

The polycomb repressor complex ubiquitylates γ-H2AX and other components of the DNA damage response pathway to facilitate genomic repair.
Abstract
Polycomb group (PcG) proteins are major determinants of cell identity, stem cell pluripotency, and epigenetic gene silencing during development. The polycomb repressive complex 1, which contains BMI1, RING1, and RING2, functions as an E3-ubuiquitin ligase. We found that BMI1 and RING2 are recruited to sites of DNA double-strand breaks (DSBs) where they contribute to the ubiquitylation of γ-H2AX. In the absence of BMI1, several proteins dependent on ubiquitin signaling, including 53BP1, BRCA1, and RAP80, are impaired in recruitment to DSBs. Loss of BMI1 sensitizes cells to ionizing radiation to the same extent as loss of RNF8. The simultaneous depletion of both proteins revealed an additive increase in radiation sensitivity. These data uncover an unexpected link between the polycomb and the DNA damage response pathways, and suggest a novel function for BMI1 in maintaining genomic stability.
Introduction
The cellular response to double-strand breaks (DSBs) is characterized by the relocalization and accumulation of DNA damage signaling/repair proteins into subnuclear domains termed ionizing radiation (IR)-induced foci (IRIF; Fernandez-Capetillo et al., 2003; Petrini and Stracker, 2003). In addition to protein accumulation, IRIF are sites of chromatin remodeling and posttranslational modifications (PTMs) of histones at DSBs (Ismail and Hendzel, 2008). One of the initial targets of DSB signaling is the phosphorylation of the histone H2A variant H2AX, which then accumulates within chromatin surrounding the sites of DSBs to generate structures termed γ-H2AX foci (Rogakou et al., 1998, 1999). Phosphorylation of H2AX by ataxia telangiectasia mutated (ATM), ATM and Rad3 related (ATR), and DNA-dependent protein kinase (DNA-PK) is an early event in response to DSBs and represents the most robust histone modification induced by IR (Burma et al., 2001; Ward and Chen, 2001; Stiff et al., 2004). Although γ-H2AX is dispensable for the initial recruitment of several mediators/repair proteins, including MDC1, BRCA1, 53BP1, and ATM, it is required for their efficient retention at the chromatin surrounding the break (Celeste et al., 2003).
Histone ubiquitylation plays an important role in DNA damage signaling. The E3 ubiquitin ligase, RNF8, and its associated E2 conjugating enzyme, UBC13, are recruited to DSBs, where they are thought to polyubiquitylate histones H2A and H2AX with K63-linked chains (Huen et al., 2007; Kolas et al., 2007; Mailand et al., 2007). K63-linked chains decorating H2A and H2AX are thought to provide binding sites for the ubiquitin-interacting motif (UIM) of RAP80, and this in turn facilitates the recruitment of BRCA1 to IRIF (Huen et al., 2007; Kolas et al., 2007; Mailand et al., 2007). A recent study revealed the crystal structure of RAP80-UIM1-UIM2 complexed with K63-linked diubiquitin. The two UIMs generate higher affinity binding through an avidity mechanism, whereas the linker region that joins the two UIMs specifies the selectivity for the K63-linked chains (Sato et al., 2009).
Two histone H2A/H2AX/H2AZ-E3 ubiquitin ligases have been identified: the polycomb repressive complex 1 (PRC1) and RNF8/RNF168 (Huen et al., 2007; Kolas et al., 2007; Mailand et al., 2007; Doil et al., 2009; Stewart et al., 2009). Knockdown of either PRC1 or RNF8 E3 ligase activity significantly reduces steady-state levels of ubiquitylated H2A (Wang et al., 2004; Cao et al., 2005; Huen et al., 2008). RNF8 contains a forkhead-associated (FHA) domain that binds to phosphorylated MDC1 to recruit this E3 ubiquitin ligase to sites of DNA damage (Huen et al., 2007; Mailand et al., 2007). Depletion of RNF8 eliminates the generation of diubiquitylated γ-H2AX (Huen et al., 2007); however, there remains a significant level of IR-induced monoubiquitylated γ-H2AX, which suggests that other E3-ubiqituin ligases ubiquitylate histone H2A at sites of DNA damage. This ubiquitylation is dynamic. Incubation of laser microirradiated cells with a proteosome inhibitor rapidly depletes ubiquitin from sites of DNA damage (Mailand et al., 2007). Furthermore, knockdown of the deubiquitylase, BRCC36, results in significant accumulation of ubiquitylated γ-H2AX in RNF8-deficient cells (Shao et al., 2009). This suggests that there is more than one H2A E3 ubiquitin ligase that responds to DSBs. In this respect, it is notable that knockdown of either RING2 or RNF8 significantly reduces the ubiquitylation of histone H2A after UV damage (Bergink et al., 2006; Marteijn et al., 2009). Thus, the PRC1 E3 ubiquitin ligase is a good candidate for the additional histone H2A/H2AX ubiquitylation at sites of DSBs.
Polycomb group (PcG) proteins are chromatin-associated proteins that maintain heritable gene repression patterns (Sparmann and van Lohuizen, 2006; Gieni and Hendzel, 2009). They are also involved in embryonic and adult stem cell maintenance and have been implicated in cancer development (Sparmann and van Lohuizen, 2006; Gieni and Hendzel, 2009). At least two distinct human PcG complexes have been identified (Sparmann and van Lohuizen, 2006). The PRC1 contains PC2/CBX4, HPH1, and RING domain–containing proteins (RING1, RING2, and BMI1). The PcG repressive complex 2 (PRC2) contains EED, EZH2, and SUZ12 proteins. Three distinct enzymatic activities—methylation, sumoylation, and ubiquitylation—have been linked to PcG complexes. The PRC2 complex is responsible for trimethylation of lysine 27 of histone H3 (H3K27me3), which is recognized by the PRC1 complex through the polycomb chromodomain of Pc2 (Sparmann and van Lohuizen, 2006). The PRC1 complex possesses E3 ubiquitin ligase activity that targets ubiquitylation of H2A on lysine 119 (uH2AK119), linking uH2AK119 to PcG-mediated gene silencing (de Napoles et al., 2004; Wang et al., 2004; Buchwald et al., 2006). The catalytic subunit has been mapped to RING2, whereas the presence of RING1 and BMI1 can enhance the enzymatic activity in vitro and target RING2 to substrates (de Napoles et al., 2004; Wang et al., 2004; Zeidler et al., 2005; Buchwald et al., 2006; Li et al., 2006). In addition, BMI1 has also been shown to play an important role in cell proliferation (Jacobs et al., 1999), stem cell self-renewal (Lessard and Sauvageau, 2003), and cancer (van Lohuizen et al., 1991; van Kemenade et al., 2001; Lessard and Sauvageau, 2003). Here, we identify BMI1 as an early DNA damage response (DDR) protein that critically regulates the cellular response to DSBs.
We find that BMI1 and RING2 are required for the accumulation of monoubiquitylated γ-H2A.X, the predominant form of ubiquitylated γ-H2A.X, at sites of DNA damage. BMI1 and RING2 recruit to IRIF and laser microirradiated DNA damage sites. Unlike the E3 ubiquitin ligases RNF168 and BRCA1 (Huen et al., 2007; Kolas et al., 2007; Mailand et al., 2007; Doil et al., 2009; Stewart et al., 2009), RNF8 is not required for the recruitment of BMI1 or RING2 to sites of DNA damage. Loss of BMI1 increases radiosensitivity and decreases DSB repair efficiency. In the absence of BMI1, proteins that require ubiquitylation are impaired in their recruitment to DNA damage sites, including 53BP1, RAP80, and BRCA1. Our results define the PcG histone H2A E3 ubiquitin ligase as a second major γ-H2A.X E3 ubiquitin ligase at sites of DSBs.
Results
Identification of the BMI1–RING2 complex as an early DDR protein
BMI1, like other PcG proteins, enriches in discrete nuclear structures termed PcG bodies (Hernández-Muñoz et al., 2005). These PcG bodies are unique nuclear structures that are reported to associate with constitutive heterochromatin (Saurin et al., 1998). In examining BMI1 nuclear foci in human bone osteosarcoma cells (U2OS), we noted similarities with the enlarged γ-H2AX foci that are frequently observed in unirradiated cells (Hao et al., 2004; McManus and Hendzel, 2005). In unirradiated cells, two distinct γ-H2AX focal populations are evident: a novel and predominant population of small foci that do not colocalize with DSB repair proteins and a small population of large foci that colocalize with many repair proteins (Fig. S1 A). To evaluate the possible localization of BMI1 at γ-H2AX foci, we examined its staining pattern relative to that of γ-H2AX foci by immunofluorescence (IF) microscopy in U2OS. In the absence of DNA damage, BMI1 concentrated in several larger foci termed PcG bodies, which is consistent with the previously described localization of BMI1 (Fig. S1 A; Saurin et al., 1998). Notably, there appeared to be two classes of these structures in U2OS cells. A subset of the cells contained larger domains enriched in BMI1 that were positive for γ-H2AX staining despite the absence of any treatment to induce DNA damage. The presence of a small number of large foci enriched in γ-H2AX has been reported before and is thought to represent persistent DSBs that may include eroded telomeres (Hao et al., 2004). This suggested a possible role for BMI1 in the DDR. To test this hypothesis, we exposed cells to IR and examined the localization of BMI1 relative to γ-H2AX foci. Upon IR treatment, BMI1 localized to a large number of foci throughout the nucleus. Most of these sites colocalized with the DSB marker γ-H2AX (Fig. 1 A and Video 1). Interestingly, a small number of sites that included the largest domains enriched in BMI1, which were similar in appearance to the γ-H2AX–positive domains in untreated cells, were not positive for γ-H2AX (Fig. S1 A). However, virtually all γ-H2AX foci in IR-treated cells were enriched in BMI1 (Fig. 1 A and Video 1). Colocalization analysis of z-stack images of 40 BMI1 and γ-H2AX double-stained cells revealed that 92% of BMI1 foci colocalized with γ-H2AX foci compared with the 98% of γ-H2AX foci that colocalized with BMI1 foci (Fig. 1 A and Video 1). This difference in colocalization percentages reflects the presence of BMI1 in structures in addition to the DNA damage foci (PcG bodies). This is seen in Video 1, where the channels are toggled on and off to more easily compare localization. In control experiments, we confirmed the specificity of our BMI1 antibody. The antibody did not stain cells lacking BMI1 (Fig. S1 B). Other DNA-damaging agents, such as UV light and H2O2, did not lead to immediate (5 min) formation of additional BMI1 foci (Fig. S2 A). Similar results were obtained when cells exposed either to UV light or H2O2 were allowed to recover for different time points (e.g., 15, 30, and 60 min; unpublished data). In contrast, BMI1 foci were observed in cells treated with calicheamicin γ1 (CLM), a radiomimetic drug that causes DSBs (Fig. S2 A; Elmroth et al., 2003). BMI1 IRIF could be visualized as early as 5 min after exposure to 2 Gy of IR, with ∼50% of cells being positive for these foci. More than 90% of cells were positive for BMI1 IRIF after 1 h (Fig. S2 B).
