphyrins, respectively. The complexes show good solubility in CHCl3, THF, diethylether, and acetone but are hardly soluble in methanol. The metalloporphyrins are air-stable in solution and in the solid state and were characterized by 1H NMR, UV−vis spectroscopy, and MALDI-TOF. The 1H NMR spectra of all compounds display the correct number of proton signals, and the mass spectra of all complexes confirm the molecular mass of the corresponding parent complex (cf. Experimental Section). As already shown for the 2NF ligand, the corresponding complexes are present as the cis isomers.
Scheme 2.

Photophysical Properties
Electronic Absorption Spectroscopy
The absorption spectra of all complexes measured in diluted toluene solutions at room temperature under ambient conditions show a subsequent bathochromic shift the more naphtho moieties are annealed to the porphyrin system. For the platinum(II) and palladium(II) complexes, the molar absorption coefficients are very high (cf. Table 1). In contrast to the minor shift of the Soret band, the Q band shifts significantly from 654 to 691 nm for the palladium(II) complexes and from 638 to 678 nm for platinum(II) porphyrins (cf. Table 1; Figure 1). In addition to the already literature-known tetra-annulated porphyrins palladium(II)-meso-tetra(4-fluorophenyl)tetrabenzoporphyrin (PdTPTBPF; λmax,abs 443 and 629 nm, solvent: toluene),10 palladium(II)-meso-tetraphenyltetranaphthoporphyrin (PdTPTNP; λmax,abs 463 and 705 nm, solvent: benzene),19 platinum(II)-meso-tetra(4-fluorophenyl)tetrabenzoporphyrin (PtTPTBPF; λmax,abs 430 and 615 nm, solvent: toluene),10 and platinum(II)-meso-tetraphenyltetranaphthoporphyrin (PtTPTNP; λmax,abs 436 and 689 nm, solvent: toluene),7 these “hybrid” porphyrins completely cover the spectral range between 615 and 705 nm. As a result, tailor-made near-infrared excitable porphyrin dyes are available for NIR oxygen sensing applications. These chromophores are particularly attractive materials for different multiplexing applications, e.g., simultaneous glucose and oxygen detection in enzymatic sensors. It should be emphasized that each additional naphtho moiety results in a red shift of the Q band of approximately 20 nm. Moreover, it should be noted that the Q-band absorption exhibits a slight red shift of approximately 2 nm in polymeric films containing 1 wt % of the indicator in polystyrene (cf. Supporting Information Figure S16).
Table 1. Photophysical Properties of the Platinum(II) and Palladium(II) Porphyrin Complexes at Room Temperature in Diluted Toluene Solutions.
| complex | abs, λmax, λ [nm] (ε [10−3 cm−1 M−1]) | emission, λmax [nm] | τ [μs] | Φ [%] |
|---|---|---|---|---|
| Pt1NF | 434 (272), 583 (25.6), 628 (147), 638 (140) | 815 | 44 | 53 |
| Pt2NF | 438 (106), 594 (14.2), 652 (108) | 835 | 28 | 27 |
| Pt3NF | 441 (108), 618 (17.2), 635 (21.9), 667 (83.5), 678 (78.9) | 870 | 21 | 25 |
| Pd1NF | 450 (202), 601 (12.7), 641 (74.0), 654 (72.6) | 849 | 203 | 18 |
| Pd2NF | 452 (190), 608 (102), 666 (142) | 868 | 138 | 12 |
| Pd3NF | 456 (138), 630 (17.0), 652 (22.0), 681 (63.8), 691 (60.0) | ∼882 | 106 | 7 |
Figure 1.

Absorption spectra of palladium(II) porphyrin complexes in toluene (*Q-band data from ref (19), c ∼ 10−3 M).
A comparative photophysical study as well as some theoretical calculations have been recently published in the work of Kobayashi et al.20 for reduced symmetry peripheral fused-ring-subtituted zinc phthalocyanines. In this context, the splitting observed in the Q bands of the trans isomer is significantly greater than is the case with the cis isomer despite the fact that the trans complex is the higher symmetry complex. The Q bands of Pd2NF and Pt2NF show a similar splitting like the zinc phthalocyanines cis isomer.20 In summary, extension of the conjugated system has only little effect on the Soret band but shifts the Q band significantly to lower energy. A question arises whether the differences in the absorption properties of these mixed benzo- and naphthoporphyrins are caused by differences in planarity of these systems, by the electronic effects of naphtho vs benzo moieties or by a combination of both of these factors. According to Finikova et al.,12 the Q-band transitions are mainly affected by the extension of π conjugation, which is in fact a substitution of the β-pyrrole carbons.
