

Abstract
Disruption of the cellular membrane by the amyloidogenic peptide IAPP (or amylin) has been implicated in β-cell death during type 2 diabetes. While the structure of the mostly inert fibrillar form of IAPP has been investigated, the structural details of the highly toxic prefibrillar membrane-bound states of IAPP have been elusive. A recent study showed that a fragment of IAPP (residues 1–19) induces membrane disruption to a similar extent as the full-length peptide. However, unlike the full-length IAPP peptide, IAPP1–19 is conformationally stable in an α-helical conformation when bound to the membrane. In vivo and in vitro measurements of membrane disruption indicate the rat version of IAPP1–19, despite differing from hIAPP1–19 by the single substitution of Arg18 for His18, is significantly less toxic than hIAPP1–19, in agreement with the low toxicity of the full-length rat IAPP peptide. To investigate the origin of this difference at the atomic level, we have solved the structures of the human and rat IAPP1–19 peptides in DPC micelles. While both rat and human IAPP1–19 fold into similar mostly α-helical structures in micelles, paramagnetic quenching NMR experiments indicate a significant difference in the membrane orientation of hIAPP1–19 and rIAPP1–19. At pH 7.3, the more toxic hIAPP1–19 peptide is buried deeper within the micelle, while the less toxic rIAPP1–19 peptide is located at the surface of the micelle. Deprotonating H18 in hIAPP1–19 reorients the peptide to the surface of the micelle. This change in orientation is in agreement with the significantly reduced ability of hIAPP1–19 to cause membrane disruption at pH 6.0. This difference in peptide topology in the membrane may correspond to similar topology differences for the full-length human and rat IAPP peptides, with the toxic human IAPP peptide adopting a transmembrane orientation and the nontoxic rat IAPP peptide bound to the surface of the membrane.
Islet amyloid polypeptide (IAPP,1 also known as amylin) is one of an increasing number of proteins in which the propensity to form a misfolded state is correlated with pathologies in tissue functioning. Physiologically, IAPP is one of a family of peptides related to calcitonin that act in the control of metabolic functions. Specifically, IAPP acts in concert with insulin to control plasma glucose levels and acts independently of insulin to slow gastric emptying and therefore food intake (1-3). For reasons that are currently poorly understood, IAPP aggregates in the pancreatic β-cells of type II diabetics to form highly ordered and extremely stable long protein fibers (amyloid deposits). Similarly, other tissue-specific amyloid deposits have also been identified in a growing list as possible pathological features in other common and devastating diseases such as Parkinson’s, Alzheimer’s, Creutzfeld-Jacob’s, and Huntington’s diseases. All of these proteins, despite the differences in amino acid sequences and in the structures of the monomeric protein, adopt a common cross-β-sheet structure consisting of parallel association of β-strands to form protofibril units and lateral association of protofibril units to form amyloid fibers (4). Amyloid deposition is generally correlated with the severity of the disease, increasing over time as the disease progresses and β-cell function decreases (5).
Amyloidogenic proteins commonly have a strong interaction with negatively charged membranes, forming oligomeric structures within the membrane that act as largely nonselective ion channels (6-9). Atomic force microscopy images show pentameric structures with a small central cavity suggestive of a well-defined ion channel when IAPP is bound to supported lipid bilayers (6). The formation of these ion channels has been proposed to disrupt calcium homeostasis and lead to mitochondrial oxidative stress (7). However, other studies have shown direct disruption of the membrane by the uptake of lipid molecules during fibrillogenesis into protofibril units (10, 11). In order to better understand the membrane interaction and function of IAPP, it is important to determine the high-resolution structure of IAPP in a membrane environment. However, the rapid aggregation of IAPP to form amyloid fibers has been a great challenge to overcome in solving the atomistic-level resolution structures of these peptides using NMR spectroscopy. In addition, the oligomeric species of IAPP that form during the aggregation process can interact with the membrane in different ways, further complicating structural studies of IAPP. To overcome the difficulties associated with IAPP, we have recently shown that the N-terminal 1–19 fragment of IAPP (the amino acid sequence is given in Figure 1) is a useful model system for the study of the IAPP-induced membrane disruption (12). The full-length IAPP peptide can be divided into three regions: an N-terminal region from residues 1 to 19 that largely determines the membrane binding (13, 14), a primary amyloidogenic region from residues 20 to 29, and a C-terminal region from residues 30 to 37 that enhances amyloid formation (13, 15, 16). Peptide array binding studies have shown that the N-terminal region of IAPP is also strongly involved in the self-association of the peptide. Using an array of decamer peptides made from hIAPP, Mazor et al. measured the binding of full-length IAPP to truncated versions of the peptide immobilized on a cellulose membrane matrix (17). The strongest peptide—peptide interactions in this study were found to extend from C7 to N21, which substantially overlaps with the 1–19 region. It is noteworthy that the binding of full-length hIAPP to truncated peptides containing the main amyloidogenic region (residues 20–29) was significantly less. The preferential affinity of full-length IAPP for truncated peptides made from the N-terminal region over those made from the amyloidogenic region suggests self-association in full-length IAPP may be initiated first by interactions in the 1–19 region before interactions within the 20–29 region can occur. This is significant because IAPP-induced toxicity and membrane damage can occur before amyloid formation as reported in the literature (7, 18). The 1–19 fragment of IAPP may therefore be an excellent structural model to investigate the early aggregation and membrane disrupting activity of IAPP that is observed before the formation of amyloid fibers.
Figure 1.

Amino acid sequences of rat and human IAPP. The 1–19 region is shown in blue, and differences between the sequences are shown in red. There is a disulfide bond between residues 2 and 7. The C-termini of the peptides used in this study are amidated.
In an effort to determine the amino acid residues responsible for the toxicity of IAPP, we recently found that the N-terminal 1–19 region of human IAPP both disrupts POPG vesicles similarly to full-length human IAPP and also disrupts β-cell homeostasis like full-length human IAPP (12, 19). Significantly, hIAPP1–19 exhibits greatly reduced amyloidogenicity, forming fibers only at very high concentrations in solution (20). While binding to membranes accelerates amyloid formation for full-length hIAPP, human IAPP1–19 is conformationally stable when bound to POPG membranes, maintaining the same α-helical conformation for at least 13 days (12). A similar effect has been noted for a longer fragment of the hIAPP (hIAPP1–24) that also contains part of the amyloidogenic region (21).
