

Summary
Since apoptosis is impaired in malignant cells overexpressing pro-survival Bcl-2 proteins, drugs mimicking their natural antagonists, BH3-only proteins, might overcome chemoresistance. Of seven putative BH3 mimetics tested, only ABT-737 triggered Bax/Bak-mediated apoptosis. Despite its high affinity for Bcl-2, Bcl-xL and Bcl-w, many cell types proved refractory to ABT-737. We show that this resistance reflects its inability to target another pro-survival relative, Mcl-1. Down-regulation of Mcl-1 by several strategies conferred sensitivity to ABT-737. Furthermore, enforced Mcl-1 expression in a mouse lymphoma model conferred resistance. In contrast, cells overexpressing Bcl-2 remained highly sensitive to ABT-737. Hence, ABT-737 should prove efficacious in tumors with low Mcl-1 levels, or when combined with agents that inactivate Mcl-1, even to treat those tumors that overexpress Bcl-2.
Significance
Targeting the pro-survival Bcl-2-like proteins for cancer therapy is attractive because their overactivity promotes tumor formation and often limits responses to cytotoxic agents. Hence, drugs mimicking their antagonists, BH3-only proteins, offer promise as anti-cancer agents. Unlike other putative BH3 mimetics tested, ABT-737 induced apoptosis by the expected mechanism. Because it targets only certain pro-survival proteins (Bcl-2, Bcl-xL, Bcl-w), the efficacy of ABT-737 as a single agent is restricted to tumors where pro-survival Mcl-1 is low. We show that resistant cells can be sensitized to ABT-737 by approaches that down-regulate, destabilize or inactivate Mcl-1. Our studies provide a rational basis for designing clinical trials of this highly promising agent and a benchmark for systematically evaluating BH3 mimetic compounds.
Introduction
Impaired apoptosis is a central step in tumor development (Hanahan and Weinberg, 2000) and renders the tumor cell more resistant to conventional cytotoxic therapy (Johnstone et al., 2002). Consequently, an attractive novel approach for anti-cancer therapeutics is to overcome this inherent resistance to apoptosis by directly activating the normal cell death machinery (Fesik, 2005).
The key regulators of apoptosis are the interacting proteins of the Bcl-2 family (Cory et al., 2003). Its pro-survival members, Bcl-xL, Bcl-w, Mcl-1, A1 (Bfl-1) as well as Bcl-2 itself, are countered by a sub-family of distantly related death ligands, the BH3-only proteins (Huang and Strasser, 2000), which share with other family members only the short BH3 interaction domain. When BH3-only proteins such as Bim, Bad or Noxa are activated by developmental cues or intracellular damage, their amphipathic α-helical BH3 domain inserts into a hydrophobic groove on their pro-survival target (Liu et al., 2003; Petros et al., 2000; Sattler et al., 1997). This key interaction initiates apoptosis, but cell death ensues only in cells that express Bax and/or Bak (Cheng et al., 2001; Lindsten et al., 2000; Zong et al., 2001), related multi-domain pro-apoptotic Bcl-2 family members. When activated, Bax and Bak oligomerize on the mitochondrial outer membrane and permeabilize it, inducing the release of apoptogenic proteins, including cytochrome c, that promote activation of the caspases that mediate cellular demolition.
In many tumors, the capacity of the Bcl-2 family to remove damaged cells is subverted, either because a pro-survival family member is overexpressed (Cory et al., 2003), or because mutations in the p53 pathway ablate induction by p53 of the BH3-only proteins Puma and Noxa, which would otherwise trigger apoptosis (Jeffers et al., 2003; Shibue et al., 2003; Villunger et al., 2003). Nevertheless, nearly all tumors retain the core apoptotic machinery. Therefore, there is great interest in the prospect of developing anti-cancer agents that directly target Bcl-2-like pro-survival proteins by mimicking the BH3 domain (Baell and Huang, 2002; Fesik, 2005; Rutledge et al., 2002). A ‘BH3 mimetic’ should readily kill tumor cells, even those lacking p53 function.
