

Abstract
While a majority of patients with refractory anemia with ring sideroblasts and thrombocytosis harbor JAK2V617F and rarely MPLW515L, JAK2/MPL-negative cases constitute a diagnostic problem. 23 RARS-T cases were investigated applying immunohistochemical phospho-STAT5, sequencing and SNP-A-based karyotyping. Based on the association of TET2/ASXL1 mutations with MDS/MPN we studied molecular pattern of these genes. Two patients harbored ASXL1 and another 2 TET2 mutations. Phospho-STAT5 activation was present in one mutated TET2 and ASXL1 case. JAK2V617F/MPLW515L mutations were absent in TET2/ASXL1 mutants, indicating that similar clinical phenotype can be produced by various MPN-associated mutations and that additional unifying lesions may be present in RARS-T.
Keywords: RARS-T, TET2, ASXL1, JAK2 V617F, MPL W515L, STAT5
Introduction
Refractory anemia with ring sideroblasts and thrombocytosis (RARS-T) has been considered a provisional subtype within the diagnostic entity of myelodysplastic/myeloproliferative neoplasms (MDS/MPN). As the JAK2 V617F mutation is present in a significant proportion of RARS-T patients(1-7), many investigators consider this entity to be more closely related to MPN, as reflected in the newest version of the 2008 revised WHO classification of myeloid neoplasms [8]. Some cases of RARS-T revealed the MPL W515L mutation typical for classical MPN [9;10]. In addition, we have previously postulated that an aberrant STAT5 phosphorylation pattern may be a characteristic feature of true RARS-T, while negative cases may result from an unrelated etiology [11]. Consequently, a significant minority of patients with RARS-T that does not harbor JAK2/MPL mutations may simply represent RARS cases with other thrombocytosis-inducing conditions, or be due to unrelated molecular lesions producing a similar phenotype. Conversely, morphologically indistinguishable phenotypes may be due to different gene mutations functionally connected by convergent networks.
Using single nucleotide polymorphism (SNP) arrays we have recently observed a frequent area of somatic uniparental disomy (UPD) at 4q24, most commonly encountered in patients with chronic myelomonocytic leukemia (CMML), mixed MDS/MPN, some typical MDS, secondary acute myeloid leukemia (sAML) and one report on RARS-T [12-18]. Overlapping microdeletions on 4q24 pointed towards the TET2 gene, in which mutations were identified by us and other groups in different myeloid malignancies, most significantly MPN and MDS/MPN [15;16;19]. Recent studies have also identified mutations of ASXL1 (additional sex combs like 1) gene in MDS and MPN [20;21] that is located in the chromosomal region 20q11. ASXL1, along with TET2, is a member of WNT pathway and possibly regulates histone modifications [21]. Based on the association with MDS/MPN, and the established correlation of RARS-T with JAK2 V617F and MPL W515L mutations, we therefore sought to evaluate the mutational status of TET2 and ASXL1 in RARS-T patients.
Marerials and Methods
Patients
Bone marrow and blood samples were collected from 23 patients with RARS-T. Informed consent was obtained according to protocols approved by the IRB of the Cleveland Clinic. The RARS-T patient characteristics are presented in Table 1. Metaphase cytogenetic (MC) analysis was performed on marrow aspirates according to standard methods.
Table 1.
Clinical and pathologic features of patients with RARS-T.