DFT calculations were performed for all complexes and reveal nonplanar equilibrium geometries in a “saddle”-like geometry. The calculated nonplanar structures (cf. Supporting Information) of the title compounds are in good agreement with the structure determinations (crystallographic data) for similar compounds described in the literature.7,12−14 The distortion from planarity observed for the metalloporphyrins can be attributed to steric repulsion between the 4-fluorophenyl groups in the meso position and the macrocycle, respectively. Calculations for the ligand 1NF also remain nonplanar, whereas naphtho annulated porphyrins without the 4-fluorphenyl groups in the meso position return to a flat geometry. In agreement with the experiment, we find in all cases that the strongest excitation occurs in the 420 nm region (approximately 429 nm for Pd1NF and 420 nm for Pt1NF) and does not shift much with increasing number of naphtho moieties. It should be stressed in this context that the Soret band in fact consists of several optical transitions which are all found between ∼350 and ∼420 nm. In this context, we will only comment on the strongest excitation in this region. In contrast, the spectral position of the Q band changes significantly. For Pd1NF, TD-DFT predicts two peaks of similar intensity located at 594 and 610 nm, however, significantly blue-shifted with respect to the experimental peaks (641 nm, 654 nm). In the case of Pd2NF, the cis and trans isomers were calculated. The optical spectra of both compounds are markedly different: while for the trans isomer the double-peak shape of the Q band is retained, in the cis isomer these two transitions coincide at the same energy, 619 nm. Thus, the optical spectra corroborate the results from NMR measurements which indicate the formation of the cis isomer. By adding yet another naphtho moiety, the double peak structure of the Q band is recovered. The TD-DFT calculation for Pd3NF predicts two peaks at wavelengths of 630 and 648 nm, i.e., again red-shifted with respect to the smaller conjugated systems. In total, between Pd1NF and Pd3NF, DFT finds a shift of 36 and 40 nm for the two Q-band peaks, in excellent agreement with the experimental findings of 40 and 37 nm, respectively. The Pt derivatives show the same trends—including the near-degenerated excitation in cis-Pt2NF—as the Pd molecules, but the transitions occur at somewhat larger energies, in accordance with the experiment. In order to discern the influence of increased conjugation and geometry changes induced by the addition of the naphtho groups, we have recalculated all molecules in a planar conformation. In all cases, this induces a rigid blue-shift with respect to the equilibrium geometry of about 10−20 nm. The blue-shift affects all transition rather equally; i.e., the Soret band is shifted as well as the Q band. In the cis-M2NF systems, the near-degeneracy of the two Q-band transitions is not lifted, underlining the stability of this result. In the planar geometries, we find the shift of the Q-band peaks between Pd1NF and Pd3NF to be 40 and 41 nm, i.e., only slightly larger than in the equilibrium geometries. It can thus be concluded that the shift is indeed based on the π extension rather than geometrical distortions, as predicted by Finikova et al.12 Comparative theoretical calculations have been recently described for Zn and Pd complexes of symmetrically extended porphyrins, which revealed that the influence of nonplanar distortion is much smaller on the red shifts of the optical transitions than that of the π conjugation.19 This behavior can be rationalized by looking at the corresponding orbitals (cf. Figure 2). The energetically lower lying excitation of the Q band corresponds mainly to a HOMO→LUMO transition, while the energetically higher lying Q-band excitation mainly involves HOMO→LUMO+1. As Figure 2 illustrates, LUMO and LUMO+1 are almost degenerated in Pt2NF, resulting in (almost) degenerated excitation energies. In Pt1NF and Pt3NF, LUMO and LUMO+1 are different energetically, giving rise to the characteristic double-peak shape. Within the series, both LUMO and LUMO+1 vary only very little in energy, and hence the strong shift of the Q band to higher wavelength is dominated by the energy increase of the HOMO, triggered by the larger size of the conjugated system. In contrast, the main contributions to the Soret band include the HOMO−1 orbital. Unlike the HOMO, the HOMO−1 shows hardly any lobes on the naphtho moieties. It is thus unaffected by the extension of the π system and remains at practically the same energy, thus leading to constant transition energies for all three molecules.
Figure 2.

Energies and isodensity representations of the Kohn−Sham orbitals HOMO−1, HOMO, LUMO, and LUMO+1 calculated for the platinum(II) porphyrins.
Luminescence Spectroscopy
Palladium(II) complexes emitted in toluene solutions under ambient conditions with maxima ranging from 849 to 882 nm (cf. Supporting Information Figure S17), and the corresponding platinum(II) compounds revealed emission maxima in the range of 815 to 870 nm (cf. Table 1, Figure 3). Compared to the emission maxima of the tetra-annulated porphyrins palladium(II)-meso-tetra(4-fluorophenyl)tetrabenzoporphyrin (PdTPTBPF; λmax,em 803 nm, solvent: toluene),10 palladium(II)-meso-tetraphenyltetranaphthoporphyrin (PdTPTNP; λmax,em 937 nm, solvent: benzene),19 platinum(II)-meso-tetra(4-fluorophenyl)tetrabenzoporphyrin (PtTPTBPF; λmax,em 773 nm, solvent: toluene),10 and platinum(II)-meso-tetraphenyltetranaphthoporphyrin (PtTPTNP; λmax,em 883 nm, solvent: toluene),7 these “hybrid” porphyrins complement the NIR emission spectral range between 773 and 937 nm. As a result, not only absorption but also emission wavelengths are tunable by systematic extension of the aromatic porphyrin framework.
Figure 3.

Emission spectra of platinum(II) porphyrins measured in toluene at room temperature.
Quantum yields (Φ) of the complexes were measured in deoxygenated toluene solutions, and palladium(II)-meso-tetraphenyltetrabenzoporphyrin (PdTPTBP) was used as the standard. For the palladium complexes, the observed luminescence quantum yields are 18% for Pd1NF, 12% for Pd2NF, and 7% for Pd3NF. The obtained values are consistent with the quantum yields known for PdTPTBPF (Φ = 23%) and PdTPTNP (Φ = 6.5%).10,20 Platinum complexes possess significantly higher luminescence quantum yields (53, 27, and 25%, respectively, for Pt1NF, Pt2NF, and Pt3NF), and the corresponding values for PtTPTBPF (Φ = 60%) and PtTPTNP (Φ = 22%)7,10 are consistent with our results. Luminescence lifetimes are in agreement with the corresponding tetrabenzoporphyrin complexes (297 μs for PdTPTBPF, 50 μs for PtTPTBPF)10 and tetranaphthoporphyrin complexes (65 μs for PdTPTNP,20 8.5 μs for PtTPTNP(7)), respectively. Similar to meso-tetraaryltetranaphtho[2,3]porphyrins (Ar4TNP) described by Finikova et al.,12 every additional fused naphtho moiety enhances radiativeless deactivation of the triplet states, which is most probably caused by lower energy levels. However, room temperature phosphorescence quantum yields of the metalloporphyrins presented in this work render these phosphorescent dyes very suitable for incorporation into near-IR light-emitting devices.2b,7