Although targeting a protein-protein interaction for therapeutics is challenging (Cochran, 2001), several candidate BH3 mimetics, both peptidic and non-peptidic, have now been reported (Baell and Huang, 2002; Oltersdorf et al., 2005; Rutledge et al., 2002; Walensky et al., 2004). The search for non-peptidyl small molecules that might act as killer BH3 ligands has included both in silico screens (e.g. Wang et al., 2000) and “wet” screening of compound libraries (e.g. Degterev et al., 2001). Most of the putative BH3 mimetics so far described, however, have an affinity for their presumed protein targets that is far lower than that of BH3-only proteins (Chen et al., 2005; Petros et al., 2000) and the mechanism of their cytotoxic action is not well established (Baell and Huang, 2002; Rutledge et al., 2002).
To establish whether putative BH3 mimetics in fact kill via the Bcl-2-regulated pathway, we have explored whether their cytotoxic action requires the expression of Bax and Bak. Surprisingly, six of the seven putative BH3 mimetics tested killed cells lacking Bax and Bak. The exception was ABT-737, a recently described compound from Abbott Laboratories (Oltersdorf et al., 2005). ABT-737 holds great promise as it avidly binds the pro-survival proteins most similar to Bcl-2 and induces Bax/Bak-dependent killing. Nevertheless, with many cells, ABT-737 was not cytotoxic on its own. Its behavior mirrored that of the BH3-only protein Bad, which we showed recently to be a relatively weak killer because it cannot engage the more divergent Bcl-2 homolog Mcl-1 (Chen et al., 2005; Willis et al., 2005). Recent studies argue that Mcl-1 has a critical, distinctive role in the control of apoptosis (Cuconati et al., 2003; Nijhawan et al., 2003; Opferman et al., 2005). Indeed, we find that Mcl-1 greatly constrains the cytotoxic action of ABT-737. Accordingly, we show that several strategies for down-regulating Mcl-1, some clinically applicable, render diverse cells highly sensitive to ABT-737, even in the face of high Bcl-2 expression. These findings have notable implications for the ways potential drugs like ABT-737 might be used for treating patients with cancer.
Results
Most putative BH3 mimetics do not kill like BH3-only proteins
BH3-only proteins require Bax or Bak to kill mouse embryo fibroblasts (MEFs) (Cheng et al., 2001; Zong et al., 2001). As expected, infection with retroviruses encoding Bim or truncated Bid (tBid) rapidly killed wild-type MEFs, but not MEFs lacking both Bax and Bak (Fig. 1A). Furthermore, we have found that MEFs lacking both Bax and Bak exhibit clonogenic survival even when a BH3-only protein such as Bim is overexpressed (Fig. 1B).
Figure 1.

Many putative BH3 mimetics do not kill like BH3-only proteins
A: The viability of wild-type MEFs (WT) or Bax- and Bak-deficient MEFs (DKO) 24 h after infection with the indicated retroviruses. Expression of the cDNA encoding the BH3-only protein BimS or tBid was linked by an IRES to that of GFP, and the viability of GFP+ve cells determined by PI exclusion.
B: Representative wells showing colony formation by wild-type (WT) or Bax/Bak-deficient (DKO) MEFs after infection with the control parental retrovirus or one expressing BimL.
C–H: The viability (% cells excluding PI) of WT or Bax- and Bak-deficient (DKO) MEFs treated for 24 h with graded doses of the indicated putative BH3 mimetics.
I: Colonies formed by wild-type (WT) or Bax/Bak-deficient (DKO) MEFs in the presence of no treatment, HA14-1 or Antimycin A.
J: The relative affinities (IC50 in nM) of a BimBH3 peptide (as previously reported; Chen et al., 2005) and several putative BH3 mimetic compounds for Bcl-2 and/or Bcl-w. The affinities were measured in solution competition assays (Chen et al., 2005).
Data in A and C–H represent means ± SD from 3 independent experiments.