A comparison of amino acid sequences of IAPP of different species yields some interesting information in this context. Full-length rat IAPP is both nontoxic and nonamyloidogenic, and significantly, rats do not develop type II diabetes. However, the human and rat versions differ in only one amino acid residue within the 1–19 sequence (Figure 1) with the majority of the differences between human and rat IAPP being in the amyloidogenic 20–29 region. Despite the fact that it differs from human IAPP1–19 by only a single residue, rat IAPP1–19 is significantly less toxic than hIAPP1–19 (19). Since the 1–19 fragments of human and rat IAPP are quite stable in a membrane environment (12), it would be useful to determine the high-resolution structures and folding of these peptides using NMR spectroscopy. In this study, we have solved the high-resolution structures of both human and rat IAPP 1–19 peptides using solution NMR experiments on well-behaved dodecylphosphocholine (DPC) detergent micelles. In most respects, DPC micelles are a better model to mimic the phospholipid membrane than organic solvents, such as trifluoroethanol and hexafluoro-2-proponal, which have been used in some previous studies of amyloid peptides (22). Organic solvents lack the hydrophobic/hydrophilic interface that is present in lipid membranes. Furthermore, the difference in solvation structure and hydrogen bonding between organic solvents and lipid membranes tends to bias the structure toward non-native helical structures (23-25). The headgroup of DPC is identical to phosphatidylcholine, the most common lipid in animal cell membranes, and is expected to be less denaturing than sodium dodecyl sulfate (SDS) due to the zwitterionic nature of the detergent. Previous studies have shown that DPC preserves the native conformations of membrane-associated peptides and enzyme activity of membrane proteins in some cases (25-30).
MATERIALS AND METHODS
NMR Sample Preparation
Rat and human IAPP1–19 were synthesized and purified by Genscript. The formation of an intramolecular disulfide bond was determined by electrospray mass spectroscopy. The absence of intermolecular disulfide bonds was confirmed by nonreducing SDS—PAGE electrophoresis, which showed a single band corresponding to the monomeric peptide (data not shown). Both peptides were first dissolved in hexafluoro-2-propanol at a concentration of 10 mg/mL and then lyophilized overnight at 1 mTorr vacuum to completely remove the solvent. The NMR sample was prepared by dissolving the lyophilized peptide to a final concentration of 1.625 mM in 20 mM phosphate buffer at pH ∼7.3 containing 10% 2H2O, 200 mM perdeuterated DPC, and 120 mM NaCl.
NMR Experiments
All NMR experiments except the paramagnetic quenching experiments were performed at 30 °C on a Bruker Avance 900 MHz spectrometer equipped with a cryoprobe. Complete assignments of the side chain and backbone resonances were obtained using 2D 1H-1H TOCSY (with a 70 ms mixing time) and 2D 1H-1H NOESY (with 100 and 300 ms mixing times) experiments with 2056 complex points in the direct dimension and 512 points in the indirect dimension (31, 32). Proton chemical shifts were referenced to the water proton signal at the carrier frequency. All 2D spectra were processed using Bruker TopSpin software and analyzed using SPARKY (33, 34). Resonance assignment was carried out using a standard approach reported elsewhere (35).
Structure Calculations
The rIAPP1–19 and hIAPP1–19 structural ensembles were calculated starting from an extended structure using the simulated annealing protocol available in the NIH-XPLOR software package to generate 100 conformers. An initial temperature of 3000 K was used with 18000 high temperature steps, 9000 cooling steps, and a step size of 2 fs (36, 37). A total of 180 and 208 NOEs from rIAPP1–19 and hIAPP1–19, respectively, were classified as strong (1.8–2.9 Å), medium (1.8–4.5 Å), or weak (1.8–6.0 Å) and utilized as structural constraints. Dihedral angle restraints were obtained from the empirical correlation between α proton chemical shifts and dihedral angles using the TALOS module in NMRPIPE (38). Final refinement of the structure ensemble was calculated using simulated annealing at an initial temperature of 500 K with 20000 cooling steps and a step size of 3 fs.
The analysis was carried out using the “accept.inp” routine included in the NIH-XPLOR software package with the requirements that NOE violations should not exceed 0.5 Å, dihedral angle restraint violations should not exceed 5°, the rms difference for bond deviations should not exceed 0.01 Å from ideality, and the rms difference for angle deviations should not exceed 2° from ideality. The 20 lowest energy conformers for each of the rat and human IAPP1–19 peptides were selected for further analysis. The analysis of Ramachandran angles for the 20 lowest energy structures was carried out using PROCHECK-NMR (39). Peptide structures were analyzed using the program MOLMOL (40).
Paramagnetic Quenching
One-dimensional 1H chemical shift spectra of IAPP1–19 in DPC micelles at a concentration of 0.4, 0.8, and 1.2 mM MnCl2 at pH 7.3 and pH 6.0 were obtained. 2D 1H—1H NOESY spectra of hIAPP1–19 in DPC micelles at pH 6.0 with and without 1.2 mM MnCl2 were also obtained on Bruker Avance 600 MHz spectrometer. All other experimental conditions were the same as described above.
RESULTS
A combination of 2D 1H—1H TOCSY and 1H—1H NOESY spectra obtained at different mixing times was used for the assignment of backbone and side chain resonances. The sequential assignments were accomplished using the amide proton to α proton region of the 2D 1H—1H NOESY spectra obtained at a 300 ms mixing time. The α proton chemical shift index plots for both peptides are given in Figure 2. Due to chemical exchange, the chemical shift values for the α protons of K1—T4 and T6 of both peptides could not be observed. The fingerprint regions of NOESY spectra obtained at a 300 ms mixing time are shown in Figure 3. From the analysis of the 2D NOESY spectra, we have identified and assigned a total of 180 (73 intraresidue and 107 interresidue) NOEs for rIAPP1–19 and 208 (79 intraresidue and 129 interresidue) NOEs for hIAPP1–19. Figure 4 shows a summary of backbone NOEs for the secondary structure assignment with a histogram indicating the number of NOEs per residue.
Figure 2.
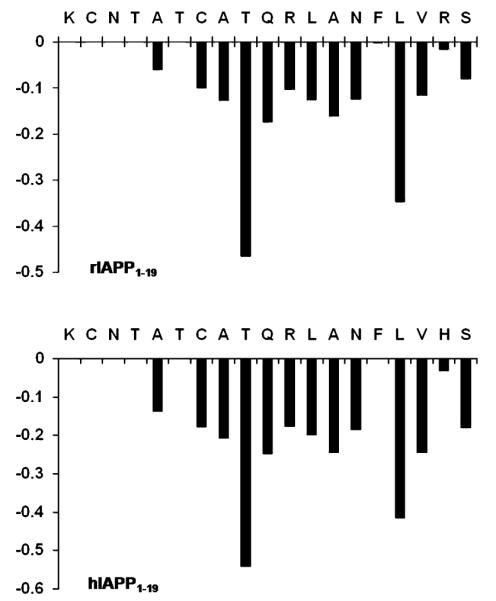
α proton chemical shift index for rat (top) and human (bottom) IAPP1–19.