Oxygen Quenching
Phosphorescence of all complexes was found to be efficiently quenched by oxygen both in toluene solutions and in polystyrene films (cf. Figure 4). Polystyrene was chosen as a rigid polymer with good optical properties and moderate diffusion and partition coefficients for oxygen,5,21 as well as a model matrix to enable better comparison with literature data.10 In the case of the heterogeneous luminescent oxygen-sensing films, the Stern−Volmer plots are not linear, which is in contrast to the behavior in solutions. This nonlinear behavior is common for most oxygen-sensitive materials, including benzoporphyrins in polystyrene.9b,9c The so-called two-site model is used to describe the quenching plots.22 It assumes localization of an oxygen-sensitive chromophore in two environments originating from microinhomogeneities in the polymeric film (e.g., in crystallinity). Since these areas of the polymer possess different gas permeabilities, two Stern−Volmer constants KSV1 and KSV2 are obtained for the same chromophore:
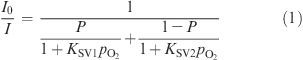 |
1 |
where P is the partition coefficient, i.e., the fraction of the chromophore located in the first environment, with 1 − P, respectively, for the second environment. The simplified equation (KSV2 is set as 0) is found to excellently fit the nonlinear Stern−Volmer plots, also for the decay time (correlation coefficient >0.998). It should be mentioned here that the two-site model approximation physically makes sense only for the luminescence intensity fit. In contrast, in the presence of oxygen, the decay times are expected to become substantially different in both environments, and the overall signal is not monoexponential. However, the decay times obtained in the frequency domain are averaged to the certain extent which can explain why the fit with the equation from the “two-site model” is adequate. The Stern−Volmer constants KSV as well as the quenching constants kq calculated from the Stern−Volmer plots (obtained from the decay time plots) are summarized in Table 2. The KSV values in polystyrene films (1% of the indicator, w/w) and in toluene solution are consistent with the luminescence decay times. Oxygen sensitivity in polystyrene decreases moving Pd1NF with a KSV1 of 0.60 kPa−1 to Pd3NF with a KSV1 of 0.47 kPa−1. Palladium(II)-meso-tetraphenyltetrabenzoporphyrin (PdTPTBP)10 exhibits the highest sensitivity (KSV1 = 0.92 kPa−1), which corroborates the trend in this series. Platinum(II) porphyrins show significantly lower sensitivity than the respective palladium(II) complexes (which correlates with the shorter decay times of the former), but a similar trend for the KSV1 values is observed. Platinum(II)-meso-tetraphenyltetrabenzoporphyrin (PtTPTBP)10 completes the series revealing the same trend as for the corresponding palladium compounds in solution as well in polymeric film. Interestingly, the quenching constants kq in polymer films (kq,film) and in toluene solution (kq,solution) show the following trends: PdTPTBP < Pd1NF < Pd2NF < Pd3NF and PtTPTBP < Pt1NF < Pt2NF < Pt3NF, which correlate well with the increased radii of the metalloporphyrin molecules. The increase in the indicator size should result in a higher probability of the collision between the excited indicator and molecular oxygen. The obtained data (cf. Table 2) clearly show that these “hybrid” porphyrin complexes are suitable as indicators for use in optical oxygen sensors. Considering the sensitivity, the materials based on the platinum(II) complexes are excellently suitable for most biological applications, and the more sensitive palladium(II) porphyrin-based sensors are more adequate for measurements under anoxic conditions.
Figure 4.

Stern−Volmer plots for the Pt(II) complexes embedded in polystyrene (at 25 °C). Curve fitting is performed according to eq 1 (KSV2 = 0).
Table 2. Quenching Constants (kq), Stern−Volmer Constants (KSV), and Partition Coefficients (P) of the Porphyrin Complexes in Polystyrene (1% of the Indicator, w/w) and in Toluene Solution.
| complex | KSV1,film [kPa−1] | P | kq,film [Pa−1s−1] | KSV,solution [kPa−1] | kq,solution [Pa−1s−1] |
|---|---|---|---|---|---|
| PdTPTBP | 0.92 ± 0.01a | 0.87a | 2.6 ± 0.1a | ||
| Pd1NF | 0.60 ± 0.02 | 0.88 | 3.6 ± 0.2 | 15.8 ± 0.3 | 85 ± 4 |
| Pd2NF | 0.50 ± 0.03 | 0.92 | 4.2 ± 0.2 | 15.4 ± 0.2 | 114 ± 6 |
| Pd3NF | 0.47 ± 0.01 | 0.92 | 4.8 ± 0.2 | 10.4 ± 0.2 | 118 ± 6 |
| PtTPTBP | 0.17 ± 0.01a | 0.87a | 3.0 ± 0.1a | ||
| Pt1NF | 0.13 ± 0.01 | 0.84 | 3.7 ± 0.2 | 2.8 ± 0.1 | 69 ± 3 |
| Pt2NF | 0.11 ± 0.01 | 0.80 | 3.9 ± 0.2 | 2.4 ± 0.1 | 93 ± 5 |
| Pt3NF | 0.08 ± 0.01 | 0.66 | 4.8 ± 0.2 | 1.9 ± 0.1 | 98 ± 5 |
Data from ref (10).
Photostability
The photostability of oxygen indicators is of particular interest for practical applications, especially in those cases where high light densities are applied or long-term measurements are performed. As theoretically predicted by Kobayashi et al.,15 π-ring expansion results in the destabilization of the third LUMOs and the first HOMOs of the porphyrins, and therefore, they become unstable against oxidation and reduction. In this work, the photostability of all complexes is investigated in solution as well as in the polystyrene films. For photostability measurements in solution, PdTPTBP and PtTPTBP were used as reference materials. The photostability of the complexes in DMF solution was determined by continuously irradiating the samples with a red LED array (λmax = 638 nm, www.led-tech.de, Flux: 6200 μmols−1 m−2 μA at 5.0 W). Data presented in Figure 5 indicate that the photostability of the hybrid systems is significantly lower than that of PtTPTBP. In fact, after 1 min of irradiation, 4% of Pt1NF, 14% of Pt2NF, and 25% of Pt3NF, respectively, is destroyed compared to 0.2% of PtTPTBP. The decrease in photostability correlates well with the number of naphtho moieties annealed to the porphyrin system. Photobleaching rates for the palladium(II) and the platinum(II) complexes are comparable (photodegradation after 1 min: 6% for Pd1NF, 15% for Pd2NF, 30% for Pd3NF, and 0.6% for PdTPTBP; cf. Supporting Information). The nature of the solvent has a pronounced influence on the photobleaching rates. For example, the photodegradation of the platinum complex Pt3NF was further investigated with toluene as the solvent. After 5 min of irradiation, the absorption of Pt3NF in toluene remained unchanged. In contrast to that, 52% of Pt3NF was destroyed under the same irradiation conditions with DMF as a solvent (cf. Supporting Information Figure S35). Finally, it should be emphasized that irradiation of the complexes in solution results in a complete degradation of the porphyrin system, and no additional chromophores are formed.