| Patient No. | Age | Plt. Count, (× 10∧9/L) | Fibrosis | WBC (× 10∧9/L) | Splenomegaly | TET2 | ASXL1 | JAK2 V617F | MPL W515L | pSTAT5 | Cytogenetics | SNP-A karyotyping | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gain | Loss | UPD | ||||||||||||
| 1*†‡ | 78 | 277§ | 2+ | 19.5 | N | - | - | - | + | + | 46,XY[20] | - | - | - |
| 2*† | 73 | 664 | 2+ | 10.14 | Y | delC 1480Sfs | NA | - | - | + | 46,XY[20] | - | - | - |
| 3† | 79 | 660 | 4+ | 23.84 | Y | - | - | + | + | + | 46,XX,inv(9)(p11;q12)[20] | - | 2p16.2; 22q11.23 | 1p11.2p-ter |
| 4† | 76 | 587 | 1+ | 28.06 | Y | - | - | + | - | + | 46,XY[20] | - | - | 9p13.1-pter |
| 5† | 71 | 766 | 4+ | 26.2 | Y | - | L1395V | - | - | + | 46,XY[20] | - | - | - |
| 6† | 57 | 600 | 4+ | 37.09 | Y | - | - | + | - | + | 46,XX[20] | - # | - # | - # |
| 7† | 78 | 894 | 2+ | 3.81 | N | - | - | + | - | + | 46,XX[20] | - | - | - |
| 8† | 58 | 771 | 0 | 9.99 | N | - | - | - | - | + | 46,XX[20] | - | - | - |
| 9† | 87 | 625 | 2+ | 15.4 | Y | - | - | - | + | + | 46,XY[20] | - | - | 1p13.3-pter 2p12-pter 6p21.2-p22.1 6p16.3-q22.33 8p21.3-q24.13 10p13-p14 |
| 10† | 78 | 483 | NA | 22.48 | N | - | - | - | - | + | 46, XY,inv(3)(q21;q26.2)[20] | 11p25 | - | - |
| 11*†‡ | 61 | 329π | 2+ | 90.34 | Y | - | - | + | - | + | 46,XY[20] | - | - | - |
| 12*† | 85 | 598 | 1+/2+ | 21.8 | N | - | NA | + | - | + | 46,XX[20] | 7q21.2 | - | - |
| 13† | 71 | 496 | 0 | 4.14 | N | - | NA | + | - | NA | 5q- | 20q13.12 | - | 1p32-pter |
| 14*† | 72 | 512 | 0 | 8.1 | N | - | - | + | - | NA | 45,X,inv(10)(q21.2;q24.3)[3] 46,XX,inv(10)(q21.2;q24.3)[17] |
- | - | - |
| 15† | 66 | 773 | NA | 5.39 | N | - | - | - | - | - | 45,X,-X[4]/46,XX[16] | 2p11.23 | Xq25 | - |
| 16† | 69 | 616 | 2+ | 4.26 | Y | NA | - | - | - | - | 46,XY,del(5)(q22;q33)[20] | - | 5q21.1q31.3 | 1p35.1-pter |
| 17† | 63 | 611 | 0 | 4.99 | N | - | - | - | - | - | 46,XX[20] | 21q22.3 | - | - |
| 18†‡ | 73 | 667 | 2+ | 33.46 | N | V1718L | - | - | - | - | 47,XX,+8[20] | 8 | - | - |
| 19 | 83 | 469 | NA | 4.6 | N | - | Q1102D | - | - | NA | 46,XX[20] | NA | NA | NA |
| 20 | 63 | 614 | NA | 14.36 | Y | - | - | - | - | - | 46,XX[20] | - | - | 2p22.3-pter 3q21.3qter |
| 21 | 61 | 457 | 0 | 5.79 | N | - | - | - | - | - | 46,XX,add(15)(p11.1),add(22)(p11.2)[3]/47,idem, +19 [19] | +19 | - | - |
| 22 | 83 | 636 | 1+ | 5.30 | N | - | - | - | - | - | 46,XY[20] | - | - | - |
| 23 | 80 | 761 | 0 | 8.27 | N | - | - | - | - | NA | 46,XX[20] | - | - | 3q21.3qter |
Patients were part of a previous series where mutation analysis was confined to JAK2 V617F/MPL W515L mutations and pSTAT5 analysis [1,11,15]; NA - not available;
documented history of thrombocytosis over previous 6 months (to 1353×109/l);
documented history of thrombocytosis over previous 6 months (546–1213×109/l);
used 50K array.
DNA extraction
The Genomic DNA Purification Kit (Gentra Systems, Inc., MN) was used for DNA isolation according to manufacturer instruction.