In contrast, Bax/Bak-deficient cells were as sensitive as wild-type ones to killing by several small chemical entities reported to be BH3 mimetics: HA14-1 (Wang et al., 2000), BH3I-1 (Degterev et al., 2001), Compound 6 (Enyedy et al., 2001), Antimycin A (Tzung et al., 2001), Chelerythrine (Chan et al., 2003) and Gossypol (Kitada et al., 2003), both in short-term (Figs. 1C–H) and clonogenic survival assays (Fig. 1I). Clearly, as their cytotoxic activity does not depend on Bax and/or Bak, none of these compounds functioned solely as a BH3 mimetic. This may reflect their affinities for pro-survival targets, which are much lower (μM range) than those of the BH3-only proteins (nM range) (Chen et al., 2005; Petros et al., 2000). Solution competition assays with an optical biosensor confirmed the weak affinities (Fig. 1J) of some of the compounds (HA14-1, BH3I-1, Antimycin A, Gossypol) for their putative targets, in accord with another recent study (Zhai et al., 2006).
ABT-737, a Bad-like BH3 mimetic compound
In contrast to these compounds, in solution competition assays (Chen et al., 2005) the BH3 mimetic ABT-737 (Oltersdorf et al., 2005) bound with high affinity to Bcl-2, Bcl-xL and Bcl-w (IC50 <10 nM), but not detectably to the more divergent Mcl-1 or A1 (Fig. S1A). Furthermore, direct binding studies using isothermal calorimetry confirmed tight stoichiometric (1:1) binding of ABT-737 to Bcl-xL (Fig. S1B), akin to the binding of Bim (Fig. S1C), whereas unlike Bim (Fig. S1D) the drug did not bind Mcl-1 (Fig. S1B). Thus, ABT-737 targets the same selected subset of pro-survival proteins as the BH3-only protein Bad (Chen et al., 2005).
ABT-737 kills through Bax/Bak, but efficient killing also requires Mcl-1 neutralization
Notably, Bax/Bak-deficient MEFs were completely resistant to ABT-737 (Fig. 2A). However, even wild-type (WT) MEFs were unexpectedly refractory to the drug; after 48 h of exposure to the maximal dose tested (10 μM), ~80% of them remained viable (Fig. 2A). We hypothesized that the limited cytotoxic action of ABT-737 reflects its restricted binding spectrum for the pro-survival proteins (Figs. 2B, S1A).
Figure 2.

ABT-737 cooperates with Noxa to induce Bax/Bak-dependent killing
A: The viability of wild-type MEFs (WT), Bax/Bak-deficient MEFs (DKO), and Bak- or Bax-singly deficient MEFs was determined by PI exclusion 48 h after exposure to ABT-737 (10 μM) or Etoposide (10 μM).
B: ABT-737 is a Bad BH3 mimetic. Based on the relative affinities (IC50 in nM) of ABT-737 for mammalian pro-survival proteins, determined in solution competition assays (Fig. S1A), ABT-737 and Bad bind to the same subset of Bcl-2 pro-survival proteins. According to our model for initiating the apoptotic program (Chen et al., 2005; Willis et al., 2005), Bad and Noxa are poor inducers of apoptosis individually because each binds only a subset of the pro-survival proteins, whereas Bim is a potent killer because it binds all of them. By this rationale, ABT-737 (like Bad) should also cooperate with Noxa to kill cells.
C: Noxa triggers Mcl-1 degradation. Immunoblots of lysates prepared from the MEFs after retroviral infection with wild-type Noxa or the 3E mutant (an inactive mutant that does not bind Mcl-1) probed for Mcl-1 and HSP70 (loading control).
D: Noxa sensitizes wild-type MEFs to ABT-737 killing. Wild-type MEFs expressing wild-type human Noxa or an inactive mutant (Noxa 3E) (Willis et al., 2005), were exposed to ABT-737 for 8 h and their viability determined.
E: Bax/Bak-deficient MEFs (DKO) are resistant to ABT-737 even when Mcl-1 is targeted. Long-term clonogenic survival of cells exposed to ABT-737. Equal numbers of the indicated MEFs, or their counterparts stably expressing Noxa or the inactive Noxa 3E, were plated in media containing vehicle or ABT-737 (1 μM, replenished after 3 d) and the colonies formed scored after 6 d. The number of colonies obtained with ABT-737 treatment is expressed as a proportion of colonies formed with the vehicle alone. - no colonies.