Figure 5.

Photodegradation curves for the platinum(II) porphyrins in DMF solution at room temperature. Irradiation is performed with a red LED array at 638 nm (flux: 6200 μmols−1 m−2 μA).
The photostability of the complexes in polystyrene under synthetic air was estimated upon continuous irradiation of the samples with a blue LED (λmax = 450 nm, Roithner Lasertechnik; Vienna, Austria, Flux: 200 μmols−1 m−2 μA) and monitoring the emission intensity as well as the decay time. According to the obtained results, the “mononaphtho” complexes Pd1NF and Pt1NF show a significantly higher photostability compared to the “dinaphto” (Pd2NF, Pt2NF) and “trinaphtho” complexes (Pd3NF, Pt3NF). In fact, after 30 min of irradiation, 18% of Pd2NF is destroyed (cf. Figure 6), compared to 3% of Pd1NF (cf. Supporting Information). Platinum(II) porphyrins show comparable photodegradation values of 2% for Pt1NF and 19% for Pt2NF. In contrast to that, the luminescence decay times remained stable for all complexes during 30 min of irradiation time (cf. Figure 6, Supporting Information). Photostability measurements under a nitrogen atmosphere revealed stable signals for luminescence intensity as well as decay time during 30 min of irradiation time (cf. Figure 6, Supporting Information). Thus, all the indicators perform well in the sensor material, and no influence of photobleaching on the luminescence decay times could be observed. In addition, no recalibration is needed for these dyes; however, the signal-to-noise ratio deteriorates under air. However, it should be mentioned that a continuous irradiation time of 30 min corresponds to 36 000 measurement points when a short light pulse of 50 ms is applied.
Figure 6.

Photodegradation curves for the metalloporphyrin Pd2NF embedded in a polystyrene film (1% of the indicator, w/w; 2.5-μm-thick film). Irradiation is performed with a blue LED (λmax = 450 nm, flux: 200 μmols−1 m−2 μA).
In summary, the photostability of these mixed benzo- and naphthoporphyrins can be a critical issue. For some potential applications, especially for those where high light intensities are used (e.g., microscopy, fiber optical microsensors) or very long acquisition is applied, photostability should be improved. Considering the significantly higher photobleaching rates in the presence of oxygen, the photo-oxydation by singlet oxygen is the most probable photodegradation pathway. Therefore, the photostability is expected to dramatically improve if the hydrogen atoms are substituted by the halogens, e.g., fluorine or chlorine. Due to the electron-withdrawing effect of the halogen atoms, such perhalogenated derivatives are expected to be more difficult to oxidize and, therefore, are likely to represent highly photostable indicators. It should be mentioned here that more sophisticated synthetic routes for the preparation of a new precursor for naphthoporphyrins bearing a perchlorinated outer ring are possible.1 In addition to the improved photostability, the chlorine atoms are expected to further improve the solubility in organic solvents and in polymers. Current efforts are directed at developing efficient synthetic strategies for perchlorination.
Conclusion
In conclusion, mixed benzo- and naphthoporphyrin complexes of palladium(II) and platinum(II) have been prepared. Absorbance and luminescence as well as lifetime measurements in solution and in the solid state have been performed to establish a qualitative relationship between structure and luminescence properties. The compounds under investigation show tunable absorption in the near-infrared region (628−691 nm) as well as controllable emission in the range of 815−882 nm. In addition, these metalloporphyrins are strongly luminescent, with quantum yields up to 53% for platinum(II) porphyrins and up to 18% for palladium(II) complexes, respectively. DFT calculations revealed nonplanar geometries for all complexes. The calculations corroborate that only the cis isomer was formed during the synthesis according to Lindsey. Moreover, they also show a strong bathochromic shift in the Q band, while the Soret band remains almost constant. This behavior is rationalized by the different energy evolution of the involved orbitals HOMO and HOMO−1, respectively, with respect to the increased size of the conjugated system. The systematic extension of the conjugated system by annealing further naphtho moieties to the porphyrin system results in “tailor-made” near-infrared absorbing indicators for oxygen. These chromophores are particularly promising candidates for different multiplexing applications, e.g., simultaneous glucose and oxygen determination in enzymatic sensors. The enlargement of the aromatic system has a significant impact on the photophysical properties. Particularly, the emission bands shift bathochromically, the luminescence lifetime becomes shorter, and the luminescence quantum yields decrease. The sensitivity of the materials to oxygen is also affected and gradually reduces upon substitution. Unfortunately, the substitution significantly increases the photobleaching rates. In this context, our future synthetic work concerning these hybrid systems will essentially focus on the improvement of the photochemical stability via halogenation of the porphyrin macrocycle.