Figure 3.
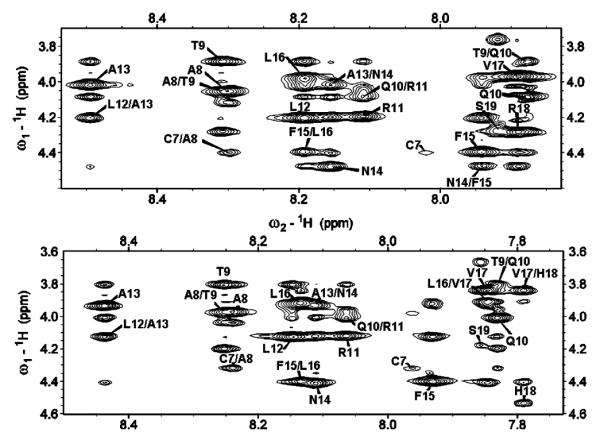
Fingerprint region of 2D 1H—1H NOESY spectra of rat (top) and human (bottom) IAPP1–19 in DPC micelles at pH 7.3 showing NOE α proton connectivities.
Figure 4.
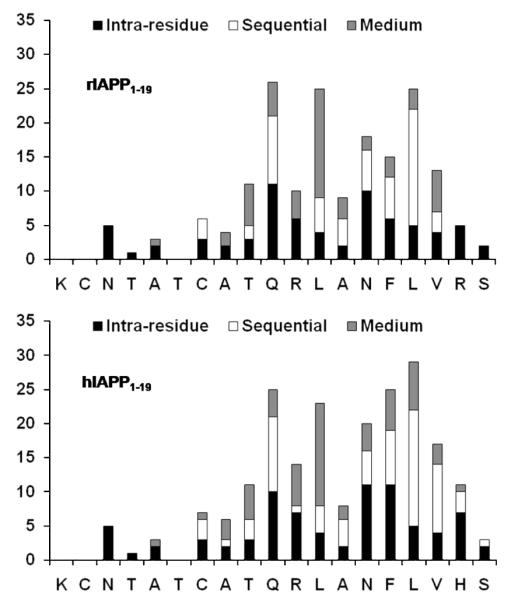
Histogram of NOEs versus residues for rat (top) and human (bottom) IAPP1–19.
The NMR spectra of rIAPP1–19 and hIAPP1–19 embedded in detergent micelles are sufficiently resolved to assign most of the resonances but not to measure the J coupling constants. All assigned NOEs were then converted into distances and modeled using the classical simulated annealing protocol built in XPLOR-NIH (6). Out of the 100 refined structures, 41 of the rat IAPP1–19 and 60 of the human IAPP1–19 did not have NOE violations >0.5 Å, dihedral angle restraint violations >5°, rms difference for bond deviations from ideality >0.01 Å, and rms difference for angle deviations from ideality >2°. Of the structures that passed the acceptance criteria, 20 structures were further selected for the final analysis. The superposition of backbone atoms from residues 1 to 19 gives an rmsd of 0.58 ± 0.12 and 0.57 ± 0.13 Å for rIAPP1–19 and hIAPP1–19 respectively, while the superposition of all heavy atoms gives an rmsd of 1.69 ± 0.28 and 1.51 ± 0.27 Å. The overlays of the backbone and side chain heavy atoms for the final selected conformers are shown in Figure 5. Analysis of the Ramachandran plot for the final conformers shows that 93.8% and 81.8% of the residues fall in the most favored region while the rest of the residues fall in the additionally allowed regions for rIAPP1–19 and hIAPP1–19, respectively (more details are given in Table 1).The secondary structure representations of rIAPP1–19 and hIAPP1–19 are shown in Figure 6.
Figure 5.
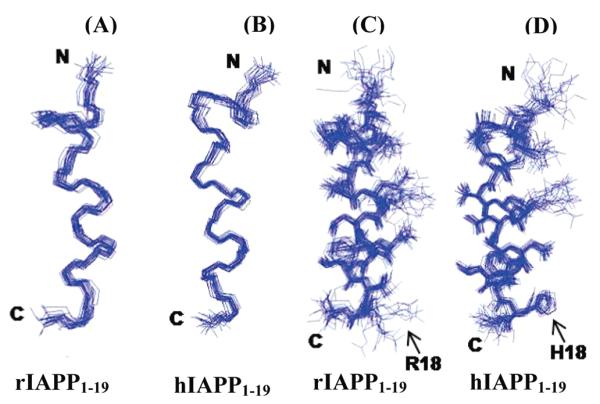
An ensemble of conformers for rat (A and C) and human (B and D) IAPP1–19 showing the convergence of the conformers for backbone atoms (A and B) and side chain atoms (C and D).
Table 1.
Statistical Information for the Structural Ensembles of rIAPP1–19 and hIAPP1–19
| rIAPP1–19 | hIAPP1–19 | ||
|---|---|---|---|
| distance constraints | total | 180 | 208 |
| intraresidual | 73 | 79 | |
| interresidual | 107 | 129 | |
| sequential (i — j = 1) | 56 | 71 | |
| medium (i — j = 2, 3, 4) | 51 | 58 | |
| structural statistics | NOE violations (Å) | 0.059 ± 0.0007 | 0.056 ± 0.0014 |
| dihedral angle restraint violations (deg) | 1.034 ± 0.104 | 1.589 ± 0.138 | |
| rmsd for bond deviations (Å) | 0.005 ± 0.0001 | 0.005 ± 0.0002 | |
| rmsd for angle deviations (deg) | 0.755 ± 0.009 | 0.643 ± 0.021 | |
| rmsd of all backbone atoms (Å) | 0.58 ± 0.12 | 0.57 ± 0.13 | |
| rmsd of all heavy atoms (Å) | 1.69 ± 0.28 | 1.51 ± 0.27 | |
| Ramachandran plot | residues in most favored region (%) | 93.8 | 81.8 |
| residues in additionally allowed region (%) | 6.2 | 18.2 |
Figure 6.