F: Either Bax or Bak can mediate killing by ABT-737 provided Mcl-1 is targeted. Viability of the indicated MEFs stably expressing Noxa was determined 8 h after exposure to ABT-737. Note that Bax/Bak-deficient MEFs (DKO) are resistant.
G: Noxa sensitizes FDC-P1 myeloid cells to ABT-737 killing. The viabilities of FDC-P1 cells, retrovirally infected to express Noxa, mutant Noxa 3E or Bad, were compared after a 24 h treatment with graded doses of ABT-737.
Data in A and D–G represent means ± SD from a representative of 3 experiments.
In this regard, we reported recently that the cytotoxic action of Bad, which ABT-737 closely resembles, can be potently augmented by co-expression of Noxa, which selectively targets Mcl-1 and A1 (Chen et al., 2005) and promotes Mcl-1 degradation (Willis et al., 2005). Hence, we tested whether enforced Noxa expression would render the WT MEFs sensitive to ABT-737. As expected (Willis et al., 2005), wild-type Noxa, but not a non-binding Noxa mutant 3E, triggered marked Mcl-1 degradation (Fig. 2C). Importantly, Noxa sensitized the WT cells to ABT-737 (Fig. 2D), but not other cell death inducers (Fig. S2 and data not shown). In striking contrast, the Bax/Bak-deficient MEFs remained entirely resistant, as assessed by either long-term clonogenicity (Fig. 2E) or short-term viability (Fig. 2F). Killing of Noxa-expressing cells required either Bax or Bak, but the killing was more efficient in the presence of both (Fig. 2F).
Sensitization to ABT-737 by Noxa is not restricted to the MEFs. The myelomonocytic cell line FDC-P1 cell line proved to be highly resistant to treatment with ABT-737 (EC50 >10 μM), but introduction of Noxa, ineffectual by itself (Chen et al., 2005; Willis et al., 2005) (data not shown), increased sensitivity over 2,000-fold (EC50 ~ 5 nM; Fig. 2G). In contrast, as anticipated from the similar binding profiles of ABT-737 and Bad (Fig. 2B), introduction of Bad did not enhance sensitivity, nor did the inert Noxa mutant 3E (Fig. 2G).
The sensitized cells died by apoptosis as the loss of plasma membrane integrity (measured by uptake of propidium iodide) required caspase activity (Fig. 3A) and cell death was associated with release of cytochrome c from mitochondria (Fig. 3B). ABT-737 also caused Bax/Bak-dependent cytochrome c release in vitro, but only when Mcl-1 had been neutralized with Noxa (Fig. 3C).
Figure 3.

ABT-737 induces cytochrome c release and caspase-dependent apoptosis when Mcl-1 is neutralized
A: Cell death triggered by ABT-737 is caspase dependent. Noxa-expressing wild-type MEFs were treated with ABT-737 (1 μM) and their viability was assessed by PI exclusion; co-incubation with the broad-spectrum caspase inhibitor zVAD. fmk (50 μM) abrogated ABT-737 killing at this time point. Data represent means ± SD from a representative of 3 experiments.
B: ABT-737 induces cytochrome c release when Mcl-1 is neutralized. Noxa-expressing wild-type (WT) or Bax/Bak-deficient MEFs (DKO) were exposed to ABT-737 (10 μM for 4 h), permeabilized with digitonin to wash out any cytochrome c released to the cytosol and then fixed. Residual mitochondrial cytochrome c was detected by immunostaining and flow cytometry (Waterhouse et al., 2004). ABT-737 triggered loss of cytochrome c from the mitochondria of WT MEFs, as indicated by the peak of weaker staining (compare filled with unfilled histogram; upper), but not from the Bax/Bak-deficient DKO MEFs (lower).
C: ABT-737 and Noxa cooperate in vitro to release cytochrome c. Lysates prepared from wild-type (left) or Bax/Bak-deficient MEFs (DKO; right) stably expressing Noxa or Bad were incubated with vehicle (−) or 5 μM ABT-737 (+), before fractionation into the pellet (P) and supernatant (S) fractions. Equivalent fractions were probed for cytochrome c, Bcl-2 (membrane fraction marker) and Apaf-1 (cytosolic marker).