Experimental Section
Materials and Methods
Ethyl isocyanoacetate, 1-nitro-1-cyclohexene, and 2,3-dicyano-5,6-dichlorobenzoquinone (DDQ) were purchased from Aldrich (www.sigmaaldrich.com). 4-Fluorobenzaldehyde, palladium(II) chloride, platinum(II) chloride, and benzonitrile were purchased from ABCR (www.abcr.de). Dimethylformamide, N,N-dimethyldiisopropylamine, and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) were obtained from Fluka (www.sigmaaldrich.com). All other solvents were obtained from Roth (www.carl-roth.de). The silica-gel 60 (0.063−0.200 mm) was purchased from Merck (www.merck.de), and the neutral aluminum oxide (50−200 μm) was purchased from Acros Organics (www.acros.com). Polystyrene (mw: 250 000) was obtained from Fischer Scientific (www.fishersci.com). Poly(ethyleneterephthalate)-Mylar-support was purchased from Goodfellow. Nitrogen and synthetic-air (all of 99.999% purity) were obtained from Air Liquide (www.airliquide.at). Unless otherwise noted, all other materials were obtained from commercial sources (Aldrich, Fluka, ABCR, and Acros) and were used without further purification. PdCl2(PhCN)2 and PtCl2(PhCN)2 were prepared according to a literature procedure.23 The complexes palladium(II)-meso-tetraphenyltetrabenzoporphyrin (PdTPTBP) and platinum(II)-meso-tetraphenyltetrabenzoporphyrin (PtTPTBP) were prepared according to a technique reported elsewhere.10
1H NMR spectra were recorded on a Varian INOVA 500 MHz spectrometer at 500 MHz or a Bruker Avance III 300 MHz spectrometer at 300 MHz. 13C NMR, 19F NMR, 2D HMBC, and 2D HSQC spectra were recorded on a Varian INOVA 500 MHz spectrometer at 500 MHz. UV−visible absorption spectra were recorded on a Cary 50 Bio UV−visible spectrophotometer and fluorescence spectra on a Hitachi F-7000 fluorescence spectrometer equipped with a red-sensitive photomultiplier, R 928, from Hamamatsu. The emission spectra were corrected for the sensitivity of the PMT, which was calibrated with a halogen lamp.24,25 Relative luminescence quantum yields were determined using a solution of PdTPTBP in toluene as a standard (Φ = 0.2110). The solutions of the dyes were thoroughly deoxygenated by bubbling nitrogen through for 20 min. The quantum yields as well as luminescence phase shifts in solution were determined using a lock-in amplifier (PreSens, www.presens.de) equipped with a silicon photodiode. Excitation of the complexes was performed with a 435 nm LED (www.roithner-laser.com), which was sinusoidally modulated at a frequency of 916 Hz (absorbances at the excitation wavelength of 435 nm were 0.1 for all complexes). The errors in measurement were estimated as ±10% for luminescence quantum yields and ±5% for luminescence decay times, respectively.
Phase angle measurements concerning oxygen sensitivity in solution were performed with a 450 nm LED (www.roithner-laser.com). For phase angle measurements in solution, the following modulation frequencies were applied: 916 Hz for Pd1NF, 1831 Hz for Pd2NF, 2747 Hz for Pd3NF, 4578 Hz for Pt1NF, 5493 Hz for Pt2NF, and 6409 Hz for Pt3NF. The polymer “cocktails” for the sensor films were prepared by dissolving the metal complex and polystyrene in chloroform (10% w/w of PS in CHCl3). The “cocktails” were knife-coated on the Mylar support to give, after solvent evaporation, phosphorescent sensor films with a thickness of 2.5 μm. The films contained ∼1% (w/w) of the dye in the polymer. For phase angle measurements in polymeric films, the following modulation frequencies were applied: 1831 Hz for Pd1NF, 4578 Hz for Pd2NF and Pd3NF, 6409 Hz for Pt1NF, 8240 Hz for Pt2NF, and 10986 Hz for Pt3NF. Excitation of the complexes was performed with a 450 nm LED (www.roithner-laser.com). A BG 12 filter (Schott, www.schott.com) was used for excitation and an RG 9 filter (Schott) for emission for all phase angle measurements. Temperature was controlled by a ThermoHaake DC50 cryostat at 25 °C. Gas calibration mixtures were obtained using a gas mixing device (MKS, www.mksinst.com).
Photostability of the dyes in polymeric films was determined using a lock-in amplifier from PreSens. Sensor foils were positioned at 30° relative to the photodiode and the light source. Continuous irradiation was performed with the light of a 450 nm LED (www.roithnerlaser.com, Flux: 200 μmols−1 m−2 μA) filtered through a BG 12 filter. An RG 9 filter (Schott) was used for the emission. The same modulation frequencies as for oxygen sensitivity measurements were applied. The bleaching rates were corrected for the amount of the absorbed light at 456 nm. Photostability of the complexes in DMF solution was determined continuously irradiating the samples with a red LED array (λmax = 638 nm, www.led-tech.de, Flux: 6200 μmols−1 m−2 μA at 5.0 W).
MALDI-TOF mass spectra were recorded on a Micromass TofSpec 2E. The instrument is equipped with a nitrogen laser (337 nm wavelength, operated at a frequency of 5 Hz) and a time lag focusing unit. Spectra were taken in reflectron mode at an accelerating voltage of +20 kV. Analysis of the data was done with MassLynx 3.4 (Micromass, Manchester, U.K.). Samples were dissolved in THF (1 mg/cm3). Dithranol or retinoic acid was used as the matrix (10 mg/cm3 in THF). Solutions were mixed in the cap of a microtube in a ratio of 1:10 μL. A total of 0.5 μL of the resulting mixture was spotted onto the target and air-dried.
Density functional theory calculations were performed using the Turbomole 5.7 software suite.26 The B3LYP exchange correlation functional27 was employed together with the SVP basis set28 and relativistic effective core potentials for Pd and Pt.29 Geometry optimizations were performed explointing symmetry where possible. After the optmization, frequency calculations without imaginary frequencies ensured that a true minimum was reached. To obtain planar geometries, a biased start geometry was constructed. The planarity was enforced by increasing the symmetry point group accordingly. Frequency calculations performed after the geometry optmization showed four imaginary frequencies. Optical excitations were calculated within the time-dependent DFT framework. For all molecules, the 10 lowest lying excitations of each irreducible representation were collected. Test calculations using the higher quality TZVP30 basis set for the exitations only yielded the same general shape of the spectra. The transitions were slightly red-shifted and therefore in even better agreement with the experiment.