Figure 1.

BMI1 and RING2 are recruited to IRIF. (A) U2OS cells were irradiated with 2 Gy, extracted with 0.5% Triton X-100, and stained with antibodies recognizing BMI1 (green in composite) and γ-H2AX (red in composite). Maximum intensity projections are shown for each image. The top row is without deconvolution; the middle row is after deconvolution of the original fluorescence z series. The arrows show examples of PcG bodies. (B) Indirect IF of RING2 after irradiation with 2 Gy IR. The top left panel is a maximum intensity projection image of RING2 (green in bottom panels), the middle panel shows the same image after enhancement of gamma, and the right panel shows γ-H2AX (red in bottom panels). BMI1 and RING2 localize to sites of DSBs in addition to the PcG bodies, which remain highly enriched in PcG proteins after IR treatment. Arrows point to RING2 foci (PcG bodies) that do not colocalize with γ-H2AX foci. Bars, 5 µm.
We confirmed these results using laser microirradiation. DNA damage induced in defined regions of cells with a 750-nm two-photon laser revealed that endogenous BMI1 localized to the laser-induced DNA damage sites that overlap with γ-H2AX and poly-ADP-ribosylation (PAR; another established marker of sites of DNA damage; Fig. S2 C; Haince et al., 2008). We observed BMI1 recruitment in a panel of cell lines ranging from mouse to human (unpublished data), which demonstrates that BMI1 accumulation at the sites of DNA damage is not cell type specific.
Because BMI1 is part of the PRC1 complex (Wang et al., 2004; Cao et al., 2005), we also evaluated the recruitment of RING2, the E3 ubiquitin ligase within the PRC1 complex, and found that endogenous RING2 also localized to sites of DNA damage (Fig. 1 B). Thus, our data indicate that the key components of the PcG E3 ubiquitin ligase complex, BMI1 and RING2, are specifically recruited to sites of DNA damage.
Recruitment kinetics of PcG proteins at sites of DNA damage
To study the dynamics of PcG proteins in living cells in response to DSBs, BMI1 and RING2 were tagged at their N termini with GFP. These constructs were stably introduced into U2OS. The protein levels of exogenously expressed GFP chimeric proteins were comparable to endogenous levels (Fig. S3 A). Laser microirradiation resulted in a clear and defined recruitment of both GFP-BMI1 and GFP-RING2 to the DSB tracks in vivo (Fig. 2 A). We found that both BMI1 and RING2 recruit with similar and rapid kinetics (11 s) to sites of DNA damage (Fig. 2 A). The rapid kinetics of BMI1–RING2 recruitment to sites of DNA damage were similar to early DDR proteins such as MRE11, RNF8, and NBS1 (Fig. 2 B), which suggests that the BMI1–RING2 E3 ubiquitin ligase complex is one of the early factors involved in the response to DSBs.
Figure 2.
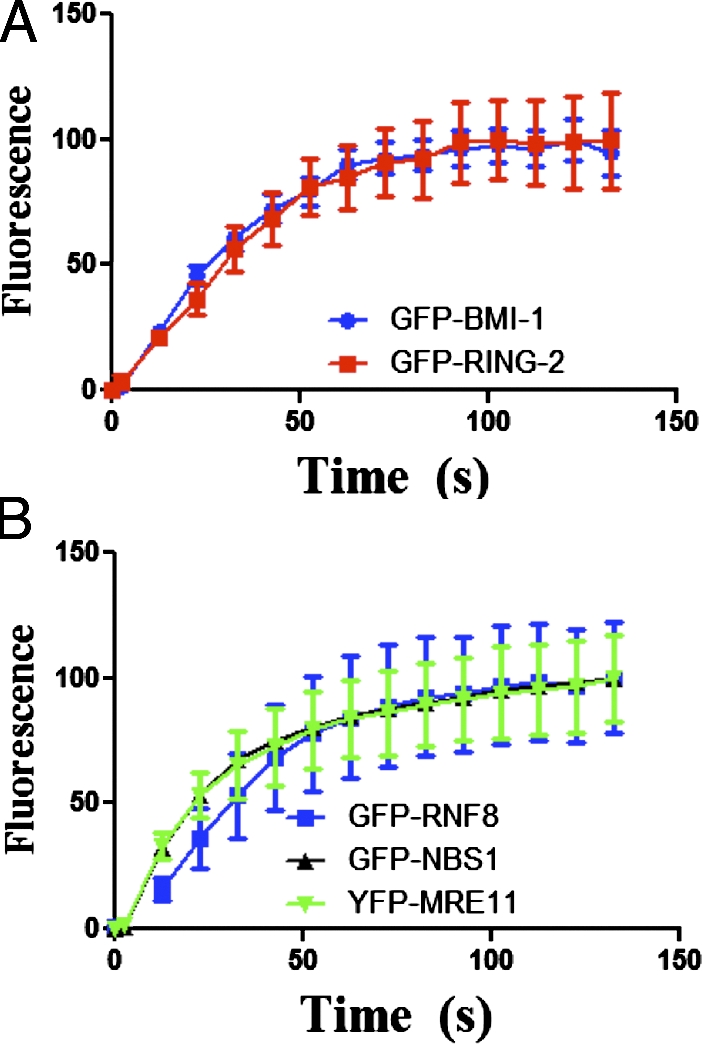
Dynamics of BMI1 and RING2 in living cells. U2OS cells expressing GFP-BMI1 or GFP-RING2 were monitored after microirradiation using time-lapse microscopy. (A) GFP-BMI1 and GFP-RING2 accumulation at laser track sites was quantified. The integrated intensity in the microirradiated areas was determined (n = 15) and the percentage of maximum value was plotted versus time. (B) The recruitment of GFP-tagged DSB repair proteins NBS1, MRE11, RNF8 was quantified for comparison. Error bars represent standard error from two independent experiments.
DNA damage causes transient immobilization of BMI1 at DNA damage sites
We next assessed the association of BMI1 with chromatin in response to DNA damage using an established chromatin fractionation assay (Li and Stern, 2005). A biochemical fractionation experiment on cells exposed or not exposed to radiation revealed that BMI1 did not show apparent chromatin enrichment upon radiation (Fig. S4 A). Rather, BMI1 was associated with chromatin with or without irradiation (Fig. S4 A). This is distinct from other DDR proteins, which are soluble in the absence of DNA damage. However, BMI1 has a very short half-life when binding to chromatin in the absence of IR treatment. This facilitates its rapid retargeting upon radiation exposure without requiring a change in how the protein fractionates. To investigate this further, we used FRAP to examine the binding affinity of GFP-BMI1 within the nucleoplasm of living cells after introduction of DSBs. To test whether or not BMI1 was more stably associated with sites of DNA damage than undamaged chromatin, we microirradiated the nucleus of U2OS cells stably expressing GFP-BMI1 and incubated the irradiated cells for 2 min at 37°C to allow accumulation of DSBs to reach a steady state. Small circular regions located either in the undamaged nucleoplasm or over the laser-generated DSB tracks were photobleached, and the fluorescence recovery in these regions was determined by repetitive image acquisition (Fig. S4 B). We found that in the absence of DSBs, GFP-BMI1 binds with lower affinity to the endogenous chromatin, as was evident by the rapid recovery of the photobleached region. In comparison, introduction of DSBs resulted in a modest increase in the binding affinity of GFP-BMI1 on the damaged nucleoplasm, resulting in an ∼23% increase in the time needed for recovery of this photobleached region (Fig. S4 C). In an additional control experiment, we found that the chromatin association profile of GFP-BMI1 is similar to that of endogenous BMI1 (Fig. S4 A). These data suggest that DNA damage results in a degree of stabilization of recruited BMI1 at DSBs sites, which may reflect a difference in the mechanism of association between BMI1 and damaged versus undamaged chromatin sites.
BMI1 targets RING2 to the sites of DSBs
We next examined whether or not BMI1 acts as an adaptor for targeting RING2 to the sites of damage. Using BMI1 wild type (BMI1 WT) and BMI1 knockout (BMI1 KO) mouse embryonic fibroblasts (MEFs), we found that RING2 accumulation at DSB sites was completely abrogated in BMI1 KO cells (Fig. 3). Importantly, the recruitment of RING2 was rescued by transfecting KO cells with GFP BMI1 (Fig. 3). These data demonstrate that RING2 requires BMI1 to be recruited to DSB sites.
Figure 3.

BMI1 is required for targeting RING2 to the DNA damage sites. BMI1 WT (top), BMI1 KO (middle), and BMI1 KO (bottom) cells reconstituted with a full-length GFP BMI1 construct were microirradiated, allowed to recover for 5 min, and stained with the indicated antibodies. Bars, 5 µm.
The RING domain targets BMI1 to DNA damage sites
BMI1 has an N-terminal RING finger (RING), a central helix-turn-helix-turn-helix-turn (H-T), and a C-terminal proline-serine rich (PEST)-like domain. The C terminus also contains an essential NLS (NLS2; Fig. 4 A). To identify the domains required for BMI1 recruitment to the break sites, we expressed myc-tagged BMI1 deletion mutants lacking the RING (dRING), H-T (dH-T), or PEST (dPEST) domains in BMI1 KO cells and immunostained for BMI1 on the damage sites. WT BMI1, dPEST, and dH-T mutants accumulated at the break sites and colocalize with PAR after laser microirradiation (Fig. 4 C). In contrast, the dRING mutant failed to localize to PAR-containing tracks. In control experiments, we found that the different BMI1 deletion expression constructs are expressed at the same levels in BMI1 KO cells (Fig. 4 B). This demonstrates that the RING finger domain of BMI1 is important for targeting BMI1 to DNA damage sites.