We conclude that ABT-737 is a bona fide BH3 mimetic, since it induces Bax/Bak mediated cell killing, but that its selective binding profile limits its cytotoxicity in some cell types. We attribute the ability of Noxa to sensitize otherwise resistant cells to its capacity to neutralize pro-survival proteins not targeted by ABT-737. Even though Noxa targets both Mcl-1 and A1 (Chen et al., 2005), absence of the latter in many cell types (see below and Willis et al., 2005) points to Mcl-1 as an important predictor of responsiveness to ABT-737.
Mcl-1 down-regulation sensitizes human carcinoma cells to ABT-737, which initiates apoptosis by inactivating pro-survival proteins
Having implicated Mcl-1, we next tested whether refractory human carcinoma cell lines could be sensitized by down-regulating Mcl-1, either by retroviral introduction of Noxa or a specific human Mcl-1 short hairpin RNA. Immunoblots showed that Mcl-1 levels were markedly down-regulated in both HeLa cervical epithelial cells (Fig. 4A, bottom) and MCF-7 breast epithelial cells (Fig. 4C, bottom). Importantly, both ways of reducing the Mcl-1 level potently sensitized these cells to ABT-737 in colony formation assays (top panels of Figs. 4A, 4C). In striking contrast, when Mcl-1 levels were unperturbed (eg. by the inert Noxa mutant or the vector control), long-term growth was not impaired by ABT-737 (Figs. 2E, 4A, 4C). Importantly, re-introduction of mouse mcl-1, which is not targeted by the human mcl-1-specific RNAi hairpin used, restored colony formation (Figs. 4B, 4D), excluding the contribution of non-specific targets.
Figure 4.

Neutralizing Mcl-1 sensitizes different cell types to ABT-737
Colony formation after continuous exposure to ABT-737 (1 μM, replenished every 3 d) of HeLa (A, B) or MCF-7 cells (C, D) infected with empty vectors, or stably expressing Noxa, mutant Noxa 3E, RNAi targeting Mcl-1, or RNAi to an irrelevant target (control RNAi). Introduction of mouse Mcl-1, which is not targeted by the human specific Mcl-1 RNAi construct, restored the resistance in HeLa (B) or MCF-7 cells (D) to ABT-737. Clonogenic survival data (after 7 d) are representative means ± SD of 3 independent experiments. (A, B) The lower panels are immunoblots for Mcl-1 or HSP70 (loading control). (C, D) The lower panels are immunoblots for human Mcl-1 (top), mouse Mcl-1 (middle: * residual signal from human Mcl-1 probe) or HSP70 (lower panel).
E: ABT-737 triggers Bax activation when Mcl-1 is neutralized. HeLa cells expressing mutant Noxa 3E, Noxa or Mcl-1 RNAi, were treated for 4 h with ABT-737 (10 μM), and Bax activation detected by flow cytometric analysis after staining permeabilized cells with an antibody (clone 3) that specifically recognizes activated Bax (Willis et al., 2005).
ABT-737 does not activate Bax directly
We next considered whether the drug could kill by directly activating Bax/Bak, as proposed for certain BH3-only proteins (Kuwana et al., 2005; Letai et al., 2002). Direct activation appeared unlikely because most cell types contain both Bax/Bak and nevertheless tolerate high concentrations of the drug with no apparent ill effects (Oltersdorf et al., 2005) (Figs. 2. 4A–D). Furthermore, we established that ABT-737 does not bind Bax (Fig. S1E) and when used on cells, only triggered Bax to undergo the conformational alteration that marks its activation (Willis et al., 2005) if Mcl-1 had been inactivated with Noxa or by mcl-1 RNAi (Fig. 4E). We therefore conclude that ABT-737 causes Bax/Bak activation indirectly, by binding tightly and selectively to Bcl-2, Bcl-xL and Bcl-w (Figs. 2, S1).