Ligand Synthesis via the Lindsey Method
4,5,6,7-Tetrahydroisoindole (529.8 mg, 4.37 mmol, 0.52 equiv) and 4,9-dihydro-2H-benz[f]isoindole (673.5 mg, 3.98 mmol, 0.48 equiv) were dissolved in dry CH2Cl2 (835 mL). The reaction mixture was degassed for 20 min with N2. The stirred mixture was protected from light and stirred under N2. 4-Fluorobenzaldehyde (896 μL, 1.04 g, 8.35 mmol, 1.0 equiv) was added, and the reaction mixture was kept in the dark under N2 and stirred for 10 min. BF3·Et2O (210 μL, 237 mg, 1.7 mmol, 0.2 equiv) was added in one portion, and the mixture was allowed to react at room temperature for 1 h. After 1 h, triethylorthoacetate (TEOAc; 1531 μL, 1355 mg, 8.35 mmol, 1.0 equiv) was added, and the reaction mixture was stirred at room temperature for 30 min. After 30 min, BF3·Et2O (40 μL, 45.2 mg, 0.32 mmol, 0.04 equiv) was added, and the reaction mixture was stirred for an additional 30 min. Finally, one last portion of BF3·Et2O (40 μL, 45.2 mg, 0.32 mmol, 0.04 equiv) was added, and the reaction mixture was stirred for 2 h. After an overall reaction time of 4 h, DDQ (9.5 g, 41.8 mmol, 5.0 equiv) was added to the mixture in one portion, and the mixture was stirred at room temperature overnight. The resulting mixture was washed with 10% aq. Na2SO3 (2 × 100 mL) and with water (2 × 100 mL). The combined organic phases were dried over Na2SO4. The solvent was evaporated, and the resulting residue was purified by column chromatography on neutral alumina (3NF: toluene, CH2Cl2/toluene = 1:1 (v/v), CH2Cl2; 2NF: CH2Cl2 + 1% THF; 1NF: CH2Cl2 + 2% THF). The three main fractions were isolated as green solids with an overall yield of 42%.
1NF
Yield: 254.8 mg, 29%. 1H NMR (δ, 20 °C, CDCl3, 300 MHz): 8.46−8.12 (m, 8H), 7.73−7.39 (m, 14H), 2.51−2.17 (m, 12H), 1.74−1.67 (m, 12H). MALDI-TOF: m/z 949.3837, calcd 949.3893. UV−vis, toluene, λmax nm (relative intensity): 460 (1.00), 548 (0.11), 586 (0.08), 628 (0.07), 690 (0.04).
2NF
Yield: 301.3 mg, 34%. 1H NMR (δ, 20 °C, CDCl3, 300 MHz): 8.33−8.08 (m, 8H), 7.72−7.39 (m, 20H), 1.69−1.47 (m, 8H), 2.54−2.24 (m, 8H). 13C NMR (δ, 20 °C, CDCl3, 500 MHz): 165.2, 164.8, 164.6, 164.5, 163.2, 162.8, 162.6, 162.5, 142.2, 138.7, 138.6, 137.88, 137.86, 137.43, 137.41, 136.5, 136.4, 136.0, 135.92, 135.88, 135.82, 131.7, 131.5, 130.78, 130.76, 130.3, 130.2, 129.4, 129.2, 128.3, 128.2, 126.98, 126.96, 126.7, 126.4, 126.2, 124.4, 123.7, 116.6, 116.5, 116.4, 116.2, 115.9, 115.8, 115.6, 115.2, 115.0, 29.9, 29.5, 27.8, 25.9, 25.8, 24.5, 23.6, 23.5. 19F NMR (δ, 20 °C, CDCl3, 500 MHz): −113.6 (2F), −109.5 (1F). MALDI-TOF: m/z 995.3709, calcd 995.3737. UV−vis, toluene, λmax nm (relative intensity): 471 (1.00), 589 (0.09), 630 (0.13), 691 (0.04).
3NF
Yield: 324.2 mg, 37%. 1H NMR (δ, 20 °C, CDCl3, 300 MHz): 8.33−8.20 (m, 8H), 7.81−7.36 (m, 22H), 7.28−7.26 (m, 4H, ovelap with solvent), 2.55−2.41 (m, 4H), 1.68−1.54 (m, 4H). MALDI-TOF: m/z 1040.3414, calcd 1040.3502. UV−vis, toluene, λmax nm (relative intensity): 475 (1.00), 558 (0.04), 599 (0.08), 641 (0.14), 718 (0.09).
Synthesis of the Pd(II) and Pt(II) Porphyrins
Pd1NF
An excess of PdCl2(PhCN)2 (36.0 mg, 0.09 mmol, 2.0 equiv) was added to a solution of porphyrin ligand 1NF (44.5 mg, 0.05 mmol, 1.0 equiv) in THF (20 mL), and the mixture was refluxed for 15 min. N,N-dimethyldiisopropylamine (20 μL) was added as a base, and the mixture was refluxed for an additional 15 min. The conversion was monitored by UV−vis spectroscopy (solvent: toluene). DDQ (106.0 mg, 0.47 mmol, 10 equiv) was added, and the mixture was refluxed for 15 min. The oxidation was monitored by UV−vis spectroscopy (solvent toluene). The solvent volume was reduced to 20 mL under vacuum conditions. CH2Cl2 (100 mL) was added, and the mixture was washed with aqueous 10% Na2SO3. The organic phase was dried over Na2SO4, and the solvent was evaporated. The crude product was purified by column chromatography on Al2O3 (removal of excess DDQ, eluent: n-hexane/toluene = 2:1 (v/v); elution of Pd1NF, eluent: toluene). Final purification was accomplished by recrystallization from CH2Cl2/MeOH after the removal of Al2O3 by filtration to yield the product as a green solid. Yield: 26.1 mg, 53%. 1H NMR (δ, 20 °C, CDCl3, 300 MHz): 8.30−8.20 (m, 8H), 7.74−7.49 (m, 14H), 7.31−7.12 (m, 12H, overlapped w/solv). MALDI-TOF: m/z 1038.1804, calcd 1038.1761.