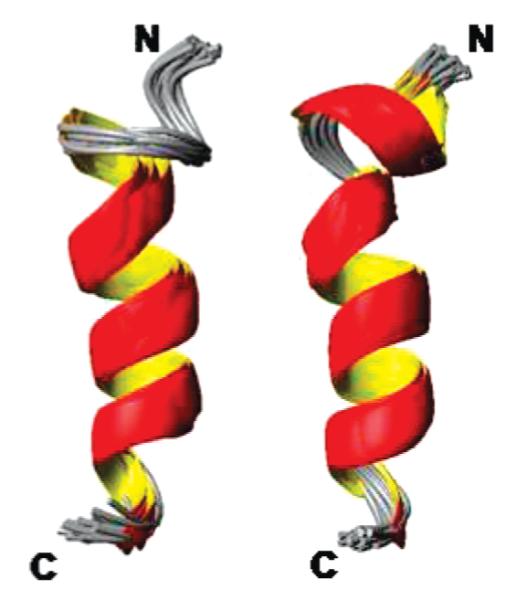
Secondary structure representations of an overlaid ensemble of conformers for rat (left) and human (right) IAPP1–19.
Since rIAPP1–19 and hIAPP1–19 differ significantly in their functions but have similar structures, it is important to determine the orientation of both peptides with respect to the membrane. As a first approximation of the membrane orientation, we have obtained the 1D 1H chemical shift spectra of both peptides in DPC micelles with varying concentrations of the paramagnetic quencher MnCl2. It is known that manganese ions affect the relaxation rate of nuclei that are in close proximity and therefore decrease the observed signal intensity. Because manganese ions cannot penetrate into the interior of the micelle, the observed changes in the signal intensities reflect the exposure of the amino acid residues of the peptide to solvent. The 1D 1H chemical shift spectra of DPC micelles containing hIAPP1–19 and rIAPP1–19 at pH 7.3 are given in Figure 7. The considerable decrease in the signal intensities and broadening of spectral lines observed for micelles containing rIAPP1–19 even at the relatively low concentration of 0.4 mM MnCl2 suggest that the peptide is well-exposed to the water phase and not in the hydrophobic core of the micelle. On the other hand, a significant reduction in signal intensities and an increase in line broadening is observed only at a high concentration of MnCl2 (1.2 mM) for micelles containing hIAPP1–19 at pH 7.3, which suggests that the peptide is inserted into the hydrophobic core of the micelle.
Figure 7.
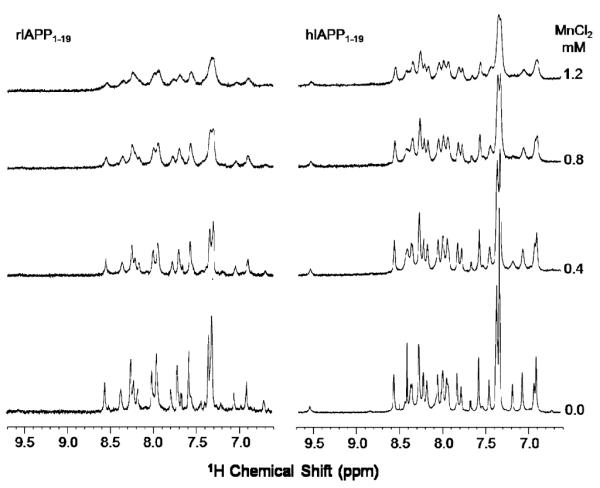
The amide proton chemical shift region of 1H NMR spectra of rat (left) and human (right) IAPP1–19 in DPC micelles at pH 7.3 with and without MnCl2.
Overall, these results suggest that hIAPP1–19 is inserted into the micelle at pH 7.3 while rIAPP1–19 binds closer to the surface. The only difference between hIAPP1–19 and rIAPP1–19 is the H18R mutation. Histidine is likely to be neutral at pH 7.3; therefore, if the origin of this effect is the difference in charge at residue 18, hIAPP1–19 should adopt a surface-associated binding mode at a lower pH where H18 is protonated. Accordingly, we have measured both the 1D 1H chemical shift (Figure 8) and 2D 1H—1H NOESY (Figure 9) spectra of hIAPP1–19 in DPC micelles at pH 6.0 in the presence of MnCl2. In contrast to the modest decrease in signal observed at pH 7.3 when MnCl2 is added (Figure 7), there is a significant decrease in signal intensity at pH 6.0 with the addition of MnCl2 (Figures 8 and 9). The strong effect of MnCl2 on the proton spectrum of hIAPP1–19 in DPC micelles at pH 6.0, but not at pH 7.3, indicates hIAPP1–19 is significantly closer to the surface of the micelle when H18 is protonated.
Figure 8.
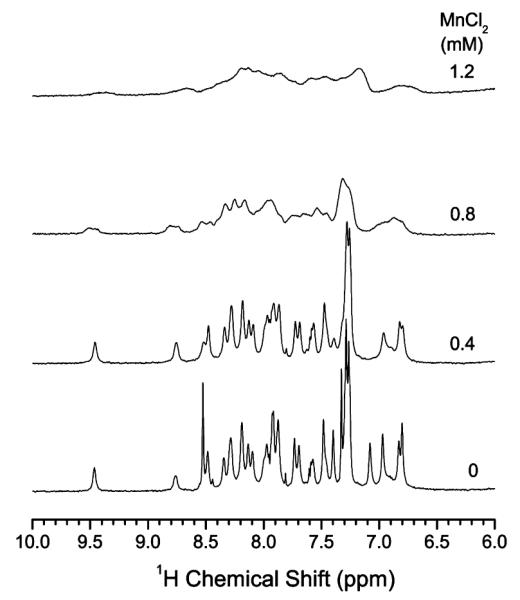
The amide proton chemical shift region of 1H NMR spectra of hIAPP1–19 in DPC micelles at pH 6.0 with and without MnCl2. A comparison of these 1H spectra with those given in Figure 7 for hIAPP1–19 in DPC micelles at pH 7.3 suggests that the peptide is more exposed to the water phase at pH 6.0 and therefore peaks are considerably broadened even at 0.8 mM MnCl2.
Figure 9.

Fingerprint region of 2D 1H—1H NOESY spectra of hIAPP1–19 in DPC micelles at pH 6.0 in the absence (A) and presence (B) of 1.2 mM MnCl2.