Figure 4.

The RING domain of BMI1 is required for its accumulation at DNA damage sites. (A) Schematic representation of BMI1 deletion constructs used in B. Amino acid numbers are indicated. (B) BMI1 KO MEFs were transfected with WT BMI1 or the individual myc-tagged BMI1 deletion constructs shown in A. Immunoblots of extracts prepared from these cells were probed with myc antibody. As a loading control, the membrane was probed for actin. (C) BMI1 KO cells reconstituted with different myc-tagged BMI1 deletion constructs were microirradiated and double-stained with the myc (top) and PAR (bottom) antibodies. Bar, 5 µm.
BMI1 mediates IR-induced monoubiquitylation of γ-H2AX
Given that BMI1 and RING2 recruit to sites of DSBs, we tested the hypothesis that BMI1 is a major histone H2A ubiquitin ligase that contributes to the previously reported IR-induced RNF8-independent monoubiquitylation of γ-H2AX (Huen et al., 2007). U2OS cells were transfected with control or one of two different BMI1 small hairpin RNAs (shRNAs). Immunoblotting revealed that BMI1 shRNA successfully knocked down the levels of BMI1 approximately fivefold (Fig. S3 B). Consistent with previously published results (Huen et al., 2007; Kolas et al., 2007; Mailand et al., 2007), we found that γ-H2AX ubiquitylation is regulated in an IR-dependent manner in vivo (Fig. 5 A). BMI1 knockdown did not have an effect on IR-induced γ-H2AX. However, we found that IR-induced γ-H2AX mono-ubiquitylation was significantly reduced in cells transfected with BMI1 shRNA (Fig. 5 A). Because knockdown of either BMI1 or RNF-8 results in a reduction in steady-state ubiquitylation, it is possible that the loss of ubiquitylated γ-H2AX reflected a reduction in H2AX ubiquitylation before DNA damage (Fig. 5 B). To test whether or not BMI1 contributed to ubiquitylation at the site of DNA damage, we measured the ratio of ubiquitylated γ-H2AX/γ-H2AX and compared the changes in this species to the reductions observed in ubiquitylation of histone H2AX. For simplification, the unirradiated WT cells were normalized to 1.0, and the irradiated WT cells were normalized to 1.0. Because the radiation was delivered randomly throughout the genome, the reduction in steady-state ubiquitylation in H2AX should be similar to the reduction of γ-H2AX ubiquitylation if the influence of BMI1 is simply on steady-state ubiquitylation. BMI1-dependent ubiquitylation of γ-H2AX after DNA damage would be expected to result in a greater loss of ubiquitylated γ-H2AX after IR than the loss of ubiquitin from H2AX before DNA damage. In the absence of DNA damage, BMI1 knockdown results in an ∼50% loss in uH2AX (Table I). In contrast, after DNA damage, the reduction in γ-H2AX ubiquitylation is ∼80% (Table I). To further confirm these results, we examined γ-H2AX ubiquitylation in RNF8-null (RNF8 KO) and BMI1 KO cells. We found that DNA damage induced γ-H2AX mono- and diubiquitylation in WT MEFs. Although γ-H2AX diubiquitylation is severely reduced, RNF8 KO cells had reduced but significant levels (65%) of IR-induced γ-H2AX monoubiquitylation (Fig. 5 C and Table I). In contrast, we found that BMI1 KO cells have 85% reduction in both IR-induced γ-H2AX mono- and diubiquitylation (Fig. 5, B and C; and Table I). Because RNF8 KO cells completely lack RNF8 (Minter-Dykhouse et al., 2008), we can exclude the possibility that the observed ubiquitylation depends on RNF8. To determine whether or not BMI1 regulates the observed IR-induced γ-H2AX monoubiquitylation in RNF8 KO cells, we transfected the cells with either control or one of two different BMI1 shRNAs. We found that knocking down BMI1 resulted in an ∼80% reduction in IR-induced γ-H2AX monoubiquitylation at the 60 min time point (Fig. 5 E and Table I), whereas BMI1 knockdown did not affect IR-induced H2AX phosphorylation. In control experiments, we found that our γ-H2AX antibody specifically recognizes phosphorylated H2AX species after DNA damage (Fig. S4 D). These data demonstrate a role for BMI1 in regulating γ-H2AX ubiquitylation. Not surprisingly, given the conservation of the ubiquitylation site between H2A and H2AX, it was previously shown that the RING2 complex can ubiquitylate H2AX in vitro (Elderkin et al., 2007).
Figure 5.

BMI1 mediates DNA damage–induced γ-H2AX monoubiquitylation. U2OS cells transfected with either control or a BMI1 shRNA were irradiated (6 Gy) and permitted to recover for 1 h at 37°C. (A and B) Histone extracts were prepared and immunoblotted with γ-H2AX (A) and H2AX antibodies (B). Black lines indicate that intervening lanes have been spliced out. RNF8 WT and RNF8 KO were treated as in A. (C and D) Histone extracts were prepared and immunoblotted with γ-H2AX (C) and H2AX (D) antibodies. (E and F) RNF8 KO cells transfected with either control shRNA or one of two different BMI1 shRNA for 24 h and were treated as in A and B.
Table I.
Quantification of changes in ubiquitylation of H2AX
| Cells and treatment | u-H2AX/H2AX no IR | uγ-H2AX/γ-H2AX + IR |
| Ctrl shRNA U2OS | 1.0 | 1.0 |
| BMI1 shRNA1 U2OS | 0.40 | 0.10 |
| BMI1 shRNA2 U2OS | 0.68 | 0.34 |
| WT MEFs | 1.0 | 1.0 |
| BMI1 KO MEFs | 1.03 | 0.14 |
| RNF8 KO MEFs | 1.04 | 0.63 |
| Ctrl shRNA RNF8 KO | 1.0 | 1.0 |
| BMI1 shRNA1 RNF8 KO | 0.68 | 0.18 |
| BMI1 shRNA2 RNF KO | 0.48 | 0.23 |
Cells were treated as in Fig. 5. The intensity of the H2AX and uH2AX were analyzed and the ratio between the two bands was determined. The ratio was then normalized relative to the control condition. In IR-treated cells, the same approach was used to quantify the ratio between ubiquitylated and parent forms of γ-H2AX.
BMI1 is required for efficient DDR
To test the function of BMI1 during the DDR, we examined the effect of BMI1 down-regulation on indices of ATM signaling. BMI1 WT and BMI1 KO cells were irradiated (2 Gy), and the subsequent activation and relocalization of several DSB downstream factors were analyzed by indirect IF. Depletion of BMI1 levels in cells exposed to IR did not have an apparent effect on the accumulation of MRE11, NBS1, p-ATM, or γ-H2AX onto IRIF (Fig. S5) at the 30 min time point. These data indicate that BMI1 is not required for the accumulation and relocalization of these proteins onto the sites of DNA damage.
Several groups have reported the localization of conjugated ubiquitin proteins at the DNA damage sites using a monoclonal antibody (FK2; Mailand et al., 2007; Doil et al., 2009). In agreement with previous reports, we found that upon exposing U2OS cells to IR (2 Gy), FK2 forms IRIF. Quantification of conjugated-Ub (FK2) focus formation 1 h after treatment with 2 Gy of IR in BMI1 WT and BMI1 KO MEFs revealed that BMI1 KO cells have approximately three times less FK2 IRIF compared with BMI1 WT MEFs (Fig. 6 A). To confirm this defect, we analyzed the ability of RAP80, a ubiquitin-binding protein, to form IRIF in BMI1 WT and BMI1 KO MEFs. Immunostaining of BMI1 WT and BMI1 KO MEFs using several commercially available RAP80 antibodies failed to detect endogenous RAP80 or RAP80 IRIF. To overcome difficulties with RAP80 immunostaining, BMI1 WT and BMI1 KO cells were transiently transfected with EGFP-RAP80, and the effect of irradiation on the cellular distribution of GFP RAP80 was examined by confocal microscopy. Consistent with previously published data (Yan et al., 2007), examination of the time course of GFP RAP80 accumulation showed that RAP80 did not rapidly relocate to DNA damage foci (20 min); however, by 120 min after radiation, translocation of RAP80 to the foci was clearly observed (Fig. 6 B). Examining GFP RAP80 IRIF in BMI1 WT and BMI1 KO cells revealed that BMI1 KO cells have approximately ninefold less RAP80 IRIF relative to BMI1 WT MEFs (Fig. 6 B). This difference is statistically significant (P = 0.0004). These data are consistent with the previous experiment (Fig. 6 A), demonstrating that BMI1 KO cells have a reduced number of FK2 IRIF-positive cells. We next examined the impact of BMI1 KO on the relocalization of 53BP1 and BRCA1 onto DNA damage foci. 53BP1 and BRCA1 targeting to DSB sites is mediated in a ubiquitin-dependent manner (Huen et al., 2007; Kolas et al., 2007; Mailand et al., 2007). We have found that the recruitment of 53BP1 into the sites of DNA damage is severely suppressed in BMI1 KO and slightly reduced in RNF8 KO MEFs (Fig. 6 C) at the 5 min time point. To confirm this defect, we analyzed the recruitment of 53BP1 onto microirradiation sites in BMI1 KO cells 30 min after irradiation. We found that 53BP1 accumulates normally onto damaged chromatin in both RNF8 KO and BMI1 KO MEFs at 30 min (Fig. 6 C). Because of the lack of a specific commercially available BRCA1 antibody, we transfected GFP-BRCA1 into BMI1 WT and BMI1 KO MEFs, and the kinetics of BRCA1 recruitment to the sites of DNA damage were monitored. We found that within 11 s after microirradiation, BRCA1 accumulated at the irradiated sites in BMI1 WT MEFs, whereas BRCA1 accumulation was delayed to 71 s in BMI1 KO MEFs (Fig. 6 D). We conclude that BMI1 regulates the initial and early ubiquitin modifications induced by DNA damage and subsequently the kinetics of recruitment of several DDR proteins onto these sites.