TET2 and ASXL1 sequencing. JAK2 V617F and MPL W515L mutation detection by ARMS-PCR
The status of the JAK2 V617F and MPL W515L mutations were determined by a DNA tetra-primer ARMS assay as previously described [11]. Genomic DNA was used for TET2 sequencing as previously described [15]. ASXL1 primers will be described elsewhere. Briefly, PCR primers were designed to amplify and sequence all coding region of TET2 and ASXL1. When needed, multiple set of primers overlapping by 100bp was used to ensure complete coverage. Sequencing was performed by standard technique using ABI 3730xl DNA analyzer (Applied Biosystems, Foster City, CA).
Immunohistochemistry
Staining was performed as previously described [1] using mouse monoclonal anti-phospho-STAT5a/b (Y694/99; Advantex BioReagents LLP, Conroe, TX) at 1:500 dilution. Phospho-STAT5 staining was defined as positive if more than 10% of the megakaryocytes examined showed nuclear staining [1].
SNP-A analysis
Analysis was performed as previously described [11]. Signal intensity and SNP genotypes were determined using GTYPE software (v.4.0). Copy number and loss of heterozygosity (LOH) were investigated using Copy Number Analyzer for Affymetrix GeneChip Mapping (CNAGv.2.0). Somatic changes were identified by analyzing paired sorted CD3+ lymphocytes.
Results and Discussion
The previously reported cohort of 20 patients with RARS-T [11] has been extended to newer cases and all patients were analyzed for the presence of ASXL1 and TET2 mutations. The extended group of 23 patients with phenotypic features consistent with RARS-T (Tab. 1) was subjected to MC and SNP-A analysis. All patients showed the presence of ring sideroblasts (>15%), some degree of reticulin fibrosis and varying degrees of thrombocytosis (>450*109/l). SNP-A facilitated detection of previously cryptic lesions: 13/22 patients showed an abnormal SNP-A-based karyotype (only 4 of these defects were also detected by MC). The new lesions seen by SNP-A included various deletions of chromosome 2p and 5q, as well as areas of UPD, including 1p, 2p, 3q, 6p, 8p, and 10p (Tab. 1). The presence of UPD9p and UPD1p suggests that homozygous mutations are involved in the progression of the disease [11;22;23]; patients having the JAK2 V617F mutation in a homozygous constellation can be characterized by a more aggressive types of MPN [24], and an increased platelet level may be preferentially associated with the W515L [25]. None of the patients showed a somatic LOH at 4q24 or 20q11, suggesting that biallelic TET2 or ASXL1 mutations were not involved in the pathogenesis of RARS-T. Simultaneously, lack of UPD11q suggested that CBL mutations were absent.
Mutational analysis showed that JAK2 V617F mutations were present in 8 of 23, and MPL W515L mutations in 3 of 23 patients. One of the patients was double positive for JAK2 V617F/MPL W515L (Tab. 1, Fig. 1). The abnormal activation of STAT5 always correlated with the presence of the upstream mutations. However, 4 patients demonstrated abnormal megakaryocytic STAT5 phosphorylation, despite the absence of both JAK2 V617F and MPL W515L mutations. Within this group, a monoallelic TET2 mutation, delC 1480Sfs and monoallelic ASXL1 L1395V were identified. Additionally, we also found a group of 7 patients without JAK2 V617F or MPL W515L mutations, and also without association of aberrant phospho-STAT5 staining typical for other cases. One of these patients had a monoallelic TET2 V1718L mutation. Interestingly, another patient harbored a novel ASXL1 Q1102D mutation, but the phospho-STAT5 staining was not available. These findings indicate involvement of both TET2 and ASXL1 mutations in RARS-T pathogenesis, and also suggest that RARS-T cases with MPN-associated mutations may not show obligatory phospho-STAT5 staining. Of note is that Pt. 21 and 22 had increased staining of granulocytic and erythroid precursors.
Figure 1.