ABT-737 effectively counters overexpression of Bcl-2
When ABT-737 is used alone, the experiments above (Figs. 2–4) identify Mcl-1 as a key factor that determines if a cell responds. A1, the other pro-survival protein that the drug fails to bind (Fig. S1A), is not expressed in most tumor cell lines, including MCF-7 and HeLa cells (Su et al., 2002), or in MEFs (Willis et al., 2005). To directly test if A1 also impairs response to ABT-737, we have exploited a variant Noxa BH3 that we have found to be highly selective for Mcl-1 over A1 and other pro-survival proteins, namely mouse Noxa BH3 B (mNoxaB), as well as a mutant of it (E74F) that binds both Mcl-1 and A1 (Fig. 5A). Each of these BH3 sequences, inserted within an inert BimS backbone, was introduced via retroviruses into MEFs engineered to overexpress A1. When treated with ABT-737, the Mcl-1-selective ligand (mNoxaB) was less effective at blocking colony growth than the E74F mutant that binds both guardians (Fig. 5B). Hence, A1 can also reduce sensitivity to ABT-737.
Figure 5.

Pro-survival proteins differ in their ability to antagonize ABT-737
A: Noxa variants that selectively neutralize Mcl-1 or both Mcl-1 and A1. Whereas the human Noxa used in Figs. 2–4 (above) binds both Mcl-1 and A1 (Chen et al., 2005) (Fig. 2), the mouse Noxa BH3 B region (mNoxaB) only binds tightly to Mcl-1 (IC50 60 nM; IC50 > 2μM for all other pro-survival proteins). The E74F mutant of mNoxaB binds tightly to both Mcl-1 and A1 (IC50Mcl-1 24 nM, IC50A1 12nM), but has weaker affinity (IC50 > 2 μM) for all other pro-survival proteins. The affinities were measured in solution competition assays (Chen et al., 2005).
B: A1 expression confers partial resistance to ABT-737. Colony formation after 6 d by parental wild-type MEFs or MEFs stably overexpressing FLAG-tagged A1 in the presence of ABT-737 (1 μM, replenished after 3 d) and the indicated BH3 domains, placed within an otherwise inert BimS backbone lacking its own BH3 (Chen et al., 2005) and expressed from retroviruses.
C, D: Killing by ABT-737 is not inhibited by Bcl-2 and only partially by Bcl-xL. Wild-type MEFs, or MEFs overexpressing FLAG-tagged Bcl-xL or Bcl-2, were tested for their sensitivity to ABT-737 (1μM) in the presence human Noxa. The Bcl-2 overexpression did not rescue any colony formation, even though it inhibited apoptosis induced by 24 h exposure to Etoposide (D). - no colonies.
Data in B–D represent means ± SD from a representative of 3 experiments.
Since tumors often overexpress Bcl-2 (Tsujimoto et al., 1985) or Bcl-xL, we also tested the impact of their overexpression. Even when Mcl-1 was inactivated (by expressing Noxa), Bcl-xL overexpression conferred limited resistance to ABT-737 (Fig. 5C), perhaps by raising the level of ABT-737 targets. Surprisingly, however, Bcl-2 overexpression did not prevent ABT-737-induced death (Fig. 5C), even though its level was sufficient to inhibit Etoposide-induced apoptosis (Fig. 5D). Thus, if Mcl-1 is inactivated, Bcl-2 overexpression does not diminish the cytotoxic activity of ABT-737 and Bcl-xL overexpression does so only moderately. This suggests that combining ABT-737 with strategies to inactivate Mcl-1 has therapeutic potential, even in the many tumors where Bcl-2 is markedly elevated.
Mcl-1 overexpression confers resistance to ABT-737 in vitro and in a mouse lymphoma model
If inactivation of Mcl-1 sensitizes cells to ABT-737 (Figs. 2–5), then overexpression of Mcl-1 might be expected to attenuate sensitivity to the drug. Unlike most other cell types that we have tested, factor-dependent myeloid (FDM) cells (Ekert et al., 2004) proved to be moderately sensitive to ABT-737. As predicted, ectopic Mcl-1 expression rendered these cells resistant to ABT-737, whereas Bcl-2 overexpression at much higher levels had no effect (Fig. S3).
