

Abstract
A stereoselective synthesis of the C1-C12 segment of the potent cytotoxic macrolide, iriomoteolide 1a, has been accomplished. The key steps involve an enzymatic kinetic resolution of a β-hydroxy amide, a Pd-catalyzed cross-coupling to a substituted allylsilane, a highly regio- and stereoselective conjugate addition of lithium dimethylcopper to an α, β-acetylenic esters and an elaboration of the C6-C7 trans-olefin geometry by a Julia-Kocienski olefination.
Macrocyclic marine natural products are a rich source of potent and structurally novel anticancer agents with clinical potential.1 Over the years, Kobayashi and co-workers have reported a variety of structurally diverse macrolides known as amphidinolides from marine dinoflagellates, Amphidinium Sp.2 Recently, Tsuda and co-workers isolated iriomoteolide 1a (1), a 20-membered macrolide from Amphidinium Sp. from benthic sea sand collected off Iriomote island in Japan.3 Iriomoteolide 1a displayed remarkably potent cytoxicity against human B lymphocyte DG-75 cells with an IC50 value of 2 ng/mL. Furthermore, it has shown cytotoxicity against Epstein-Barr virus – infected human B lymphocyte Raji cells with IC50 value of 3 ng/mL. Despite its potent activity, the biological mechanism of action of iriomoteolide 1a is currently unknown. The gross structure of 1 was established by extensive mass spectroscopy and NMR studies.3 The unique structural features of iriomoteolide 1a coupled with its potent antitumor activity attracted our interest in its synthesis and structure-activity studies. Herein we report synthesis of the C1-C12 segment of iriomoteolide 1a in which the key steps involve lipase catalyzed kinetic resolution of a β-hydroxy amide, a highly stereoselective conjugate addition, and a Julia-Kocienski olefination to install the C6-C7 trans olefin geometry. Thus far, only Yang and coworkers reported the synthesis of C1-C12 fragment of iriomoteolide 1a and the total synthesis of iriomoteolide has not yet been achieved.4
As shown in figure 1, our synthetic strategy of iriomoteolide 1a is convergent and involves the assembly of fragments 2 (C1-C12 segment) and 3 (C13- C23 segment) by a Sakurai reaction5 and subsequent macrolactonization between the C19-hydroxyl group and the C1-carboxylic acid. Segment 2 was planned to be synthesized by a Julia-Kocienski olefination reaction6 between sulfone 4 and aldehyde 5. This reaction is expected to establish the C6-C7 trans-olefin geometry.
Figure 1.

Retrosynthetic analysis of iriomoteolide 1a
The synthesis of sulfone 4 was carried out as shown in Scheme 1. Deprotonation of N-methoxy-N-methylacetamide by lithium diisopropylamide followed by reaction of the resulting enolate with acrolein at −78 °C gave racemic alcohol 6 in 91% yield. The racemic alcohol 6 was then exposed to enzymatic acylation reaction using lipase PS-30 in pentane in the presence of excess vinyl acetate at 25 °C for 30 h to provide enantio-enriched acetate derivative 7 in 49% yield and alcohol (R)-(+)-6 in 45% yield.7 The alcohol was converted to its corresponding Mosher’s ester and optical purity of 97% ee was determined by 19F NMR analysis.8 Protection of alcohol R(+)-6 with tert-butyldimethylsilyl chloride and imidazole provided silyl ether 8. Reaction of 8 with methylmagnesium bromide furnished methyl ketone 9 in 96% yield. Treatment of 9 with borane dimethylsulfide complex resulted in the hydroboration of the olefin as well as reduction of the ketone providing a diol. The resulting diol was selectively protected to give the bis-silyl ether. Swern oxidation of the resulting alcohol furnished methyl ketone 10. Treatment of methyl ketone 10 with KHMDS and phenyl triflimide in THF from −100 °C to −78 °C gave the corresponding vinyl triflate. Cross-coupling9 of the triflate and trimethylsilylmethylmagnesium chloride in the presence of a catalytic amount of Pd(PPh3)4 (7 mol%) afforded the allyl silane 11 in 69% yield in two steps. Treatment of silyl ether 11 with pyridinium p-toluenesulfonate in methanol at 23 °C for 4 h resulted in the deprotection of the primary silyl ether to provide the corresponding alcohol. A Mitsunobu reaction of the alcohol with 1-phenyl-1H-tetrazole-5-thiol furnished the sulfide 13. It was oxidized by hydrogen peroxide in the presence of ammonium molybdate to furnish sulfone 4, one of the Julia-Kocienski olefination precursors.