Figure 6.

BMI1 is required for efficient DDR. (A) BMI1 WT and BMI1 KO MEFs were irradiated (2 Gy) and allowed to recover for 60 min. Cells were stained with γ-H2AX and FK2 antibodies. The number of foci per cell was counted manually on the microscope using a 63× lens. The mean number of FK2 foci per cell is shown from 40 cells counted. The arrow points to micronuclei that are commonly observed in BMI1 KO MEFs. (B) BMI1 WT and BMI1KO MEFs expressing GFP RAP80 were irradiated (2 Gy, 120 min), and the number of GFP-RAP80 IRIF per cell was counted. (C) BMI1 WT, BMI1 KO, and RNF8 KO MEFs were microirradiated, allowed to recover for 5 or 30 min, and stained with the indicated antibodies. The relative intensity of 53BP1 recruited to the sites of DNA damage was quantified and shown as the mean of at least five cells. (D) BMI1 WT and BMI1KO MEFs expressing GFP BRCA1 were monitored after microirradiation using time-lapse microscopy. (D, left) Representative images of BRCA1 time lapse. (D, right) The integrated intensity in the microirradiated area is plotted as a percentage of maximum versus time. Error bars represent standard error from two independent experiments. Bars, 5 µm.
The initial recruitment of BMI1 to the sites of DNA damage does not require H2AX
γ-H2AX has been shown to be required for the recruitment of RNF8 to sites of DNA damage (Huen et al., 2007; Kolas et al., 2007; Mailand et al., 2007). Therefore, we examined whether or not H2AX is required for BMI1 recruitment to DSBs. Strikingly, using H2AX proficient (H2AX WT) and H2AX KO (H2AX KO) MEFs, we observed clear and rapid accumulation of BMI1 at the laser-induced damage sites in both H2AX WT and H2AX KO MEFs (Fig. 7 A). These results show that H2AX is not required for the initial recruitment of BMI1 to DSB sites.
Figure 7.

BMI1 knockdown inhibits DSB repair and sensitizes cells to IR. (A) The recruitment of BMI1 in H2AX WT and H2AX KO cells. Cells were microirradiated, allowed to recover for 5 min, and stained with the indicated antibodies. Bar, 5 µm. (B) BMI1 WT, BMI1 KO, and BMI1 KO cells reconstituted with WT BMI1 were either left untreated or exposed to 30 nM of CLM. The initial DNA damage levels and levels remaining at 1 h and 5 h at 37°C were measured with CFGE. (C) Survival curve of U2OS cells transfected with either control shRNA or a BMI1 shRNA. Cells were transfected with the indicated shRNA for 24 h, plated at low density, and exposed to the indicated doses of IR. The experiment was performed in triplicate. Error bars represent standard error from two independent experiments (n = 6).
Depletion of BMI1 inhibits DSB repair and increases radiation sensitivity
A common feature in cells lacking a DDR protein is that they repair only a fraction of DSBs induced by x rays, which results in hypersensitivity to IR. Therefore, if BMI1 is a DDR protein, the depletion of BMI1 expression is expected to reduce DSB repair and sensitize cells to IR. To assess the effect on DSB repair, we examined radiation sensitivity in BMI1 WT, BMI1 KO, and BMI1 KO cells reconstituted with a full-length BMI1 expression construct. The initial DSB levels and the levels remaining after 1 h and 5 h of repair at 37°C were measured with constant-field gel electrophoresis (CFGE; Ismail et al., 2005). As a control, we used human glioblastoma cells with normal DNA-PK levels (M059K) or a matching cell line from the same patient lacking DNA-PKcs expression (M059J; Lees-Miller et al., 1995). DSBs were introduced by the DSB-inducing drug CLM. In agreement with a previous study, we have found that cells lacking DNA-PKCs are essentially unable to repair CLM-induced DSBs (Ismail et al., 2004). We found that BMI1-KO cells have 10% more DNA damage remaining after 1 h of treatment with 30 nM CLM than BMI1 WT cells (Fig. 7 B). This compromised repair capacity of BMI1 KO cells was more pronounced (twofold) after 5 h of repair. This difference is statistically significant (P = 0.003). The compromised repair capacity of BMI1 KO cells was rescued by the expression of the full-length BMI1 construct (Fig. 7 B). These results were confirmed in similar experiments where DNA repair in IR-treated cells was monitored by a γ-H2AX foci based assay (Fig. S5 C). We conclude that cells depleted of BMI1 are unable to efficiently repair CLM-induced DSBs.
We next determined whether or not cellular depletion of BMI1 would render cells sensitive to IR. We used the colony formation assay to determine the survival of cells after IR. At 2 Gy, 83% of cells transfected with control shRNA survived. Survival of cells was reduced to 37% by BMI1 shRNA (Fig. 7 C). This reduction in survival was statistically significant (P = 0.0002). The effect of BMI1 knockdown on cellular IR sensitivity was more pronounced (fivefold) at higher doses. These data indicate that depletion of BMI1 levels in U2OS increased the sensitivity of these cells to IR.
The PRC2 complex is not required for recruitment of BMI1 to DNA damage sites
We next investigated whether or not members of the PRC2 complex are crucial for the recruitment of the BMI1–RING2 complex to DSB sites. To study this possibility, specific GFP-labeled shRNA against EZH2 or EED were used in U2OS cells. Subsequently, GFP-labeled shRNA-expressing U2OS cells were exposed to radiation (2 Gy), and the ability of endogenous BMI1 to form IRIF was examined. Immunoblotting analysis of extracts from U2OS cells transfected with specific shRNA against EZH2 or EED revealed that each shRNA efficiently knocked down the relevant protein in cells (Fig. 8, A and B). Down-regulation of endogenous EZH2 or EED proteins did not disrupt localization of BMI1 into IRIF (Fig. 8 C). Consistent with this data, we found that pretreatment of cells with the general methylation inhibitor adenosine dialdehyde (AdOx, 250 µM) also did not affect the formation of BMI1 IRIF (unpublished data). Collectively, these data suggest that the mechanism of BMI1–RING2 complex recruitment to IRIF is distinct from the mechanism used by these proteins to execute their well-known PcG protein-mediated gene silencing function.
Figure 8.

EZH2 and EED are not required for BMI1 IRIF formation. U2OS cells were transfected with control shRNA (Ctr shRNA) or GFP-tagged shRNA targeted to EZH2 (EZH2 shRNA) or EED (EED shRNA). (A and B) Nuclear extracts were prepared from these cells, and EED or EZH2 levels were assayed by Western blotting. As a loading control, membranes were probed for actin. (C) 48 h after GFP-tagged shRNA transfection, cells were exposed to IR (2 Gy, 30 min). Cells were fixed and costained with BMI1 and γ-H2AX. Representative BMI1 immunostaining images are shown in C. The GFP channel identifies cells that were transfected with shRNA and coexpress a GFP marker. Bar, 5 µm.
The FHA and BRCT domains of NBS1 promote BMI1 recruitment to the break sites
BMI1 accumulation onto IRIF was examined in several DNA repair factor–deficient cell lines to more fully define the molecular mechanism of BMI1 recruitment. BMI1 accumulation was monitored in cells lacking FANCD2, BRCA1, BRCA2, Ku80, NBS1, or MDC1. Among those tested, we found that BMI1 failed to accumulate at the damage sites only in NBS1-deficient cells (Fig. 9). Additionally, immunoprecipitation experiments demonstrated that BMI1 coimmunoprecipitates with NBS1 (unpublished data). Together, the data support the notion that NBS1 is required for targeting BMI1 to the break sites. Consistent with such a role, we found that NBS1 recruits normally to laser microirradiation sites in BMI1 KO cells (Fig. S5 B). To further address this hypothesis, a series of NBS1 deletion mutants (Fig. 9 A) were expressed in NBS1-deficient cells. Deletion of NBS1 FHA–BRCT domains abolished BMI1 recruitment to the break sites. Importantly, other NBS1 deletion mutants did not disrupt BMI1 accumulation at DSB tracks (Fig. 9 B). Collectively, these results suggest that the FHA and BRCT domains of NBS1 are required for the initial recruitment of BMI1 to sites of DNA damage.
Figure 9.

The FHA and BRCT domain of NBS1 is required for targeting BMI1 to the DNA damage sites. (A) Schematic diagram of NBS1 deletion constructs used in B. Amino acid numbers are indicated. (B) NBS1-deficient cells reconstituted with empty vector, full-length, or deletion mutants of NBS1 were microirradiated and stained with PAR and BMI1 antibodies. Bars, 5 µm.
The BMI1- and RNF-8–dependent pathways are distinct
Recently, the ubiquitin ligase RNF8 has been shown to mediate the accumulation of E3 ubiquitin ligases RNF168 and BRCA1 to the sites of DNA damage (Huen et al., 2007; Kolas et al., 2007; Mailand et al., 2007). Therefore, we examined whether the recruitment of either protein was dependent on the other. Using RNF8 WT and RNF8 KO MEFs, we found that BMI1 accumulates at break sites normally in RNF8-null cells (Fig. 10 A). Similarly, cellular depletion of BMI1 did not impair RNF8 accumulation at the sites of damage (Fig. 10 B). We conclude that BMI1 and RNF8 are not dependent on each other for recruitment to sites of DNA damage. This distinguishes BMI1 from other E3 ubiquitin ligases that have been characterized at sites of DSBs. RNF168, BRCA1, and Rad18, in contrast to BMI1, are all E3 ubiquitin ligases that require RNF8 to be recruited to sites of DSBs (Huen et al., 2007; Kolas et al., 2007; Mailand et al., 2007; Doil et al., 2009; Watanabe et al., 2009). Consistent with the notion that BMI1 and RNF8 are distinct pathways, a recent study showed that 53BP1 IRIF is partially reduced in RNF8 KO B-cells (Li et al., 2010). These data indicate that another E3 ubiquitin ligase complex exists in RNF8 KO cells that can compensate for the loss of RNF8 function. We next tested whether or not BMI1 and RNF8 were part of the same or distinct genetic pathways contributing to radiation resistance. The survival of either RNF8 KO cells transfected with one of two different BMI1 shRNA or BMI1 KO cells transfected with one of two different RNF8 shRNA were quantified. We found that BMI1 KO cells and RNF8 KO cells had similar radiation sensitivity (Fig. 10, C and D). Knockdown of RNF8 in BMI1-null cells or knockdown of BMI1 in RNF8-null cells additively increases radiation sensitivity beyond the loss of either E3 ligase alone, implying that these E3 ligases are not part of the same genetic pathway (Fig. 10, C and D).