The preparation of Pd2NF was performed similarly to Pd1NF starting from PdCl2(PhCN)2 (38.5 mg, 0.10 mmol, 2.0 equiv) and 2NF (50.0 mg, 0.05 mmol, 1.0 equiv). Yield: 26.4 mg, 48%. 1H NMR (δ, 20 °C, CDCl3, 300 MHz): 8.35−8.19 (m, 8H), 7.75−7.50 (m, 20H), 7.30−7.19 (m, 6H, ovelapped w/solv), 7.14−7.11 (m, 2H). MALDI-TOF: m/z 1088.2034, calcd 1088.1919.
Pd3NF was synthesized analogously to Pd1NF starting from PdCl2(PhCN)2 (37.9 mg, 0.10 mmol, 2.0 equiv) and 3NF (51.5 mg, 0.05 mmol, 1.0 equiv). Yield: 27.5 mg, 49%. 1H NMR (δ, 20 °C, CDCl3, 300 MHz): 8.35−8.24 (m, 8H), 7.75−7.49 (m, 26H), 7.31−7.19 (m, 4H, ovelapped w/solv). MALDI-TOF: m/z 1138.2065, calcd 1138.2075.
Pt1NF
An excess of PtCl2(PhCN)2 (90.4 mg, 0.19 mmol, 3.6 equiv) was added to a solution of porphyrin ligand 1NF (50.0 mg, 0.05 mmol, 1.0 equiv) in 1,2,4-trimethylbenzene (20 mL), and the mixture was refluxed for 10 min. N,N-dimethyldiisopropylamine (40 μL) was added as a base, and the mixture was refluxed for an additional 30 min. The conversion was monitored by UV−vis spectroscopy (solvent: toluene). The reaction mixture was cooled to room temperature and filtered over neutral alumina to remove colloidal platinum black. The solvent was evaporated, and the crude product was dried. Oxidation was performed in THF (20 mL) with DDQ (134.6 mg, 0.59 mmol, 12 equiv) by refluxing the mixture for 15 min. The oxidation was monitored by UV−vis spectroscopy (solvent: toluene). The solvent volume was reduced to 20 mL. CH2Cl2 (100 mL) was added, and the mixture was washed with aqueous 10% Na2SO3. The organic phase was dried over Na2SO4, and the solvent was evaporated. The remaining material was purified by column chromatography on Al2O3 (removal of excess of DDQ, eluent: n-hexane/toluene = 2:1 (v/v); elution of Pt1NF, eluent: toluene). Final purification was accomplished by recrystallization from CH2Cl2/MeOH after the removal of Al2O3 by filtration to yield the product as a green solid. Yield: 20.0 mg, 32%. 1H NMR (δ, 20 °C, CDCl3, 300 MHz): 8.30−8.19 (m, 8H), 7.73−7.50 (m, 14H), 7.32−7.26 (m, 7H, ovelapped w/solv), 7.20−7.17 (m, 1H), 7.12−7.09 (m, 4H). MALDI-TOF: m/z 1128.2311, calcd 1128.2346.
The preparation of Pt2NF was performed similarly to that of Pt1NF starting from PtCl2(PhCN)2 (79.0 mg, 0.17 mmol, 2.8 equiv) and 2NF (59.4 mg, 0.06 mmol, 1.0 equiv). Yield: 17.4 mg, 25%. 1H NMR (δ, 20 °C, CDCl3, 300 MHz): 8.35−8.19 (m, 8H), 7.73−7.51 (m, 20H), 7.31−7.08 (m, 8H, ovelapped w/solv). MALDI-TOF: m/z 1178.256, calcd 1178.250.
Pt3NF was synthesized analogously to Pt1NF starting from PtCl2(PhCN)2 (99.0 mg, 0.21 mmol, 3.0 equiv) and 3NF (50.0 mg, 0.07 mmol, 1.0 equiv). Yield: 18.5 mg, 21%. 1H NMR (δ, 20 °C, CDCl3, 300 MHz): 8.35−8.25 (m, 8H), 7.73−7.53 (m, 26H), 7.33−7.18 (m, 4H, ovelapped w/solv). MALDI-TOF: m/z 1228.2660, calcd 1228.2631.
Acknowledgments
Financial support by the Austrian Science Fund (FWF; Research Project No. P21192−N17) is gratefully acknowledged.
Supporting Information Available
Detailed synthetic procedures, comprehensive photophysical data (tables, absorption, excitation and emission spectra, oxygen quenching, photostability), NMR spectra, MALDI-TOF data, orbitals, and DFT calculated geometries. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Cheprakov A. V.; Filatov M. A. J. Porphyrins Phthalocyanines 2009, 13, 291–303. [Google Scholar]
- a Perez M. D.; Borek C.; Forrest S. R.; Thompson M. E. J. Am. Chem. Soc. 2009, 131, 9281–9286. [DOI] [PubMed] [Google Scholar]; b Borek C.; Hanson K.; Djurovich P.; Thompson M.; Aznavour K.; Bau R.; Sun Y.; Forrest S.; Brooks J.; Michalski L.; Brown J. Angew. Chem., Int. Ed. 2007, 46, 1109–1112. [DOI] [PubMed] [Google Scholar]; c Brunel M.; Chaput F.; Vinogradov S. A.; Campagne B.; Canva M.; Boilot J. P.; Brun A. Chem. Phys. 1997, 218, 301–307. [Google Scholar]; d Guha S.; Kang K.; Porter P.; Roach J. F.; Remy D. E.; Aranda F. J.; Rao D. V. G. L. N. Opt. Lett. 1992, 17, 264–266. [DOI] [PubMed] [Google Scholar]
- a Finikova O.; Galkin A.; Rozhkov V.; Cordero M.; Haggerhall C.; Vinigradov S. J. Am. Chem. Soc. 2003, 125, 4882–4893. [DOI] [PubMed] [Google Scholar]; b Apraleva S. V.; Wilson D. F.; Vinogradov S. A. Appl. Opt. 2006, 45, 8547–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg J. S.; Skema C.; Baum E. D.; Burdick J.; Vinogradov S. A.; Wilson D. F.; Horan A. D.; Nachamkin I. J. Antimicrob. Chemother. 2001, 48, 105–107. [DOI] [PubMed] [Google Scholar]