Scheme 1.
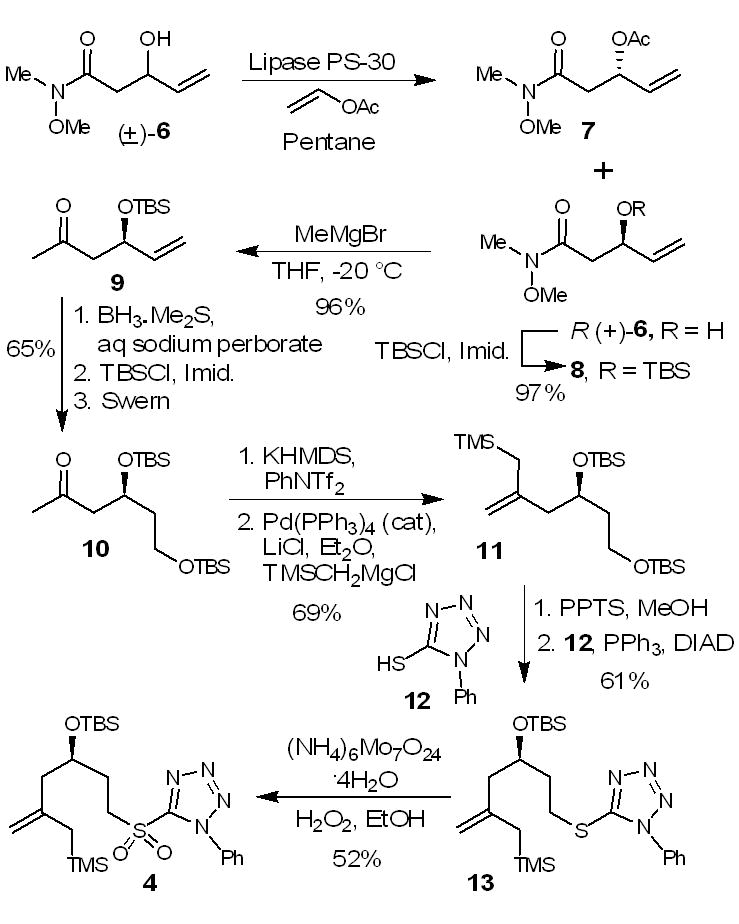
Synthesis of sulfone 4
The synthesis of aldehyde 5 and its subsequent conversion to C1-C12 segment (2) is outlined in Scheme 2. Enolization of tert-butylacetate using lithium diisopropylamide followed by reaction of the resulting enolate with acrolein at −78 °C gave racemic alcohol 14 in 90% yield.10 The racemic alcohol 14 was then exposed to lipase PS-30 in pentane in the presence of excess vinyl acetate at 30 °C for 19 h to provide acetate derivative 15 and enantioenriched alcohol 16 in 47% and 44% yields, respectively.11 The alcohol was converted to the corresponding Mosher ester and 19F NMR analysis revealed optical purity to be 98% ee.8 Treatment of alcohol 16 with lithium diisopropylamide followed by reaction of the resulting dianion with methyl iodide as described by Seebach and co-workers afforded the anti-alcohol 17 as a single isomer by 1H-NMR analysis.12 Protection of alcohol as TBS-ether followed by DIBAL-H reduction afforded alcohol 18. Swern oxidation of 18 followed by subjection of the resulting aldehyde to Corey-Fuchs’ homologation aldehyde 5 using carbon tetrabromide and triphenyl-phosphine in dichloromethane at 0 °C to 23 °C for 30 min afforded the corresponding dibromo olefin in 90% yield for two steps. Treatment of the dibromide with butyl lithium followed by reaction of the derived alkynyl anion with methyl chloroformate furnished the alkynyl ester 19 in near quantitative yield. Removal of the TBS-ether by exposure to HF·pyridine followed by protection of the alcohol as MOM-ether with MOMCl and diisopropylethylamine afforded 20 in 95% yield. Alkynyl ester 20 was treated with freshly prepared Me2CuLi14 to provide the Z-olefin 21 as a single product in 96% isolated yield. The observed NOESY among the protons are consistent with the assigned Z-olefin geometry in ester 21. DIBAL-H reduction followed by protection with tert-butyldimethylsilyl chloride furnished the silyl ether 22. Selective oxidative cleavage of the terminal olefin provided the other Julia-Kocienski olefination precursor, aldehyde 5.