Figure 10.

The BMI1 and RNF8 recruitment to sites of DNA damage are independent of each other. Cells were treated and stained as in Fig. 7 A. (A) RNF8 WT and RNF8 KO cells. (B) BMI1 WT and BMI1 KO. Survival curves of MEFs transfected with either control shRNA or one of two different specific shRNA. Cells were treated as in Fig. 7 C. (C) RNF8 KO cells transfected with one of two different BMI1 shRNA. (D) BMI1 KO cells transfected with one of two different RNF8 shRNA. The experiment was performed in triplicate. Error bars represent standard error from two independent experiments (n = 6). Bars, 5 µm.
Discussion
In this report, we have uncovered a novel role for BMI1 in the DDR. Our data reveal that BMI1 and RING2 are rapidly recruited to sites of DSBs and that a BMI1\RING2 complex regulates H2AX ubiquitylation at DNA lesions. The demonstration that BMI1 is necessary for DNA repair, an efficient DDR, and cell viability upon exposure to irradiation suggests that BMI1 plays an early critical role in the DDR. This role for BMI1 in the DDR suggests broader functions than its documented function as a transcriptional repressor.
Recently, cells from mice lacking BMI1 have been shown to have significant mitochondrial dysfunction accompanied by a sustained increase in reactive oxygen species (ROS) that are sufficient to engage the DDR pathway (Liu et al., 2009). In that study, treatment with an antioxidant or interruption of the DDR by Chk2 deletion substantially improved some, but not all, aspects of the severely defective BMI1 KO mouse phenotype. They observed an induction of the DDR, and this was mediated by increased oxidative stress. Our results indicate the BMI1-null and BMI1-depeleted cells also have defects in efficiently repairing DNA when exogenous DNA-damaging agents such as IR or CLM induce damage. The defect that we have observed may synergize with increased oxidative stress to lead to the accumulation of DNA damage with subsequent effects on viability.
Animals with defective repair pathways are increasingly found to display radiosensitivity and immunodeficiency. BMI1 KO mice develop some phenotypes that are associated with poor DNA repair. For example, BMI1 KO mice share overlapping features with Nijmegen breakage syndrome (NBS) and Fanconi anemia. These are autosomal recessive chromosomal instability disorders characterized by developmental defects, progressive bone marrow failure, and cancer susceptibility (Taniguchi and D’Andrea, 2006). BMI1 KO mice, born with a hypocellular bone marrow, have a normal number of myeloid cells in the peripheral blood but lower numbers of lymphocytes (van der Lugt et al., 1994). Also, BMI1-depleted mice are infertile, and develop ataxia and die within 2 mo after birth (van der Lugt et al., 1994). Consistent with a new role of BMI1 in DSB repair, we found that in the absence of DNA damage, BMI1 KO cells have higher levels of γ-H2AX foci per cell, and we observed an increase in H2AX phosphorylation upon transfecting cells with BMI1-specific shRNA (Fig. 5 and not depicted). Consequently, it may be that some of the phenotypes observed in BMI1 KO mice are related to the new function of BMI1 in DSB repair described here.
Several recent studies implicated other PcG proteins in DNA repair. Overexpression of EZH2 in breast epithelial cells reduced the expression of Rad51 paralogues, which are required for proper repair of DSBs by homologous recombination (Zeidler et al., 2005). However, the survival of BRCA1-deficient tumor cells requires EZH2 expression, and pharmacological disruption of EZH2 is synthetic lethal in combination with BRCA1 deficiency (Puppe et al., 2009). The PcG protein PHF1 was found to recruit to DNA damage sites and contribute to the DDR (Hong et al., 2008). Our data extend these results and suggest a direct role for the BMI1–RING2 E3 ubiquitin ligase complex in DSB repair.
Interplay between PcG complexes and modified histones has been proposed to mediate stable transcriptional repression (Gieni and Hendzel, 2009). In the prevailing model, PRC2 is recruited to specific genomic locations where it catalyzes H3K27me3. The modified histones, in turn, recruit PRC1, which ubiquitylates H2A histones at lysine 119, which then inhibits RNA polymerase II elongation. We found that PRC2 is not required for the recruitment of BMI1–RING2 complexes to DSB sites. This indicates that the recruitment of the PRC1 complex to the sites of DNA damage does not involve the same mechanism that is involved in gene regulation. Rather, screening several mutant cell lines revealed a requirement for NBS1. The normal recruitment of NBS1 in BMI1 WT and BMI1 KO cells is consistent with NBS1 being upstream of BMI1 in the DDR.
Chromatin regulators with well-established functions in gene regulation such as the ATP-dependent chromatin remodeling complexes INO80, SWR1, RSC, and SWI/SNF, and the histone acetyltransferase complex TRAP-TIP60 also function in DNA repair (Tsukuda et al., 2005; Murr et al., 2006). Our results demonstrate that BMI1 is an H2A/H2AX E3 ubiquitin ligase that also has functions in both transcriptional regulation and DNA repair.
It remains formally possible that BMI1 and RING2 function indirectly in the DSB repair process and alter the ubiquitylation levels of γ-H2AX by decreasing the pool of u-H2AX throughout the nucleus. In this case, when cells are treated with IR, DSBs occurring in regions of the genome that contain u-H2AX will generate uγ-H2AX as the H2AX in the vicinity of the DSB is phosphorylated. A reduction in uH2AX throughout the genome will then lead to less uγ-H2AX. This explanation is less likely than the reduced uγ-H2AX observed in BMI1-null or depleted cells to reflect the direct contribution of BMI1–RING2 to ubiquitylation after the induction of DSBs. DSBs are known to be sites of ongoing ubiquitylation of γ-H2AX and H2A. This ubiquitylation turns over rapidly. For example, treatment of cells for 60 min with a proteosome inhibitor, which results in the accumulation of ubiquitin in the cytoplasm, is sufficient to virtually eliminate nuclear ubiquitin and nuclear ubiquitylated chromatin (Dantuma et al., 2006). Similarly, ubiquitin accumulating at sites of laser microirradiation–induced DNA damage is completely depleted within 60 min of treatment with proteosome inhibitor (Mailand et al., 2007). Thus, to affect the ubiquitylation levels of γ-H2AX 60 min after IR would appear to necessitate that the E3 ubiquitin ligase activity being measured is predominantly ubiquitylation that has taken place after the induction of DNA damage.
The recruitment of BMI1 and RING2 to IRIF and laser-induced DNA damage support the conclusion that the substantial reduction of uγ-H2AX in the absence of BMI1 reflects the absence of an activity that contributes to ubiquitylation directly at the sites of DSBs. The reduced recruitment of proteins that are known to involve ubiquitin signaling at DSBs further supports a direct contribution of BMI1–RING2 to ubiquitin signaling at DSBs and in response to DNA damage. Whether the reduced rate of DSB repair and the increased sensitivity to IR are determined solely through ubiquitin-dependent mechanisms or involve additional functions of BMI1–RING2 in DSB repair remains to be determined. Regardless of the underlying mechanism, it is clear that BMI1–RING2 function is important for DNA DSB repair.
There are additional unidentified H2AX E3 ubiquitin ligases that are present in cells that are responsible for γ-H2AX ubiquitylation at sites of DNA damage. There is persistent monoubiquitylation of γ-H2AX in RNF8-deficient cells (Huen et al., 2007; Mailand et al., 2007; Shao et al., 2009). Experiments that deplete a deubiquitylase involved in the DDR, BRCC36, in RNF8-null cells enhance the accumulation of γ-H2AX ubiquitylation (Shao et al., 2009).
RNF8 is also responsible for the generation of uγ-H2AX. We found that BMI1 and RNF8 independently contribute to radiation resistance but have overlapping roles in the ubiquitylation of H2A/H2AX. The coordinated crosstalk and overlapping activities of ATM and DNA-PK serve as an outstanding precedent for two enzymes with overlapping specificity both being functionally important in the DDR. There is substrate redundancy in the kinase activities of DNA-PK and ATM, including the phosphorylations of histone H2AX, RPA, and even DNA-PKCs itself (Wang et al., 2001; Chan et al., 2002; Stiff et al., 2004; Huen et al., 2007). Our results show that cells deficient in both BMI1 and RNF8 are more sensitive to radiation than cells lacking either protein individually. This suggests that these proteins represent distinct pathways even though they may share common substrates such as γ-H2AX. The sharing of substrates (H2AX) that is observed between BMI1 and RNF8 without the capacity for functional replacement, therefore, is analogous to the roles that ATM and DNA-PK play in the DDR.
Materials and methods
Plasmids and shRNA interference
A cDNA for human BMI1 (a gift from M. Vidal, Cell and Developmental Biology, Centro de Investigaciones Biológicas, CSIC, Madrid, Spain) and RING2 was inserted by PCR into pEGFP-C1 (Takara Bio Inc.). pEGFP-RAP80 (a gift from A. Jetten, Laboratory of Respiratory Biology, Research Triangle Park, NC) has been described previously (Yan et al., 2007). Control and one of two different BMI1 shRNA plasmids were obtained from OriGene. All shRNA transfections were performed with 2 µg DNA using Effectene as a transfection kit (QIAGEN) according to the manufacturer’s instructions. All deletion mutants were generated by using the QuikChange site-directed mutagenesis kit (Agilent Technologies) and verified by sequencing. The BMI1 expression construct was a gift from P. Dimri (NorthShore University HealthSystem Research Institute, Evanston, IL).