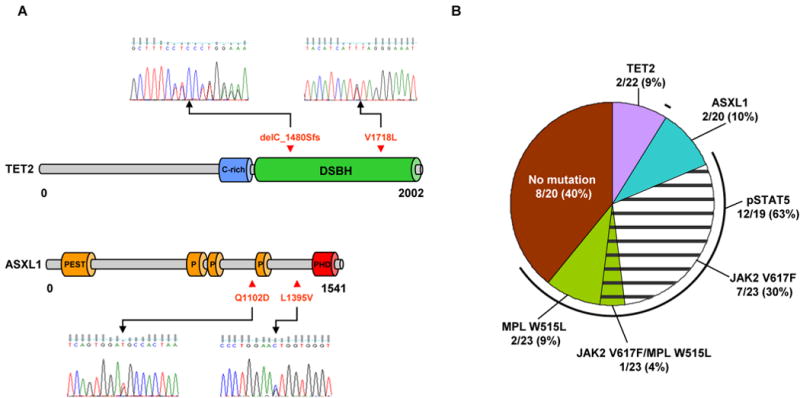
A) Schematic representation of the topographic distribution of the individual mutations in the TET2 (isoform A NM_0011270208) and ASXL1 proteins. Genomic sequencing revealed frame shift delC_1480Sfs and missense mutation V1718L in TET2 as well two missense mutations L1395V and Q1102D in ASXL1 gene. B) Frequency of TET2 and ASXL1 mutations, JAK2 V617F, MPL W515L and abnormal phospho-STAT5 activation in RARS-T patients. 2 monoallelic TET2 mutations were found in 9% of patients; 2 monoallelic ASXL1 mutations in 10%, 7 monoallelic and 1 biallelic JAK2 V617F mutations in 35%; MPL W515L mutations, 1 mono- and 2 biallelic, in 13%. Abnormal phospho-STAT5 activation was found in 63% of cases. The frequencies were calculated based on 22, 20, 23, 23, and 19 patients, respectively. Abbreviations: C-rich - cysteine-rich region; DSBH - double-stranded β helix; PEST, P - proline (P), glutamic acid (E), serine (S), and threonine (T); PHD - the plant homeodomain finger.
We also performed phospho-STAT3 staining including patients with activated STAT5 (Pt. 1, 7, 8, 10,) and those without activation (Pt. 19, 21, 22). Our results show that despite the pSTAT5 status, STAT3 pathway remains inactive in RARS-T patients. Furthermore, we performed CBL ring finger domain mutational screening in the remaining cohort of patients that showed both wild type JAK2 V617F, MPL W515L, TET2, ASXL1 and either normal or abnormal STAT5 phosphorylation. No mutation was found, and this finding was consistent with the fact that most mutated CBL cases are biallelic and associated with UPD11q; in the case of our RARS-T cohort UPD11q was not detected. The majority of patients were characterized by lack of splenomegaly, decreased white blood cell (WBC) counts, increased thrombocytosis, and a normal karyotype, although SNP-A analysis revealed additional lesions including gain of chromosome 2p, 11p and 21q, as well as deletion of chromosome X. We are not able to explain the pathogenesis of RARS-T in these patients. Theoretically, it is possible that instead of inactivating TET2/ASXL1 mutations, in some patients, expression of these genes is impaired. However, we were not able to detect any significant methylation of C residues in TET2 [15] or ASXL1 promoter (data not shown). Because of the lack of material we were unable to formally check mRNA levels of these genes; in CMML however wt TET2 mRNA levels were not decreased.