- Amao Y. Microchim. Acta 2003, 143, 1–12. [Google Scholar]
- Papkovsky D.; O’Riordan T. J. Fluoresc. 2005, 15, 569–584. [DOI] [PubMed] [Google Scholar]
- Sommer J. R.; Farley R. T.; Graham K. R.; Yang Y.; Reynolds J. R.; Xue J.; Schanze K. S. Appl. Mater. Interfaces 2009, 1, 274–278. [DOI] [PubMed] [Google Scholar]
- Currie M. J.; Mapel J. K.; Heidel T. D.; Goffri S.; Baldo M. A. Science 2008, 321, 226–228. [DOI] [PubMed] [Google Scholar]
- a Borisov S. M.; Zenkl G.; Klimant I. Appl. Mater. Interfaces 2010, 2, 366–374. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Borisov S.; Papkovsky D.; Ponomarev G.; DeToma A.; Saf R.; Klimant I. J. Photochem. Photobiol. A 2009, 206, 87–92. [Google Scholar]; c Borisov S. M.; Nuss G.; Klimant I. Anal. Chem. 2008, 80, 9435–9442. [DOI] [PubMed] [Google Scholar]; d Dunphy I.; Vinogradov S. A.; Wilson D. F. Anal. Biochem. 2002, 310, 191–198. [DOI] [PubMed] [Google Scholar]; e Kumar R.; Ohulchanskyy T. Y.; Roy I.; Gupta S. K.; Borek C.; Thompson M. E.; Prasad P. N. Appl. Mater. Interfaces 2009, 1, 1474–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Vinogradov S. A.; Lo L. W.; Jenkins W. T.; Evans S. M.; Koch C.; Wilson D. F. Biophys. J. 1996, 70, 1609–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Evans R.; Douglas P.; Williams J. A. G.; Rochester D. J. Fluoresc. 2006, 16, 201–206. [DOI] [PubMed] [Google Scholar]
- Borisov S.; Nuss G.; Haas W.; Saf R.; Schmuck M.; Klimant I. J. Photochem. Photobiol. A 2009, 201, 128–135. [Google Scholar]
- a Russell R. J.; Pishko M. V.; Gefrides C. C.; McShane M. J.; Cote G. L. Anal. Chem. 1999, 71, 3126–3132. [DOI] [PubMed] [Google Scholar]; b Chinnayelka S.; McShane M. J. Diabetes Technol. Ther. 2006, 8, 269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Brown J. Q.; McShane M. J. Biosens. Bioelectron. 2006, 21, 1760–1769. [DOI] [PubMed] [Google Scholar]
- Finikova O. S.; Aleshchenkov S. E.; Briñas R. P.; Cheprakov A. V.; Carroll P. J.; Vinogradov S. A. J. Org. Chem. 2005, 70, 4617–4628. [DOI] [PubMed] [Google Scholar]
- Finikova O. S.; Cheprakov A. V.; Beletskaya I. P.; Carroll P. J.; Vinogradov S. A. J. Org. Chem. 2004, 69, 522–535. [DOI] [PubMed] [Google Scholar]
- Lebedev A. Y.; Cheprakov A. V.; Sakadžić S.; Boas D. A.; Wilson D. F.; Vinogradov S. A. Appl. Mater. Interfaces 2009, 1, 1292–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kobayashi N.; Konami H. J. Porphyrins Phthalocyanines 2001, 5, 233–255. [Google Scholar]; b Mack J.; Asano Y.; Kobayashi N.; Stillman M. J. J. Am. Chem. Soc. 2005, 127, 17697–17711. [DOI] [PubMed] [Google Scholar]
- Shanmugathasan S.; Edwards C.; Boyle R. W. Tetrahedron 2000, 56, 1025–1046. [Google Scholar]
- Roberts J. D.; Weigert F. J. J. Am. Chem. Soc. 1971, 93, 2361–2369. [Google Scholar]
- The 13C NMR spectrum in CDCl3 of 4-fluorobenzaldehyde from Sigma-Aldrich was taken as a reference.
- Rogers J. E.; Nguyen K. A.; Hufnagle D. C.; McLean D. G.; Su W. J.; Gossett K. M.; Burke A. R.; Vinogradov S. A.; Pachter R.; Fleitz P. A. J. Phys. Chem. A 2003, 107, 11331–11339. [Google Scholar]
- Kobayashi N.; Mack J.; Ishii K.; Stillman M. J. Inorg. Chem. 2002, 41, 5350–5363. [DOI] [PubMed] [Google Scholar]
- Papkovsky D. B.; Olah J.; Troyanovsky I. V.; Sadovsky N. A.; Rumyantseva V. D.; Mironov A. F.; Yaropolov A. I.; Savitsky A. P. Biosens. Bioelectron. 1992, 7, 199–206. [Google Scholar]
- Carraway E. R.; Demas J. N.; DeGraff B. A.; Bacon J. R. Anal. Chem. 1991, 63, 337–342. [Google Scholar]
- Borisov S.; Klimant I. Dyes Pigm. 2009, 83, 312–316. [Google Scholar]
- Argauer R. J.; White C. E. Anal. Chem. 1964, 36, 368–371. [Google Scholar]
- Emanuel N. M.; Kuzmin M. G.. Experimental Methods of Chemical Kinetics (in Russian); MGU: Moscow, 1985; pp 152−157. [Google Scholar]
- Ahlrichs R.; Bär M.; Häser M.; Horn H.; Kölmel C. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar]; TURBOMOLE 5.7. http://www.turbomole.com (accessed Sept 2010).
- a Becke A. D. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]; b Stephens P. J.; Devlin F. J.; Chabalowski C. F.; Frisch M. J. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar]
- Schäfer A.; Horn H.; Ahlrichs R. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar]
- Weigend F.; Ahlrichs R. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- Schäfer A.; Huber C.; Ahlrichs R. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.