Cell culture
U2OS cells and U2OS cells stably expressing GFP-BMI1 (a gift from M. van Lohuizen, The Netherlands Cancer Institute, Antoni van Leeuwenhoek Hospital, Amsterdam, Netherlands) were cultured in Mycos 5A medium containing 10% FCS at 37°C and 5% CO2. RNF8 WT and RNF KO (a gift from X. Yu, Division of Molecular Medicine and Genetics, University of Michigan Medical School, Ann Arbor, MI), BMI1 WT, and BMI1 KO (a gift from M. van Lohuizen) cells were grown in DME supplemented with 10% FCS. NBS1-deficient (780816; P. Concanno, Benaroya Research Institute at Virginia Mason, Seattle, WA) fibroblasts were grown in DME supplemented with 10% FCS. Mouse H2AX WT and H2AX KO embryonic fibroblasts (a gift from A. Nussenzweig, Center for Cancer Research, National Cancer Institute, Bethesda, MD) were cultured in DME supplemented with 10% FCS. Unless otherwise stated, cells were irradiated in ambient air using a model CS-600 137 CS irradiator (Picker) at a dose rate of 1 Gy/min. CLM was a gift from G. Ellestad (Wyeth-Ayers Research, Monmouth Junction, NJ). The drug was dissolved at 2 mM in DMSO and stored at −70°C.
Fluorescence microscopy
Fluorescence microscopy images using MetaMorph (Molecular Devices, Inc.) controlling an Axiovert 200 M (Carl Zeiss, Inc.) equipped and acquired with a 12-bit charge-coupled device camera (Sensicam; Cooke Corp.) or a 14-bit charge-coupled device camera (Cascade; Photometrics). In some cases, confocal sections were acquired using a laser-scanning confocal microscope (LSM510; Carl Zeiss, Inc.) and a pinhole aperture setting of 1 airy unit. For confocal microscopy, GFP was excited using a 488-nm laser line, and Cy3 was excited using a 514-nm laser line. The spatial sampling ranged from 0.07 to 0.15 µm per pixel in the xy plane and 0.2 µm in the z plane. For images acquired on fields of cells, a 1.3 NA Plan-Fluor 40× objective lens (Carl Zeiss, Inc.) was used. For higher-magnification images, a 1.4 NA Plan-Apochromat 63× or 100× objective lens (Carl Zeiss, Inc.) was used. Time-lapse experiments involving living cells were typically acquired at 37°C in standard DME with added fetal calf serum.
IF microscopy
IF staining was performed as described previously (Haince et al., 2008). Cells were fixed at the indicated time points after treatment with DNA-damaging agents (laser microirradiation or IR). Cells were fixed with 4.0% paraformaldehyde in PBS, pH 7.5, for 5 min at room temperature. Cells were permeabilized with PBS containing 0.5% Triton X-100 for 5 min. Next, cells were washed twice with PBS, inverted onto 30-µl aliquots of an appropriate primary antibody, and incubated at room temperature for 30 min. Coverslips were rinsed with PBS containing 0.1% Triton X-100 and washed twice with PBS before a 30-min incubation with an appropriate secondary antibody conjugated to a fluorophore. Cells were rinsed with PBS containing 0.1% Triton X-100 and washed twice with PBS. Coverslips were mounted onto slides containing ∼10 µl of a 90% glycerol-PBS–based medium containing 1 mg of paraphenylenediamine/ml and 0.5 µg DAPI/ml. A panel of commercially available primary antibodies, directed against various DNA damage proteins and the polycomb group proteins BMI1 (Bethyl Laboratories, Inc.), PAR (Abcam), RING2 (GenWay), γ-H2AX (Millipore), and RNF8 (Abcam) were used to detect colocalization of these proteins at the break sites. Proteins were visualized with anti–rabbit Cy3-conjugated secondary antibody (Jackson Laboratory) and an anti–mouse Alexa Fluor 488 secondary antibody (Invitrogen). Cells were observed using a microscope (Axiovert 200 M; Carl Zeiss, Inc.), and composite figures of collected images were assembled in Photoshop CS3 (Adobe). Images in Fig. 1 were deconvolved with a constrained iterative deconvolution using a theoretical point spread function with Huygens software (Scientific Volume Imaging).
Two-photon microirradiation
Cells were cultured on 35-mm culture dishes containing a coverslip mounted on the bottom of the dish (MatTek Corporation) 24 h before the experiment. Cells were treated with 1 µg/ml Hoechst 33258 for 15 min and then placed on the stage of a laser-scanning confocal microscope (LSM510; Carl Zeiss, Inc.). DSBs were generated along a 0.2–1 µm-wide region across the nucleus of a single living cell by excitation of the Hoechst 33258 dye using a near-infrared 750-nm titanium-sapphire laser line. The laser output was set to 10% (unless stated otherwise), and we used 10 iterations to generate localized DSB with a Plan-Neofluar 40×/1.3 NA oil immersion objective. GFP fluorescence imaging was recorded after excitation with a 488-nm argon laser and a 515–540-nm band-pass filter. For IF staining of microirradiated cells, cells were permitted to recover for the indicated time points in a 37°C humidified incubator containing 5% CO2 before 4% paraformaldehyde fixation and indirect IF staining as detailed previously. The mean accumulation ± standard error of fluorescently tagged proteins from at least 10 cells was plotted.
Image processing and figure construction
Where necessary, images were subjected to a 3 × 3 median filter to reduce pixel noise. LSM510 and MetaMorph images were exported as 16-bit TIFF files. These were subsequently rescaled over an 8-bit data range in Photoshop. In most cases, the background fluorescence of the medium and the base signal from the detector were subtracted to better represent the dynamic range of the data content in the image. In some instances, 3D image sets were imported into Imaris 7 (BitPlane), and 3D image sets were generated. In this instance, the image was scaled to map the data over the range of the display, and the screen capture function in Imaris 7 was used to capture the image used in the figure. Because the relative intensity of PcG bodies was significantly higher than IRIFs, gamma adjustment was sometimes used. When this was done, this is clearly indicated in the figure legend. Image sets were not intended to be compared quantitatively (e.g., γ-H2AX before and after IR treatment) unless stated otherwise. Rather, independent image sets were scaled to best represent the subcellular distribution of the fluorescent stain. Figures were prepared in Photoshop CS3 (Adobe) for Windows. In general, images were scaled to span the 8-bit data depth, reducing background in the process, and then pasted into a composite canvas that was either 8-bit grayscale or 24-bit RGB color. If necessary, images were interpolated to 300 dpi using Photoshop.
Statistical analysis
A two-tailed Student’s t test was used to calculate the statistically significance. Calculations were performed using the GraphPad online t test calculator (http://www.graphpad.com/quickcalcs/ttest1.cfm).
Nuclear extractions
U2OS cells were subcultured the previous day and used at 70–80% confluency on the day of the experiment. Cells, when necessary, were exposed to drugs or IR, then harvested using 0.53 mM EDTA in PBS and washed once with cold PBS. Nuclear extracts were prepared as per the procedure described previously (Andrin and Hendzel, 2004). All steps were performed on ice or at 4°C unless stated otherwise. Protease inhibitors and a reducing agent were added to each buffer just before use (1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 10 µg/ml aprotinin, and 10 µg/ml leupeptin). In brief, cells were incubated in 5 volumes of hypotonic buffer A (20 mM Hepes, pH 7.9, 1.5 mM MgCl2, and 10 mM KCl) on ice for 15 min. Cells were centrifuged at 450 g and resuspended at 1.5 × 106 cells/ml in buffer A + 300 mm sucrose. Cells were homogenized using a Dounce homogenizer until >90% of the cells were disrupted (tested by examining trypan blue dye uptake in an aliquot of the cells and viewed under a light microscope). Nuclei were recovered by centrifugation at 3,000 g for 15 min, and the supernatant was kept as the cytoplasmic extract. The nuclei were washed once using nuclear wash buffer (10 mM Hepes, pH 7.9, 0.2 mM MgCl2, and 10 mM KCl). Washed nuclei were extracted using buffer C (20 mM Hepes, pH 7.9, 25% glycerol, 420 mM NaCl, 0.2 mM EDTA, and 1.5 mM MgCl2) for 30 min on ice. Insoluble material was removed by centrifugation at 21,000 g for 10 min. The supernatant was used as the nuclear extract. The insoluble material was kept, resuspended directly in 3× SDS-PAGE sample loading buffer, and sonicated to facilitate resuspension.
Immunoblotting
Histones extraction was prepared using 0.4 N sulfuric acid from cells exposed to radiation and incubated for 60 min at 37°C. After washing with cold PBS, cells were scraped off the plates and H2AX phosphorylation was measured as described previously (Elmroth et al., 2003). Immunoblotting was performed as previously described (Andrin and Hendzel, 2004), with one modification. In brief, nuclear extracts were separated on 6–18% SDS-PAGE (depending on the molecular weight of the protein) and transferred to 0.2-µm nitrocellulose blotting membrane (Bio-Rad Laboratories) according to standard protocols. The distribution of various nuclear proteins was examined by immunoblotting according to standard procedures using 5% BSA in TBST (TBS with 0.05% Tween) as the blocking buffer and antibody incubation buffer. The primary antibodies used were: BMI1 (Abcam), γ-H2AX (Millipore), H2AX, RNF8 (Abcam), and H2A (Millipore). Secondary antibodies used were conjugated with infrared-specific dyes (Alexa Fluor 680, Alexa Fluor 750, or IRDye 800), and all immunoblots were imaged on the Odyssey Infrared Imaging system (LI-COR Biosciences).
CFGE
The initial DSB levels and the levels remaining after 1 h and 5 h of repair at 37°C were measured with the CFGE as described previously (Ismail et al., 2005). In brief, cells were exposed to 30 nM of CLM for 30 min on ice. The cells were centrifuged, then resuspended in PBS supplemented with 0.2 mg/ml of sheared herring sperm DNA and 56 mM β-mercaptoethanol to inactivate excess CLM. Cells were either processed immediately to measure the initial DNA damage levels or incubated at 37°C for 1 h or 5 h to allow time for repair. The cells were then washed with PBS, and 150,000 cells were mixed with melted agarose (1.25% type VII in PBS with 5 mM EDTA) and transferred to a plug mold. The cells in the plug were then lysed at 4°C for a minimum of 24 h in lysis buffer (25 mM EDTA, pH 8.5, 0.5% SDS, and 3 mg/ml proteinase K added just before lysis). The cells were then resolved using agarose gel electrophoresis (0.7%) in 1× TAE (0.04 mM Tris acetate and 1 mM EDTA, pH 8) at 4°C for 17 h at 2 V/cm. The relative amount of cellular DNA migrating into the gel was quantified using laser scanning equipment (Typhoon 9200 Variable Mode Imager; ImageQuant 5.2 software; GE Healthcare) to calculate the number of DSBs.