The TET2 gene comprises 11 exons [26;27] and is widely expressed in myeloid cells [15]. Along with the other members of the family (TET1 and TET3), TET2 contains two highly conserved regions. TET2 gene mutations were recently identified in different myeloid malignancies, but their impact on prognosis remains unresolved, and the mechanisms by which TET2 leads to transformation remain unclear. Based on its homology to TET1, it is possible that TET2 may play a role in epigenetic regulation. TET1 has been shown to be involved in the mixed-lineage leukemia (MLL) gene in the chromosomal translocation t(10;11)(p12;q23)(26;27), as well as in conversion of methylcytosine to hydroxymethylcytosine, thereby preventing maintenance hypermethylation, as implicated by a recent report [28]. Mutations in TET2 could, by this mechanism, lead to inactivation of a specific tumor suppressor gene and activation of pro-proliferative pathways, resulting in activation of STAT5 in some instances. Conversely, the presence of a DSBH-2OG-depenedent dioxygenase domain in TET2 may indicate other, not yet identified, function. Another member of the WNT family, ASXL1, encodes a poorly characterized protein regulating chromatin remodeling [21], contains a C-terminal PHD (plant homeodomain) finger and belong to the polycomb and mixed lineage leukemia/trithorax chromatin modifier complexes. A mutation in ASXL1 gene could possibly truncate the protein, removing its PHD domain and thus compromising the function of the associated chromatin modifiers.
TET2 mutations were identified as a pre-JAK2 mutation in 14% of JAK2 V617F positive MPN patients [14]. When hematopoietic progenitors from patients who had MPN features were analyzed, both mutations were present in clones containing lymphoid and myeloid cells, and the JAK2 V617F mutation was observed in the presence of the TET2 mutation. In addition, recent studies of ASXL1 in myeloid malignancies suggest that acquisition of ASXL1 mutations might precede the acquisition of JAK2 in some MPN patients, similar to TET2 [20;21]. However, in our cohort of patients with RARS-T, we did not find patients with concurrent mutations affecting both JAK2 and TET2 or ASXL1. Moreover, patients that carried TET2 or ASXL1 mutations were negative for MPL W515L. Based on identification of TET2 and ASXL1 at the time of diagnosis, we hypothesize that TET2 and ASXL1 mutations constitute an early marker of RARS-T evolution that precedes either JAK2 or MPL mutations typically occurring in the later stages of disease In addition, lack of a clear relationship between activated STAT5 signaling and TET2/ASXL1 mutations could suggest that TET2/ASXL1 are not the causative mutations that activate signaling, and argues more for the TET2/ASXL1 serving as a cooperating allele in a pathway parallel to JAK2/STAT5. This however needs to be evidenced by further studies.
The majority of RARS-T patients harbor JAK2 V617F. However the number of positive JAK2 V617F mutants has dropped in our cohort of RARS-T patients. This might be a consequence of the latest WHO criteria and classification of RARS-T [29] where the platelet count threshold was decreased to >450*109/l. To our knowledge, JAK2 mutation analysis has been reported in 45 patients with RARS-T [30]. Overall, among 33 RARS patients with platelet counts between 400*109/l and 600*109/l, only 3 (9%) patients were positive for JAK2 V617F vs. 27/45 (60%) of patients with thrombocytosis above 600*109/l(30). Additionally some patients may harbor MPL W515L mutations which, along with JAK2 V617F mutants, strongly activate STAT5 phosphorylation. Overall, the TET2 mutations were present in 2/22 (9%) and ASXL1 in 2/20 (10%) of RARS-T patients.
In summary, we describe herein novel TET2 and ASXL1 mutations, which might contribute to the pathogenesis of RARS-T in some patients, and eventually could serve as additional important markers in prognosis and disease progression.
Acknowledgments
This work was supported by NIH R01 HL082983 (JPM), U54 RR019391 (JPM), K24 HL077522 (JPM), Award from AA & MDS International Foundation and a charitable donation from the Robert Duggan Cancer Research Fund.
Footnotes
Authors' contributions
HS designed and performed molecular assays, wrote the manuscript; AMJ performed molecular assays and analyzed results; HM performed molecular assays, JB performed immunohistochemistry, NB classification of patients and database; EDH reviewed pathologic specimens, interpretation of results, MAS classified and identified patients, corrected manuscript; JPM conceived the idea, designed the trial.