DISCUSSION
Since the toxicity of amyloid peptides to cells is correlated with the role of amyloid peptides in aging-related diseases (9, 41, 42), there is considerable current interest in solving high-resolution structures of amyloid peptides in a membrane environment. Several studies have reported global changes in the secondary structures of amyloid peptides upon binding to membrane and their significance in the conversion of nontoxic to toxic amyloids (18, 43-45). While the importance of IAPP/lipid interactions is well documented, the only available high-resolution structure of membrane IAPP is for the 20–29 fragment in sodium dodecyl sulfate (SDS) micelles (46). However, the toxicity of the 20–29 fragment is significantly lower than that of the full-length IAPP peptide (47). In this study, we report the first atomic-level resolution structures of a membrane-bound IAPP peptide that exhibits significant toxicity (12, 19). A previous NMR study has reported the low-resolution structure of nontoxic rat IAPP in water (48). While this study was in progress, a recent study reported the structure of IAPP using distance constraints measured from EPR experiments on liposomes containing selectively spin-labeled IAPP peptides (21).
The structures of hIAPP1–19 and rIAPP1–19 in DPC micelles are similar, consisting of a single helix extending from C7 to V17 and a distorted helical turn from C7 to the N-terminus (Figure 10). The N-terminal region is prevented from adopting a canonical α-helix conformation by the disulfide bridge from C2 to C7. Residues 2–5 are likely to be dynamic and poorly structured in both peptides, as shown by the lack of NOE restraints in this region (Figure 4). This region may be more structured in hIAPP1–19, as HA—NH connectivity was seen from C7 to Q10 and A8 to Q10 for hIAPP1–19 but not in rIAPP1–19. As these residues border the disulfide bridge from residues 2 to 7, a change in the dynamics at one end of the disulfide bridge is likely to be transmitted throughout the ring from residues 2 to 7.
Figure 10.

(A) Overlay of the average structures of rIAPP1–19 (blue) and hIAPP1–19 (red). The average of the 20 structures of each peptide was aligned using the STAMP algorithm present in the Multiseq utility in VMD (54, 55). (B) View of rIAPP1–19 and hIAPP1–19 viewed end-on from the N-terminus. (C) View of rIAPP1–19 showing the curve in the helical region. (D) View of hIAPP1–19 showing the absence of a curve in the helical region.
This finding is supported by recent NMR data on full-length rat and human IAPP in solution (48, 49). While both rat and human IAPP are predominantly unstructured in solution, both peptides transiently sample α-helical states in solution (48, 49). In agreement with the additional restraints found here for hIAPP1–19, full-length hIAPP in solution has a greater α-helical propensity at the N-terminus than rIAPP (49). Yonemoto et al. reported the presence of strong (i, i + 1) amide—amide NOEs and negative Hα chemical shift deviations across the entire sequence of hIAPP, indicating all residues of hIAPP sample α-helical dihedral angles (49). Analysis of temperature coefficients and chemical shift deviations show that the corresponding stretch of rat IAPP with a propensity to form α-helical states is shorter, stretching only from A5 to S19 (48). Notably, the chemical shift deviations in the α-helical forming region are larger for full-length hIAPP than rIAPP in solution (48, 49).
Although high-resolution structures of membrane-bound full-length IAPP are not available, both lower resolution studies on full-length IAPP in liposomes and in solution suggest the conformation determined here may represent the structured part of the full-length protein. The majority of the remaining residues on the C-terminal end of the full-length protein are likely to be unstructured both in solution and when bound to the membrane. A recent EPR study using spin-labeled full-length hIAPP has indicated that residues 9–22 form a helix when bound to anionic liposomes, while the surrounding N-terminal and C-terminal regions are less ordered without a defined secondary structure (21). In accordance with these data, circular dichroism has shown that approximately 40% of full-length human IAPP and 35% of full-length rIAPP are in a helical conformation when bound to the membrane (18, 43). The NMR studies mentioned above on full-length IAPP in solution also support the model of a C-terminal region with weaker helix propensity than the N-terminal region (48, 49).
While the structures of rIAPP1–19 and hIAPP1–19 are quite similar, the H18R substitution has a strong effect on the orientation of the peptide with respect to the membrane. The difference in the location of hIAPP1–19 and rIAPP1–19 in the membrane can be seen in Figure 7. The signal for rIAPP1–19 is diminished substantially upon the addition of the paramagnetic quencher MnCl2 even at low concentrations, while the signal for hIAPP1–19 is hardly affected. Since the Mn2+ ion cannot penetrate into the hydrophobic core of the micelle, the absence of quenching by Mn2+ for hIAPP1–19 indicates rIAPP1–19 is located closer to the surface of the membrane than hIAPP1–19 (50). The structural basis for this difference is most likely due to the anchoring of R18 in rIAPP1–19 to the phosphate group of the DPC molecule by a charge—charge interaction. The anchoring of rIAPP1–19 to the surface of the membrane at two points by the charges on K1 and R18 would likely fix the position of the peptide within the micelle (51). The effect of anchoring of rIAPP1–19 to the surface of the micelle can be seen in the structure of rIAPP1–19. The helix in the rIAPP1–19 structure has a noticeable curve with the hydrophobic residues of the amphipathic helix on the concave side and the polar and charged residues on the opposite side that is absent in the corresponding helix of the hIAPP1–19 structure (Figure 10C). Distorted helices usually occur when the peptide binds near the surface of the micelle, with the structure adopting some of the curvature of the micelle surface. Because hIAPP1–19 at pH 7.3 lacks the C-terminal charge of R18 in rIAPP1–19, it possesses a greater degree of orientational flexibility. Without the requirement that residue 18 must be anchored to the surface, hIAPP1–19 can adopt an orientation that buries it deeper within the micelle. The importance of charge in determining the binding topology of hIAPP1–19 can be clearly seen by comparing the effect of MnCl2 on the proton spectrum of hIAPP1–19 at pH 7.3 and at pH 6.0. In contrast to the modest quenching observed at pH 7.3 (Figure 7), MnCl2 strongly quenches the signal at pH 6.0 (Figures 8 and 9). This is a strong indication that hIAPP1–19 adopts a surface-associated binding mode similar to rIAPP1–19 when H18 is protonated.