Chromatin fractionation
Chromatin fraction was performed as described previously (Li and Stern, 2005). In brief, cells were washed twice with PBS (Ca2+ and Mg2+-free) and resuspended in solution A (10 mM Hepes, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 mM sucrose, 10% glycerol, 1 mM dithiothreitol, 10 mM NaF, 1 mM Na2VO3, and protease inhibitors; Roche). Triton X-100 was added to a final concentration of 0.1%, and the cells were incubated for 5 min on ice. Cytosolic proteins were separated from nuclei by centrifugation (4 min at 1,300 g). Nuclei were washed once in solution A, then lysed in solution B (3 mM EDTA, 0.2 mM EGTA, 1 mM dithiothreitol, and protease inhibitors) for 30 min. Insoluble chromatin was then separated from soluble nuclear proteins by centrifugation (4 min, 1,700 × g), washed once in solution B, and collected by centrifugation (1 min at 10,000 g). The final chromatin pellet was resuspended in SDS sample buffer.
IR sensitivity
IR sensitivity was performed as described previously (Ismail et al., 2005; Xu et al., 2002). In brief, U2OS were transfected with control or one of two different BMI1 shRNA for 24 h. The next day, cells were plated at low density and incubated at 37°C for 5 h before irradiation with different doses (2, 4, or 6 Gy) of γ rays. Cells were left to grow for 10 d, fixed, and stained. Then colonies were counted to assess the colony-forming ability. Error bars are from duplicate samples.
Online supplemental material
Fig. S1 shows immunostaining of BMI1 and RING2 in the absence of DNA damage and the specificity of the BMI1 antibody used in this study. Fig. S2 shows BMI1 localization in response to H2O2 (200 µM), UV light or CLM (1 pM), BMI1 recruitment to laser-induced DNA damage, and the time course of BMI1 IRIF formation. Fig. S3 shows the siRNA-mediated knockdown efficiency of BMI1 and characterization of the stable GFP-BMI1 and GFP-RING2 cell lines. Fig. S4 shows biochemical fractionation of BMI1in response to DNA damage, FRAP analysis of GFP BMI1, and specificity of the γ-H2AX antibody used in this work. Fig. S5 shows normal recruitment of p-ATM, GFP- NBS1, and YFP-MRE11 in BMI1 KO cells and the inability of BMI1 KO cells to efficiently repair DSBs as measured by the disappearance of γ-H2AX foci. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201003034/DC1.
Acknowledgments
We thank Drs. Andre Nussenzweig, Xiaochun Yu, Maarten van Lohuizen, Anton Jetten, Goberdhan P. Dimri, and Miguel Vidal for providing valuable reagents.
This work was supported by a grant from the Canadian Institute of Health Research (CIHR; MOP15334). M.J. Hendzel is an Alberta Innovates Health Solutions (AIHS) Senior Scholar. I.H. Ismail is a recipient of AIHS and Alberta Cancer Board (ACB) postdoctoral fellowships. We thank the Cell Imaging Facility at the Cross Cancer Institute for instrument support.
Footnotes
Abbreviations used in this paper:
- ATM
- ataxia telangiectasia mutated
- ATR
- ATM and Rad3 related
- CFGE
- constant-field gel electrophoresis
- CLM
- calicheamicin γ1
- DDR
- DNA damage response
- DNA-PK
- DNA-dependent protein kinase
- DSB
- double-strand break
- FHA
- forkhead associated
- IF
- immunofluorescence
- IR
- ionizing radiation
- IRIF
- IR-induced foci
- KO
- knockout
- MEF
- mouse embryonic fibroblast
- NBS
- Nijmegen breakage syndrome
- PAR
- poly-ADP-ribosylation
- PcG
- polycomb group
- shRNA
- small hairpin RNA
- UIM
- ubiquitin-interacting motif
- WT
- wild type
References
- Andrin C., Hendzel M.J. 2004. F-actin-dependent insolubility of chromatin-modifying components. J. Biol. Chem. 279:25017–25023 10.1074/jbc.M401805200 [DOI] [PubMed] [Google Scholar]
- Bergink S., Salomons F.A., Hoogstraten D., Groothuis T.A., de Waard H., Wu J., Yuan L., Citterio E., Houtsmuller A.B., Neefjes J., et al. 2006. DNA damage triggers nucleotide excision repair-dependent monoubiquitylation of histone H2A. Genes Dev. 20:1343–1352 10.1101/gad.373706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchwald G., van der Stoop P., Weichenrieder O., Perrakis A., van Lohuizen M., Sixma T.K. 2006. Structure and E3-ligase activity of the Ring-Ring complex of polycomb proteins Bmi1 and Ring1b. EMBO J. 25:2465–2474 10.1038/sj.emboj.7601144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burma S., Chen B.P., Murphy M., Kurimasa A., Chen D.J. 2001. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 276:42462–42467 10.1074/jbc.C100466200 [DOI] [PubMed] [Google Scholar]
- Cao R., Tsukada Y., Zhang Y. 2005. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol. Cell. 20:845–854 10.1016/j.molcel.2005.12.002 [DOI] [PubMed] [Google Scholar]
- Celeste A., Fernandez-Capetillo O., Kruhlak M.J., Pilch D.R., Staudt D.W., Lee A., Bonner R.F., Bonner W.M., Nussenzweig A. 2003. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat. Cell Biol. 5:675–679 10.1038/ncb1004 [DOI] [PubMed] [Google Scholar]
- Chan D.W., Chen B.P., Prithivirajsingh S., Kurimasa A., Story M.D., Qin J., Chen D.J. 2002. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev. 16:2333–2338 10.1101/gad.1015202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantuma N.P., Groothuis T.A., Salomons F.A., Neefjes J. 2006. A dynamic ubiquitin equilibrium couples proteasomal activity to chromatin remodeling. J. Cell Biol. 173:19–26 10.1083/jcb.200510071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Napoles M., Mermoud J.E., Wakao R., Tang Y.A., Endoh M., Appanah R., Nesterova T.B., Silva J., Otte A.P., Vidal M., et al. 2004. Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev. Cell. 7:663–676 10.1016/j.devcel.2004.10.005 [DOI] [PubMed] [Google Scholar]
- Doil C., Mailand N., Bekker-Jensen S., Menard P., Larsen D.H., Pepperkok R., Ellenberg J., Panier S., Durocher D., Bartek J., et al. 2009. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 136:435–446 10.1016/j.cell.2008.12.041 [DOI] [PubMed] [Google Scholar]
- Elderkin S., Maertens G.N., Endoh M., Mallery D.L., Morrice N., Koseki H., Peters G., Brockdorff N., Hiom K. 2007. A phosphorylated form of Mel-18 targets the Ring1B histone H2A ubiquitin ligase to chromatin. Mol. Cell. 28:107–120 10.1016/j.molcel.2007.08.009 [DOI] [PubMed] [Google Scholar]
- Elmroth K., Nygren J., Mårtensson S., Ismail I.H., Hammarsten O. 2003. Cleavage of cellular DNA by calicheamicin gamma1. DNA Repair (Amst.). 2:363–374 10.1016/S1568-7864(02)00235-5 [DOI] [PubMed] [Google Scholar]
- Fernandez-Capetillo O., Celeste A., Nussenzweig A. 2003. Focusing on foci: H2AX and the recruitment of DNA-damage response factors. Cell Cycle. 2:426–427 [PubMed] [Google Scholar]
- Gieni R.S., Hendzel M.J. 2009. Polycomb group protein gene silencing, non-coding RNA, stem cells, and cancer. Biochem. Cell Biol. 87:711–746 10.1139/O09-057 [DOI] [PubMed] [Google Scholar]
- Haince J.F., McDonald D., Rodrigue A., Déry U., Masson J.Y., Hendzel M.J., Poirier G.G. 2008. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 283:1197–1208 10.1074/jbc.M706734200 [DOI] [PubMed] [Google Scholar]
- Hao L.Y., Strong M.A., Greider C.W. 2004. Phosphorylation of H2AX at short telomeres in T cells and fibroblasts. J. Biol. Chem. 279:45148–45154 10.1074/jbc.M403924200 [DOI] [PubMed] [Google Scholar]
- Hernández-Muñoz I., Taghavi P., Kuijl C., Neefjes J., van Lohuizen M. 2005. Association of BMI1 with polycomb bodies is dynamic and requires PRC2/EZH2 and the maintenance DNA methyltransferase DNMT1. Mol. Cell. Biol. 25:11047–11058 10.1128/MCB.25.24.11047-11058.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Z., Jiang J., Lan L., Nakajima S., Kanno S., Koseki H., Yasui A. 2008. A polycomb group protein, PHF1, is involved in the response to DNA double-strand breaks in human cell. Nucleic Acids Res. 36:2939–2947 10.1093/nar/gkn146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huen M.S., Grant R., Manke I., Minn K., Yu X., Yaffe M.B., Chen J. 2007. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 131:901–914 10.1016/j.cell.2007.09.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huen M.S., Huang J., Yuan J., Yamamoto M., Akira S., Ashley C., Xiao W., Chen J. 2008. Noncanonical E2 variant-independent function of UBC13 in promoting checkpoint protein assembly. Mol. Cell. Biol. 28:6104–6112 10.1128/MCB.00987-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail I.H., Hendzel M.J. 2008. The gamma-H2A.X: is it just a surrogate marker of double-strand breaks or much more? Environ. Mol. Mutagen. 49:73–82 10.1002/em.20358 [DOI] [PubMed] [Google Scholar]
- Ismail I.H., Mårtensson S., Moshinsky D., Rice A., Tang C., Howlett A., McMahon G., Hammarsten O. 2004. SU11752 inhibits the DNA-dependent protein kinase and DNA double-strand break repair resulting in ionizing radiation sensitization. Oncogene. 23:873–882 10.1038/sj.onc.1207303 [DOI] [PubMed] [Google Scholar]
- Ismail I.H., Nyström S., Nygren J., Hammarsten O. 2005. Activation of ataxia telangiectasia mutated by DNA strand break-inducing agents correlates closely with the number of DNA double strand breaks. J. Biol. Chem. 280:4649–4655 10.1074/jbc.M411588200 [DOI] [PubMed] [Google Scholar]