Conflict of interest: None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Szpurka H, Tiu R, Murugesan G, Aboudola S, Hsi ED, et al. Refractory anemia with ringed sideroblasts associated with marked thrombocytosis (RARS-T), another myeloproliferative condition characterized by JAK2 V617F mutation. Blood. 2006 Oct 1;108(7):2173–81. doi: 10.1182/blood-2006-02-005751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang SA, Hasserjian RP, Loew JM, Sechman EV, Jones D, et al. Refractory anemia with ringed sideroblasts associated with marked thrombocytosis harbors JAK2 mutation and shows overlapping myeloproliferative and myelodysplastic features. Leukemia. 2006 Sep;20(9):1641–4. doi: 10.1038/sj.leu.2404316. [DOI] [PubMed] [Google Scholar]
- 3.Boissinot M, Garand R, Hamidou M, Hermouet S. The JAK2-V617F mutation and essential thrombocythemia features in a subset of patients with refractory anemia with ring sideroblasts (RARS) Blood. 2006 Sep 1;108(5):1781–2. doi: 10.1182/blood-2006-03-008227. [DOI] [PubMed] [Google Scholar]
- 4.Ceesay MM, Lea NC, Ingram W, Westwood NB, Gaken J, et al. The JAK2 V617F mutation is rare in RARS but common in RARS-T. Leukemia. 2006 Nov;20(11):2060–1. doi: 10.1038/sj.leu.2404373. [DOI] [PubMed] [Google Scholar]
- 5.Renneville A, Quesnel B, Charpentier A, Terriou L, Crinquette A, et al. High occurrence of JAK2 V617 mutation in refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Leukemia. 2006 Nov;20(11):2067–70. doi: 10.1038/sj.leu.2404405. [DOI] [PubMed] [Google Scholar]
- 6.Gattermann N, Billiet J, Kronenwett R, Zipperer E, Germing U, et al. High frequency of the JAK2 V617F mutation in patients with thrombocytosis (platelet count>600×109/L) and ringed sideroblasts more than 15% considered as MDS/MPD, unclassifiable. Blood. 2007 Feb 1;109(3):1334–5. doi: 10.1182/blood-2006-05-022491. [DOI] [PubMed] [Google Scholar]
- 7.Schmitt-Graeff AH, Teo SS, Olschewski M, Schaub F, Haxelmans S, et al. JAK2V617F mutation status identifies subtypes of refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Haematologica. 2008 Jan;93(1):34–40. doi: 10.3324/haematol.11581. [DOI] [PubMed] [Google Scholar]
- 8.Schmitt-Graeff A, Thiele J, Zuk I, Kvasnicka HM. Essential thrombocythemia with ringed sideroblasts: a heterogeneous spectrum of diseases, but not a distinct entity. Haematologica. 2002 Apr;87(4):392–9. [PubMed] [Google Scholar]
- 9.Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006 Nov 15;108(10):3472–6. doi: 10.1182/blood-2006-04-018879. [DOI] [PubMed] [Google Scholar]
- 10.Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006 Jul;3(7):e270. doi: 10.1371/journal.pmed.0030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Szpurka H, Gondek LP, Mohan SP, Hsi ED, Theil KS, et al. UPD1p indicates the presence of MPL W515L mutation in RARS-T, a mechanism analogous to UPD9p and JAK2 V617F mutation. Leukemia. 2009 Mar;23(3):610–4. doi: 10.1038/leu.2008.249. Epub 2008 Sep 25. [DOI] [PubMed] [Google Scholar]
- 12.Mohamedali AM, Smith AE, Gaken J, Lea NC, Mian SA, et al. Novel TET2 Mutations Associated With UPD4q24 in Myelodysplastic Syndrome. J Clin Oncol. 2009 Aug 20;27(24):4002–6. doi: 10.1200/JCO.2009.22.6985. Epub 2009 Jun 15. [DOI] [PubMed] [Google Scholar]
- 13.Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet. 2009 Jul;41(7):838–42. doi: 10.1038/ng.391. Epub 2009 May 31. [DOI] [PubMed] [Google Scholar]