Interestingly, neutron diffraction experiments have suggested a similar difference in topologies for the full-length rat and human IAPP peptides (52). A surface-associated peptide is expected to strongly disorder the packing of the bilayer by creating a void beneath the peptide that must be filled by the acyl chains of nearby phospholipids; such effects have been reported for membrane-disrupting antimicrobial peptides based on solid-state NMR studies in lipid bilayers (56, 57). A transmembrane peptide, on the other hand, does not create a void volume in the membrane and can be expected to have little effect on the packing of the bilayer if it does not alter the thickness of the membrane. The density profile of the bilayer measured by neutron diffraction in the presence of full-length hIAPP is remarkably similar to that of the bilayer in the absence of the peptide (52). The density profile of bilayers containing full-length rIAPP, on the other hand, showed a significant reduction in density in the hydrophobic core of the bilayer, consistent with the splaying of acyl chains to occupy the void volume created with a surface-associated peptide (52). Critically, water was detected in the interior of the bilayer in this experiment when human but not rat IAPP was added, indicating that a transmembrane orientation is related to the membrane disruption induced by human IAPP but not rat IAPP (52). The importance of peptide binding topology for membrane disruption is in agreement with our results reported in the companion paper, which show that the membrane disruption by hIAPP1–19 is reduced at pH 6.0 and is approximately equal to that of rIAPP1–19 at pH 6.0 (19).
An alternative topology for full-length hIAPP has been proposed by Apostolidou et al. in studies using spin-labeled IAPP bound to anionic liposomes in the presence of quenchers with different lipid penetration profiles (21). They found that full-length human IAPP was oriented parallel to the membrane surface in these conditions with the center of the helix located 6–9 Å below the phosphate group. In this EPR study, a very low (1:1000) peptide-to-lipid ratio was used, and it is therefore likely the peptide was in the monomeric state. It can clearly be seen from Figure 5 that a monomeric IAPP peptide in a transmembrane orientation would have unfavorable electrostatic interactions due to the presence of the charged R11 residue in the hydrophobic core of the membrane. The small size of the spherical micelle used in this study allows the charged side chain of R11 to easily extend outside/near the surface of the micelle regardless of the overall orientation of the peptide. In larger liposomes, a correct orientation of the peptide is necessary to stabilize this charge. In the surface-associated orientation, the charge on a monomeric peptide can be stabilized by interactions with either the headgroups or phosphates of the interfacial region (21). In order to avoid this unfavorable interaction in a transmembrane orientation, oligomerization of the peptide is necessary. The binding of hIAPP to the membrane and the membrane disruption induced by hIAPP are both strongly cooperative, and saturation of IAPP binding occurs at a higher density than is possible with a surface-associated orientation (18). This suggests a model in which IAPP first binds to the membrane in a monomeric form, recruits other IAPP monomers to the surface, and then inserts into the membrane as an oligomer with a water-filled channel that stabilizes the unfavorable charge (18). It should be noted that a transmembrane orientation would imply a hydrophobic mismatch for both hIAPP1–19 and full-length hIAPP if the remaining residues in full-length hIAPP are unfolded as suggested by EPR, CD, and NMR studies on homologous peptides (18, 21, 43, 53).
In conclusion, we have solved the high-resolution structures and found the membrane orientations of human and rat IAPP1–19 using NMR experiments on dodecylphosphocholine micelles in an effort to understand the functional differences between these two peptides. The 1–19 fragment was chosen because evidence suggests this region is responsible for most of the membrane damage induced by hIAPP (12-14, 19). The structure determined here may hold insights into both the normal physiological action of IAPP and the damage to β-cell membranes caused by the pathological aggregation of IAPP. Future studies are needed to determine the structure of the oligomeric forms of IAPP at high resolution and to understand the role of membrane composition in the structural folding of oligomeric IAPP.
ACKNOWLEDGMENT
We thank Raffaello Verardi for help with initial sample preparation, Dr. Vivekanandan Subramanian for help with NMR data analysis, Dr. Aizhuo Liu for assistance with the 900 MHz NMR facility at Michigan State University, and Dr. Al-Hashimi and Alexandar Hansen for help with the Bruker 600 MHz NMR spectrometer at the University of Michigan.
Footnotes
This research was supported by funds from the National Institutes of Health (AI054515) and the Michigan Diabetes Research Training Center at the University of Michigan.
- DPC
- dodecylphosphocholine
- EPR
- electron paramagnetic resonance
- hIAPP
- human islet amyloid polypeptide
- IAPP
- islet amyloid polypeptide
- NMR
- nuclear magnetic resonance
- NOE
- nuclear Overhauser effect
- rIAPP
- rat islet amyloid polypeptide
- SDS—PAGE
- sodium dodecyl sulfate—polyacrylamide gel electrophoresis.
REFERENCES
- 1.Bailey CJ. New pharmacological approaches to glycemic control. Diabetes ReV. 1999;7:94–113. [Google Scholar]
- 2.Cooper GJS. Amylin compared with calcitonin-gene-related peptide—Structure, biology, and relevance to metabolic disease. Endocr. ReV. 1994;15:163–201. doi: 10.1210/edrv-15-2-163. [DOI] [PubMed] [Google Scholar]
- 3.Reda TK, Geliebter A, Pi-Sunyer FX. Amylin, food intake, and obesity. Obes. Res. 2002;10:1087–1091. doi: 10.1038/oby.2002.147. [DOI] [PubMed] [Google Scholar]
- 4.Makin OS, Serpell LC. Structures for amyloid fibrils. FEBS J. 2005;272:5950–5961. doi: 10.1111/j.1742-4658.2005.05025.x. [DOI] [PubMed] [Google Scholar]
- 5.Clark A, Nilsson MR. Islet amyloid: a complication of islet dysfunction or an aetiological factor in type 2 diabetes? Diabetologia. 2004;47:157–169. doi: 10.1007/s00125-003-1304-4. [DOI] [PubMed] [Google Scholar]