- Jacobs J.J., Kieboom K., Marino S., DePinho R.A., van Lohuizen M. 1999. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 397:164–168 10.1038/16476 [DOI] [PubMed] [Google Scholar]
- Kolas N.K., Chapman J.R., Nakada S., Ylanko J., Chahwan R., Sweeney F.D., Panier S., Mendez M., Wildenhain J., Thomson T.M., et al. 2007. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science. 318:1637–1640 10.1126/science.1150034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees-Miller S.P., Godbout R., Chan D.W., Weinfeld M., Day R.S., III, Barron G.M., Allalunis-Turner J. 1995. Absence of p350 subunit of DNA-activated protein kinase from a radiosensitive human cell line. Science. 267:1183–1185 10.1126/science.7855602 [DOI] [PubMed] [Google Scholar]
- Lessard J., Sauvageau G. 2003. Polycomb group genes as epigenetic regulators of normal and leukemic hemopoiesis. Exp. Hematol. 31:567–585 10.1016/S0301-472X(03)00081-X [DOI] [PubMed] [Google Scholar]
- Li J., Stern D.F. 2005. DNA damage regulates Chk2 association with chromatin. J. Biol. Chem. 280:37948–37956 10.1074/jbc.M509299200 [DOI] [PubMed] [Google Scholar]
- Li Z., Cao R., Wang M., Myers M.P., Zhang Y., Xu R.M. 2006. Structure of a Bmi-1-Ring1B polycomb group ubiquitin ligase complex. J. Biol. Chem. 281:20643–20649 10.1074/jbc.M602461200 [DOI] [PubMed] [Google Scholar]
- Li L., Halaby M.J., Hakem A., Cardoso R., El Ghamrasni S., Harding S., Chan N., Bristow R., Sanchez O., Durocher D., Hakem R. 2010. Rnf8 deficiency impairs class switch recombination, spermatogenesis, and genomic integrity and predisposes for cancer. J. Exp. Med. 207:983–997 10.1084/jem.20092437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Cao L., Chen J., Song S., Lee I.H., Quijano C., Liu H., Keyvanfar K., Chen H., Cao L.Y., et al. 2009. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature. 459:387–392 10.1038/nature08040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailand N., Bekker-Jensen S., Faustrup H., Melander F., Bartek J., Lukas C., Lukas J. 2007. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 131:887–900 10.1016/j.cell.2007.09.040 [DOI] [PubMed] [Google Scholar]
- Marteijn J.A., Bekker-Jensen S., Mailand N., Lans H., Schwertman P., Gourdin A.M., Dantuma N.P., Lukas J., Vermeulen W. 2009. Nucleotide excision repair-induced H2A ubiquitination is dependent on MDC1 and RNF8 and reveals a universal DNA damage response. J. Cell Biol. 186:835–847 10.1083/jcb.200902150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus K.J., Hendzel M.J. 2005. ATM-dependent DNA damage-independent mitotic phosphorylation of H2AX in normally growing mammalian cells. Mol. Biol. Cell. 16:5013–5025 10.1091/mbc.E05-01-0065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minter-Dykhouse K., Ward I., Huen M.S., Chen J., Lou Z. 2008. Distinct versus overlapping functions of MDC1 and 53BP1 in DNA damage response and tumorigenesis. J. Cell Biol. 181:727–735 10.1083/jcb.200801083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murr R., Loizou J.I., Yang Y.G., Cuenin C., Li H., Wang Z.Q., Herceg Z. 2006. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat. Cell Biol. 8:91–99 10.1038/ncb1343 [DOI] [PubMed] [Google Scholar]
- Petrini J.H., Stracker T.H. 2003. The cellular response to DNA double-strand breaks: defining the sensors and mediators. Trends Cell Biol. 13:458–462 10.1016/S0962-8924(03)00170-3 [DOI] [PubMed] [Google Scholar]
- Puppe J., Drost R., Liu X., Joosse S.A., Evers B., Cornelissen-Steijger P., Nederlof P., Yu Q., Jonkers J., van Lohuizen M., Pietersen A.M. 2009. BRCA1-deficient mammary tumor cells are dependent on EZH2 expression and sensitive to polycomb repressive complex 2-inhibitor 3-deazaneplanocin A. Breast Cancer Res. 11:R63 10.1186/bcr2354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou E.P., Pilch D.R., Orr A.H., Ivanova V.S., Bonner W.M. 1998. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 273:5858–5868 10.1074/jbc.273.10.5858 [DOI] [PubMed] [Google Scholar]
- Rogakou E.P., Boon C., Redon C., Bonner W.M. 1999. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 146:905–916 10.1083/jcb.146.5.905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y., Yoshikawa A., Mimura H., Yamashita M., Yamagata A., Fukai S. 2009. Structural basis for specific recognition of Lys 63-linked polyubiquitin chains by tandem UIMs of RAP80. EMBO J. 28:2461–2468 10.1038/emboj.2009.160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saurin A.J., Shiels C., Williamson J., Satijn D.P., Otte A.P., Sheer D., Freemont P.S. 1998. The human polycomb group complex associates with pericentromeric heterochromatin to form a novel nuclear domain. J. Cell Biol. 142:887–898 10.1083/jcb.142.4.887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao G., Lilli D.R., Patterson-Fortin J., Coleman K.A., Morrissey D.E., Greenberg R.A. 2009. The Rap80-BRCC36 de-ubiquitinating enzyme complex antagonizes RNF8-Ubc13-dependent ubiquitination events at DNA double strand breaks. Proc. Natl. Acad. Sci. USA. 106:3166–3171 10.1073/pnas.0807485106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparmann A., van Lohuizen M. 2006. Polycomb silencers control cell fate, development and cancer. Nat. Rev. Cancer. 6:846–856 10.1038/nrc1991 [DOI] [PubMed] [Google Scholar]
- Stewart G.S., Panier S., Townsend K., Al-Hakim A.K., Kolas N.K., Miller E.S., Nakada S., Ylanko J., Olivarius S., Mendez M., et al. 2009. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell. 136:420–434 10.1016/j.cell.2008.12.042 [DOI] [PubMed] [Google Scholar]
- Stiff T., O’Driscoll M., Rief N., Iwabuchi K., Löbrich M., Jeggo P.A. 2004. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 64:2390–2396 10.1158/0008-5472.CAN-03-3207 [DOI] [PubMed] [Google Scholar]
- Taniguchi T., D’Andrea A.D. 2006. Molecular pathogenesis of Fanconi anemia: recent progress. Blood. 107:4223–4233 10.1182/blood-2005-10-4240 [DOI] [PubMed] [Google Scholar]
- Tsukuda T., Fleming A.B., Nickoloff J.A., Osley M.A. 2005. Chromatin remodelling at a DNA double-strand break site in Saccharomyces cerevisiae. Nature. 438:379–383 10.1038/nature04148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Lugt N.M., Domen J., Linders K., van Roon M., Robanus-Maandag E., te Riele H., van der Valk M., Deschamps J., Sofroniew M., van Lohuizen M., et al. 1994. Posterior transformation, neurological abnormalities, and severe hematopoietic defects in mice with a targeted deletion of the bmi-1 proto-oncogene. Genes Dev. 8:757–769 10.1101/gad.8.7.757 [DOI] [PubMed] [Google Scholar]
- van Kemenade F.J., Raaphorst F.M., Blokzijl T., Fieret E., Hamer K.M., Satijn D.P., Otte A.P., Meijer C.J. 2001. Coexpression of BMI-1 and EZH2 polycomb-group proteins is associated with cycling cells and degree of malignancy in B-cell non-Hodgkin lymphoma. Blood. 97:3896–3901 10.1182/blood.V97.12.3896 [DOI] [PubMed] [Google Scholar]
- van Lohuizen M., Verbeek S., Scheijen B., Wientjens E., van der Gulden H., Berns A. 1991. Identification of cooperating oncogenes in E mu-myc transgenic mice by provirus tagging. Cell. 65:737–752 10.1016/0092-8674(91)90382-9 [DOI] [PubMed] [Google Scholar]
- Wang H., Guan J., Wang H., Perrault A.R., Wang Y., Iliakis G. 2001. Replication protein A2 phosphorylation after DNA damage by the coordinated action of ataxia telangiectasia-mutated and DNA-dependent protein kinase. Cancer Res. 61:8554–8563 [PubMed] [Google Scholar]
- Wang H., Wang L., Erdjument-Bromage H., Vidal M., Tempst P., Jones R.S., Zhang Y. 2004. Role of histone H2A ubiquitination in Polycomb silencing. Nature. 431:873–878 10.1038/nature02985 [DOI] [PubMed] [Google Scholar]
- Ward I.M., Chen J. 2001. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 276:47759–47762 10.1074/jbc.M009785200 [DOI] [PubMed] [Google Scholar]
- Watanabe K., Iwabuchi K., Sun J., Tsuji Y., Tani T., Tokunaga K., Date T., Hashimoto M., Yamaizumi M., Tateishi S. 2009. RAD18 promotes DNA double-strand break repair during G1 phase through chromatin retention of 53BP1. Nucleic Acids Res. 37:2176–2193 10.1093/nar/gkp082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B., Kim S.T., Lim D.S., Kastan M.B. 2002. Two molecularly distinct G(2)/M checkpoints are induced by ionizing irradiation. Mol. Cell. Biol. 22:1049–1059 10.1128/MCB.22.4.1049-1059.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J., Kim Y.S., Yang X.P., Li L.P., Liao G., Xia F., Jetten A.M. 2007. The ubiquitin-interacting motif containing protein RAP80 interacts with BRCA1 and functions in DNA damage repair response. Cancer Res. 67:6647–6656 10.1158/0008-5472.CAN-07-0924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidler M., Varambally S., Cao Q., Chinnaiyan A.M., Ferguson D.O., Merajver S.D., Kleer C.G. 2005. The Polycomb group protein EZH2 impairs DNA repair in breast epithelial cells. Neoplasia. 7:1011–1019 10.1593/neo.05472 [DOI] [PMC free article] [PubMed] [Google Scholar]