- 14.Delhommeau F, Dupont S, Della VV, James C, Trannoy S, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009 May 28;360(22):2289–301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 15.Jankowska AM, Szpurka H, Tiu RV, Makishima H, Afable M, et al. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood. 2009 Jun 18;113(25):6403–10. doi: 10.1182/blood-2009-02-205690. Epub 2009 Apr 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tefferi A, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, et al. Detection of mutant TET2 in myeloid malignancies other than myeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML. Leukemia. 2009 Jul;23(7):1343–5. doi: 10.1038/leu.2009.59. Epub 2009 Mar 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. 2009 Jul 2;114(1):144–7. doi: 10.1182/blood-2009-03-210039. Epub 2009 May 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flach J, Dicker F, Schnittger S, Kohlmann A, Haferlach T, et al. Mutations of JAK2 and TET2, but not CBL are detectable in a high portion of patients with refractory anemia with ring sideroblasts and thrombocytosis (RARS-T) Haematologica. 2009 Nov 10; doi: 10.3324/haematol.2009.013631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tefferi A, Pardanani A, Lim KH, Abdel-Wahab O, Lasho TL, et al. TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia. 2009 May;23(5):905–11. doi: 10.1038/leu.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gelsi-Boyer V, Trouplin V, Adelaide J, Bonansea J, Cervera N, et al. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol. 2009 Jun;145(6):788–800. doi: 10.1111/j.1365-2141.2009.07697.x. [DOI] [PubMed] [Google Scholar]
- 21.Carbuccia N, Murati A, Trouplin V, Brecqueville M, Adelaide J, et al. Mutations of ASXL1 gene in myeloproliferative neoplasms. Leukemia. 2009 Nov;23(11):2183–6. doi: 10.1038/leu.2009.141. [DOI] [PubMed] [Google Scholar]
- 22.Gondek LP, Dunbar AJ, Szpurka H, McDevitt MA, Maciejewski JP. SNP Array Karyotyping Allows for the Detection of Uniparental Disomy and Cryptic Chromosomal Abnormalities in MDS/MPD-U and MPD. PLoS ONE. 2007;2(11):e1225. doi: 10.1371/journal.pone.0001225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kralovics R, Guan Y, Prchal JT. Acquired uniparental disomy of chromosome 9p is a frequent stem cell defect in polycythemia vera. Exp Hematol. 2002 Mar;30(3):229–36. doi: 10.1016/s0301-472x(01)00789-5. [DOI] [PubMed] [Google Scholar]
- 24.Vannucchi AM, Antonioli E, Guglielmelli P, Pardanani A, Tefferi A. Clinical correlates of JAK2V617F presence or allele burden in myeloproliferative neoplasms: a critical reappraisal. Leukemia. 2008 Jul;22(7):1299–307. doi: 10.1038/leu.2008.113. [DOI] [PubMed] [Google Scholar]
- 25.Vannucchi AM, Antonioli E, Guglielmelli P, Pancrazzi A, Guerini V, et al. Characteristics and clinical correlates of MPL 515W>L/K mutation in essential thrombocythemia. Blood. 2008 Aug 1;112(3):844–7. doi: 10.1182/blood-2008-01-135897. [DOI] [PubMed] [Google Scholar]
- 26.Ono R, Taki T, Taketani T, Taniwaki M, Kobayashi H, et al. LCX, leukemia-associated protein with a CXXC domain, is fused to MLL in acute myeloid leukemia with trilineage dysplasia having t(10;11)(q22;q23) Cancer Res. 2002 Jul 15;62(14):4075–80. [PubMed] [Google Scholar]
- 27.Lorsbach RB, Moore J, Mathew S, Raimondi SC, Mukatira ST, et al. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23) Leukemia. 2003 Mar;17(3):637–41. doi: 10.1038/sj.leu.2402834. [DOI] [PubMed] [Google Scholar]
- 28.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009 May 15;324(5929):930–5. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC; 2008. [Google Scholar]
- 30.Zipperer E, Wulfert M, Germing U, Haas R, Gattermann N. MPL 515 and JAK2 mutation analysis in MDS presenting with a platelet count of more than 500 x 10(9)/l. Ann Hematol. 2008 May;87(5):413–5. doi: 10.1007/s00277-007-0409-0. [DOI] [PubMed] [Google Scholar]