- 6.Quist A, Doudevski L, Lin H, Azimova R, Ng D, Frangione B, Kagan B, Ghiso J, Lal R. Amyloid ion channels: A common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. U.S.A. 2005;102:10427–10432. doi: 10.1073/pnas.0502066102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem. 2005;280:17294–17300. doi: 10.1074/jbc.M500997200. [DOI] [PubMed] [Google Scholar]
- 8.Kayed R, Sokolov Y, Edmonds B, McIntire TM, Milton SC, Hall JE, Glabe CG. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 2004;279:46363–46366. doi: 10.1074/jbc.C400260200. [DOI] [PubMed] [Google Scholar]
- 9.Ferreira ST, Vieira MNN, De Felice FG. Soluble protein oligomers as emerging toxins in Alzheimer’s and other amyloid diseases. IUBMB Life. 2007;59:332–345. doi: 10.1080/15216540701283882. [DOI] [PubMed] [Google Scholar]
- 10.Engel MF, Khemtemourian L, Kleijer CC, Meeldijk HJ, Jacobs J, Verkleij AJ, de Kruijff B, Killian JA, Hoppener JW. Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc. Natl. Acad. Sci. U.S.A. 2008;105:6033–6038. doi: 10.1073/pnas.0708354105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sparr E, Engel MFM, Sakharov DV, Sprong M, Jacobs J, de Kruijff B, Hoppener JWM, Killian JA. Islet amyloid polypeptide-induced membrane leakage involves uptake of lipids by forming amyloid fibers. FEBS Lett. 2004;577:117–120. doi: 10.1016/j.febslet.2004.09.075. [DOI] [PubMed] [Google Scholar]
- 12.Brender JR, Lee EL, Cavitt MA, Gafni A, Steel DG, Ramamoorthy A. Amyloid fiber formation and membrane disruption are separate processes localized in two distinct regions of IAPP. The type-2-biabetes-related peptide. J. Am. Chem. Soc. 2008;130:6424–6429. doi: 10.1021/ja710484d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engel MFM, Yigittop H, Elgersma RC, Rijkers DTS, Liskamp RMJ, de Kruijff B, Hoppener JWM, Killian JA. Islet amyloid polypelptide inserts into phospholipid monolayers as monomer. J. Mol. Biol. 2006;356:783–789. doi: 10.1016/j.jmb.2005.12.020. [DOI] [PubMed] [Google Scholar]
- 14.Lopes DHJ, Meister A, Gohlke A, Hauser A, Blume A, Winter R. Mechanism of islet amyloid polypeptide fibrillation at lipid interfaces studied by infrared reflection absorption spectroscopy. Biophys. J. 2007;93:3132–3141. doi: 10.1529/biophysj.107.110635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Westermark P, Engstrom U, Johnson KH, Westermark GT, Betsholtz C. Islet amyloid polypeptide—Pinpointing amino-acid-residues linked to amyloid fibril formation. Proc. Natl. Acad. Sci. U.S.A. 1990;87:5036–5040. doi: 10.1073/pnas.87.13.5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaikaran E, Higham CE, Serpell LC, Zurdo J, Gross M, Clark A, Fraser PE. Identification of a novel human islet amyloid polypeptide beta-sheet domain and factors influencing fibrillogenesis. J. Mol. Biol. 2001;308:515–525. doi: 10.1006/jmbi.2001.4593. [DOI] [PubMed] [Google Scholar]
- 17.Mazor Y, Gilead S, Benhar I, Gazit E. Identification and characterization of a novel molecular-recognition and self-assembly domain within the islet amyloid polypeptide. J. Mol. Biol. 2002;322:1013–1024. doi: 10.1016/s0022-2836(02)00887-2. [DOI] [PubMed] [Google Scholar]
- 18.Knight JD, Hebda JA, Miranker AD. Conserved and cooperative assembly of membrane-bound α-helical states of islet amyloid polypeptide. Biochemistry. 2006;45:9496–9508. doi: 10.1021/bi060579z. [DOI] [PubMed] [Google Scholar]
- 19.Brender JR, Hartman K, Reid KR, Kennedy RT, Ramamoorthy A. A single mutation in the nonamyloidogenic region of islet amyloid polypeptide greatly reduces toxicity. Biochemistry. 2008;47:12680–12688. doi: 10.1021/bi801427c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Radovan D, Smirnovas V, Winter R. Effect of pressure on islet amyloid polypeptide aggregation: Revealing the polymorphic nature of the fibrillation process. Biochemistry. 2008;47:6352–6360. doi: 10.1021/bi800503j. [DOI] [PubMed] [Google Scholar]
- 21.Apostolidou M, Jayasinghe SA, Langen R. Structure of α-helical membrane-bound hIAPP and its implications for membrane-mediated misfolding. J. Biol. Chem. 2008;283:17205–17210. doi: 10.1074/jbc.M801383200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crescenzi O, Tomaselli S, Guerrini R, Salvadori S, D’Ursi AM, Temussi PA, Picone D. Solution structure of the Alzheimer amyloid beta-peptide (1–42) in an apolar microenvironment—Similarity with a virus fusion domain. Eur. J. Biochem. 2002;269:5642–5648. doi: 10.1046/j.1432-1033.2002.03271.x. [DOI] [PubMed] [Google Scholar]
- 23.Buck M. Trifluoroethanol and colleagues: cosolvents come of age. Recent studies with peptides and proteins. Q. ReV. Biophys. 1998;31:297–355. doi: 10.1017/s003358359800345x. [DOI] [PubMed] [Google Scholar]
- 24.Sanders CR, Sonnichsen F. Solution NMR of membrane proteins: practice and challenges. Magn. Reson. Chem. 2006;44:S24–S40. doi: 10.1002/mrc.1816. [DOI] [PubMed] [Google Scholar]
- 25.Zamoon J, Mascioni A, Thomas DD, Veglia G. NMR solution structure and topological orientation of monomeric phospholamban in dodecylphosphocholine micelles. Biophys. J. 2003;85:2589–2598. doi: 10.1016/s0006-3495(03)74681-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Traaseth NJ, Thomas DD, Veglia G. Effects of Ser16 phosphorylation on the allosteric transitions of phospholamban/Ca2+-ATPase complex. J. Mol. Biol. 2006;358:1041–1050. doi: 10.1016/j.jmb.2006.02.047. [DOI] [PubMed] [Google Scholar]
- 27.Zamoon J, Nitu F, Karim C, Thomas DD, Veglia G. Mapping the interaction surface of a membrane protein: Unveiling the conformational switch of phospholamban in calcium pump regulation. Proc. Natl. Acad. Sci. U.S.A. 2005;102:4747–4752. doi: 10.1073/pnas.0406039102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vinogradova O, Sonnichsen F, Sanders CR. On choosing a detergent for solution NMR studies of membrane proteins. J. Biomol. NMR. 1998;11:381–386. doi: 10.1023/a:1008289624496. [DOI] [PubMed] [Google Scholar]
- 29.Porcelli F, Buck-Koehntop BA, Thennarasu S, Ramamoorthy A, Veglia G. Structures of the dimeric and monomeric variants of magainin antimicrobial peptides (MSI-78 and MSI-594) in micelles and bilayers, determined by NMR spectroscopy. Biochemistry. 2006;45:5793–5799. doi: 10.1021/bi0601813. [DOI] [PubMed] [Google Scholar]