Scheme 2.
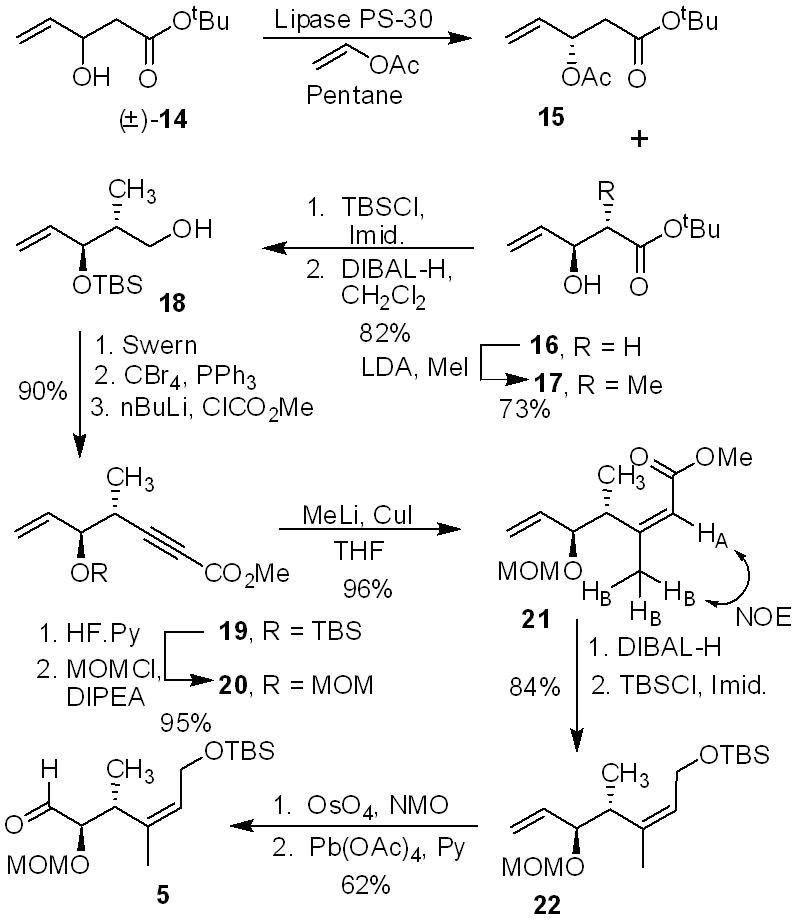
Synthesis of aldehyde 5
With the aldehyde and sulfone in hand, we then carried out Julia-Kocienski olefination as shown in Scheme 3. Thus, treatment of sulfone 4 with KHMDS in THF followed by addition of aldehyde 5 provided 2 (C1-C12 segment) in 71% isolated yield.15 To test the feasibility of the Sakurai reaction, we have investigated reaction of allyl silane 2 with isobutylaldehyde as a model. As shown, the reaction of 2 with 1.5 equiv of isobutylaldehyde in the presence of 1.5 equiv of SnCl4 and 0.5 equiv of Et3N at −78 °C for 10 min in CH2Cl2 afforded alcohol 23 as a mixture (1:1.5) of diastereoisomer in 43% yield. Dess-Martin Periodinane oxidization of the alcohol mixture furnished ketone 24 in 85% yield.15
Scheme 3.

Synthesis of ketone 24
In summary, a highly stereocontrolled synthesis of the C1-C12 fragment of iriomoteolide 1a has been achieved. Lipase catalyzed kinetic resolution of β-hydroxy amide provided the key starting material for the synthesis. Other important steps involve a Pd-catalyzed cross-coupling reaction, a highly regioselective and stereoselective conjugate addition of methylcuprate to an α, β-acetylenic esters and elaboration of the C6-C7 trans-olefin geometry by a Julia-Kocienski olefination reaction. Sakurai reaction of 2 with isobutylaldehyde followed by oxidation of the resulting alcohol provided ketone 24 in modest yield. Further work toward the total synthesis of iriomoteolide 1a is in progress.