- 30.Porcelli F, Verardi R, Shi L, Henzler-Wildman KA, Ramamoorthy A, Veglia G. NMR structure of the cathelicidin-derived human antimicrobial peptide LL-37 in dodecylphosphocholine micelles. Biochemistry. 2008;47:5565–5572. doi: 10.1021/bi702036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bax A, Davis DG. MLEV-17-based two-dimensional homonuclear magnetization transfer spectroscopy. J. Magn. Reson. 1985;65:355–360. [Google Scholar]
- 32.Kumar A, Ernst RR, Wuthrich K. A two-dimensional nuclear overhauser enhancement (2D NOE) experiment for the elucidation of complete proton-proton cross-relaxation networks in biological macromolecules. Biochem. Biophys. Res. Commun. 1980;95:1–6. doi: 10.1016/0006-291x(80)90695-6. [DOI] [PubMed] [Google Scholar]
- 33.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRpipe—a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 34.Kneller DG, Kuntz ID. UCSF Sparky—an NMR display, annotation and assignment tool. J. Cell. Biochem. 1993:254–254. [Google Scholar]
- 35.Wuthrich K. NMR of Proteins and Nucleic Acids. John Wiley and Sons; New York: 1986. [Google Scholar]
- 36.Nilges M, Gronenborn AM, Brunger AT, Clore GM. Determination of 3-dimensional structures of proteins by simulated annealing with interproton distance restraints—application to crambin, potato carboxypeptidase inhibitor and barley serine proteinase inhibitor-2. Protein Eng. 1988;2:27–38. doi: 10.1093/protein/2.1.27. [DOI] [PubMed] [Google Scholar]
- 37.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 38.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 39.Laskowski RA, Rullmann JAC, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 40.Koradi R, Billeter M, Wuthrich K. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 41.Lashuel HA, Lansbury PT. Are amyloid diseases caused by protein aggregates that mimic bacterial pore-forming toxins? Q. ReV. Biophys. 2006;39:167–201. doi: 10.1017/S0033583506004422. [DOI] [PubMed] [Google Scholar]
- 42.Haataja L, Gurlo T, Huang CJ, Butler PC. Islet Amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr. ReV. 2008;29:302–316. doi: 10.1210/er.2007-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jayasinghe SA, Langen R. Lipid membranes modulate the structure of islet amyloid polypeptide. Biochemistry. 2005;44:12113–12119. doi: 10.1021/bi050840w. [DOI] [PubMed] [Google Scholar]
- 44 (a).Bokvist M, Lindstrom F, Watts A, Grobner G. Two types of Alzheimer’s beta-amyloid (1–40) peptide membrane interactions: Aggregation preventing transmembrane anchoring versus accelerated surface fibril formation. J. Mol. Biol. 2004;335:1039–1049. doi: 10.1016/j.jmb.2003.11.046. [DOI] [PubMed] [Google Scholar]; (b) Ilangovan U, Ramamoorthy A. Conformational studies of human islet amyloid peptide using molecular dynamics and simulated annealing methods. Biopolymers. 1998;45:9. doi: 10.1002/(SICI)1097-0282(199801)45:1<9::AID-BIP2>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 45.Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- 46.Mascioni A, Porcelli F, Ilangovan U, Ramamoorthy A, Venglia G. Conformational preferences of the amylin nucleation site in SDS micelles: An NMR study. Biopolymers. 2003;69:29–41. doi: 10.1002/bip.10305. [DOI] [PubMed] [Google Scholar]
- 47.Tenidis K, Waldner M, Bernhagen J, Fischle W, Bergmann M, Weber M, Merkle ML, Voelter W, Brunner H, Kapurniotu A. Identification of a penta- and hexapeptide of islet amyloid polypeptide (IAPP) with amyloidogenic and cytotoxic properties. J. Mol. Biol. 2000;295:1055–1071. doi: 10.1006/jmbi.1999.3422. [DOI] [PubMed] [Google Scholar]
- 48.Williamson JA, Miranker AD. Direct detection of transient α-helical states in islet amyloid polypeptide. Protein Sci. 2007;16:110–117. doi: 10.1110/ps.062486907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yonemoto IT, Kroon GJ, Dyson HJ, Balch WE, Kelly JW. Amylin proprotein processing generates progressively more amyloidogenic peptides that initially sample the helical state. Biochemistry. 2008;47:9900–9910. doi: 10.1021/bi800828u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jarvet J, Danielsson J, Damberg P, Oleszczuk M, Graeslund A. Positioning of the Alzheimer Abeta(1–40) peptide in SDS micelles using NMR and paramagnetic probes. J. Biomol. NMR. 2007;39:63–72. doi: 10.1007/s10858-007-9176-4. [DOI] [PubMed] [Google Scholar]
- 51.Daily AE, Greathouse DV, van der Wel PCA, Koeppe RE. Helical distortion in tryptophan- and lysine-anchored membrane-spanning alpha-helices as a function of hydrophobic mismatch: A solid-state deuterium NMR investigation using the geometric analysis of labeled alanines method. Biophys. J. 2008;94:480–491. doi: 10.1529/biophysj.106.097543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Balali-Mood K, Ashley RH, Hauss T, Bradshaw JP. Neutron diffraction reveals sequence-specific membrane insertion of pre-fibrillar islet amyloid polypeptide and inhibition by rifampicin. FEBS Lett. 2005;579:1143–1148. doi: 10.1016/j.febslet.2004.12.085. [DOI] [PubMed] [Google Scholar]
- 53.Motta A, Andreotti G, Amodeo P, Strazzullo G, Morelli MAC. Solution structure of human calcitonin in membrane-mimetic environment: The role of the amphipathic helix. Proteins. 1998;32:314–323. [PubMed] [Google Scholar]
- 54.Roberts E, Eargle J, Wright D, Luthey-Schulten Z. MultiSeq: Unifying sequence and structure data for evolutionary analysis. BMC Bioinf. 2006;7:382–392. doi: 10.1186/1471-2105-7-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J. Mol. Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 56.Henzler-Wildman KA, Martinez GV, Brown MF, Ramamoorthy A. Perturbation of the hydrophobic core of lipid bilayers by the human antimicrobial peptide LL-37. Biochemistry. 2004;43:8459–8469. doi: 10.1021/bi036284s. [DOI] [PubMed] [Google Scholar]
- 57.Ramamoorthy A, Thennarasu S, Tan A, Lee DK, Clayberger C, Krensky AM. Cell selectivity correlates with membrane interactions: a case study on the antimicrobial peptide G15 derived from granulysin. Biochim. Biophys. Acta. 2006;1758:154–163. doi: 10.1016/j.bbamem.2006.02.014. [DOI] [PubMed] [Google Scholar]