Acknowledgments
This research is supported in part by the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Harris CR, Danishefsky SJ. J Org Chem. 1999;64:8434. [Google Scholar]
- 2.Kobayashi J, Kubota T. J Nat Prod. 2007;70:451. doi: 10.1021/np0605844. [DOI] [PubMed] [Google Scholar]
- 3.Tsuda M, Oguchi K, Iwamoto R, Okamoto Y, Kobayashi J, Fukushi E, Kawabata J, Ozawa T, Masuda A, Kitaya Y, Omasa K. J Org Chem. 2007;72:4469. doi: 10.1021/jo070414b. [DOI] [PubMed] [Google Scholar]
- 4.Fang L, Xue H, Yang J. Org Lett. 2008;10:4645. doi: 10.1021/ol801940r. [DOI] [PubMed] [Google Scholar]
- 5.Hosomi A, Sakurai H. Tetrahedron Lett. 1976:1295. [Google Scholar]
- 6.Blakemore PR, Cole WJ, Kocienski PJ, Morley A. Synlett. 1998:26. [Google Scholar]
- 7.Enzymatic resolution of β-hydroxy amide, particularly with Weinreb amide, has not been reported. For enzymatic resolution of -hydroxy ester, see: Vrielynck S, Vandewalle M, García AM, Mascareñas JL, Mouriño A. Tetrahderon Lett. 1995;36:9023.
- 8.Dale JA, Dull DL, Mosher HS. J Org Chem. 1969;34:2543. [Google Scholar]
- 9.Enev VS, Kachlig H, Mulzer J. J Am Chem Soc. 2001;123:10764. doi: 10.1021/ja016752q. [DOI] [PubMed] [Google Scholar]
- 10.Ghosh AK, Kulkarni S. Org Lett. 2008;10:3907. doi: 10.1021/ol8014623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pollini GP, Risi CD, Lumento F, Marchetti P, Zanirato V. Synlett. 2005:164. [Google Scholar]
- 12.(a) Seebach D, Aebi J, Wasmuth D. Collect. III. John Wiley and Sons; New York: 1990. Organic Synthesis; pp. 153–159. [Google Scholar]; (b) Hermann JL, Schlessinger RA. Tetrahedron Lett. 1973;14:2429. [Google Scholar]
- 13.Corey EJ, Fuchs PL. Tetrahedron Lett. 1972;13:3769. [Google Scholar]
- 14.Corey EJ, Katzenel J. J Am Chem Soc. 1969;91:1851. [Google Scholar]
- 15.All new compounds gave satisfactory spectroscopic and analytical results. Compound 2: 1H NMR (CDCl3): δ 5.66 (dt, J = 15.5, 7.0 Hz, 1H), 5.38 (t, J = 5 Hz, 1H), 5.25 (dd, J = 15.5, 9.0 Hz, 1H), 4.65 (d, J = 7.0 Hz, 1H), 4.60 (d, J = 2.5 Hz, 1H), 4..57 (d, J = 2.5 Hz, 1H), 4.37 (d, J = 7.0 Hz, 1H), 4.31 (dd, J = 7.2, 13.0 Hz 1H), 4.23 - 4.16 (m, 1H), 3.85 - 3.79 (m, 1H), 3.31 (s, 3H), 2.67 – 2.64 (m, 1H), 2.31 – 2.20 (m, 2H), 1.68 (brs, 3H), 0.92 (d, J = 6.9 Hz, 3H), 0.89 (s, 9H), 0.88 (s, 9H), 0.06 - 0.01 (m, 21H). Compound 24: 1H NMR (CDCl3): δ 5.67 (dt, J = 15, 7.0 Hz, 1H), 5.42 (t, J = 4.5 Hz, 1H), 5.28 (dd, J = 15, 8.0 Hz, 1H), 5.00 (s, 1H), 4.94 (s, 1H), 4.67 (d, J = 7.0 Hz, 1H), 4.41 (d, J = 7.0 Hz, 1H), 4.33 (dd, J = 13.0, 5Hz, 1H), 4.20 (dd, J = 13.0 Hz, 1H), 3.87 (t, J = 5.0 Hz, 1H), 3.83 (t, J = 9.0 Hz, 1H), 3.34 (s, 3H), 3.24 (AB, JAB = 16.0Hz, ΔVAB = 32.5Hz, 2H), 2.75-2.71 (m, 1H), 2.72-2.67 (m, 1H), 2.33-2.26 (m, 2H), 2.25-2.20 (m, 2H), 1.72 (brs, 3H), 1.13 (s, 3H), 1.12 (s, 3H), 0.92 (d, J = 7.0 Hz, 3H), 0.89 (s, 9H), 0.88 (s, 9H), 0.06 - 0.01 (m, 12H); MS (EI), m/z = 619 (M+Na)+.