To assess the impact of Mcl-1 expression on the response to ABT-737 in vivo, we engineered lymphomas that stably express Mcl-1 or Bcl-2. Lymphoma cells derived from two Eμ-myc/bcl-2 bi-transgenic mice (Strasser et al., 1990) were infected with retroviruses expressing Bcl-2 or Mcl-1, or a control virus. When the infected cells were transplanted into syngeneic mice, the recipients became moribund ~30 days later if left untreated or treated with vehicle alone (Figs. 6A, 6B and data not shown). Significantly, ABT-737 therapy prolonged the survival of recipient mice transplanted with the control or Bcl-2-transduced tumors by up to 30 days (Figs. 6B, 6C). Strikingly, however, the Mcl-1-transduced tumors proved highly refractory to ABT-737. Indeed, the mice bearing these tumors succumbed between 20 and 30 days after transplantation, like the vehicle control group (compare Figs. 6C with 6A, and 6D with 6B).
Figure 6.

Mcl-1 expression blunts the in vivo response of Eμ-myc/bcl-2 bi-transgenic lymphomas to ABT-737
Two independent progenitor B cell lymphomas (#9: A, C and #16: B, D) derived from Eμ-myc/bcl-2 bi-transgenic mice (Strasser et al., 1990) were infected with the control GFP expressing retrovirus, or ones co-expressing Bcl-2 or Mcl-1 and GFP. The mice were injected with 106 infected tumor cells before initiating therapy 4 d later with (C, D) ABT-737 (75 mg/kg given daily for two weeks by intraperitoneal injection) or the (A, B) vehicle alone. ABT-737 improved the survival of mice transplanted with both tumors even when Bcl-2 was overexpressed. However, Mcl-1 overexpressing lymphomas were highly resistant to ABT-737 and these mice died rapidly, akin to their untreated counterparts. Kaplan-Meier survival curves were derived from an experiment with 3 mice in each cohort.
Thus, our data identifies Mcl-1 as a critical barrier to responsiveness to ABT-737. Its increased expression renders sensitive cells resistant in vitro and in vivo (Figs. S3, 6), whereas its inactivation sensitizes resistant cells (Figs. 2–5).
Synergy between ABT-737 and genotoxic agents, even in the face of Bcl-2 overexpression
As most tumor cells do not die when treated with ABT-737 alone (Oltersdorf et al., 2005), we next explored potential strategies to sensitize them to it by countering Mcl-1. One therapeutic strategy would be to combine ABT-737 with genotoxic agents, as several lead to Mcl-1 down-regulation (Cuconati et al., 2003; Nijhawan et al., 2003; Willis et al., 2005), in part by p53-induced up-regulation of Noxa (Shibue et al., 2003; Villunger et al., 2003). Therefore, ABT-737 and genotoxic drugs should exhibit synergy. Indeed, in accord with results in other cell types (Oltersdorf et al., 2005), ABT-737 sensitized FDC-P1 cells, by at least 100-fold, to apoptosis induced by Cytosine Arabinoside (Ara-C), Etoposide or γ-irradiation (Fig. S4A–C).
As chemoresistance mediated by overexpression of Bcl-2 or Bcl-xL is a major clinical problem (Cory et al., 2003; Kaufmann and Vaux, 2003), we also assessed whether the synergy persisted in FDC-P1 cells engineered to overexpress these guardians. As expected (Huang et al., 1997a), these cells were now resistant to Ara-C or Etoposide (Fig. 7A). Notably, even in the face of the overexpressed Bcl-2 or Bcl-xL, ABT-737 showed striking synergy with all three genotoxic agents (Figs. 7B, 7C, S4A–C). The Bcl-2 expressing cells were sensitized ~100-fold and the Bcl-xL expressing ones at least 5-fold. As reported with other triggers of DNA damage (Cuconati et al., 2003; Nijhawan et al., 2003; Willis et al., 2005), all three genotoxic agents reduced Mcl-1 levels in the myeloid cells (Fig. 7D). Similar effects were observed in Eμ-myc B lymphoma cells engineered to overexpress Bcl-2 or Bcl-xL (data not shown). In every case, the sensitization was greater in cells overexpressing Bcl-2 than Bcl-xL, even though Bcl-2 was expressed at higher levels than Bcl-xL (Fig. S4D; see Discussion).
Figure 7.
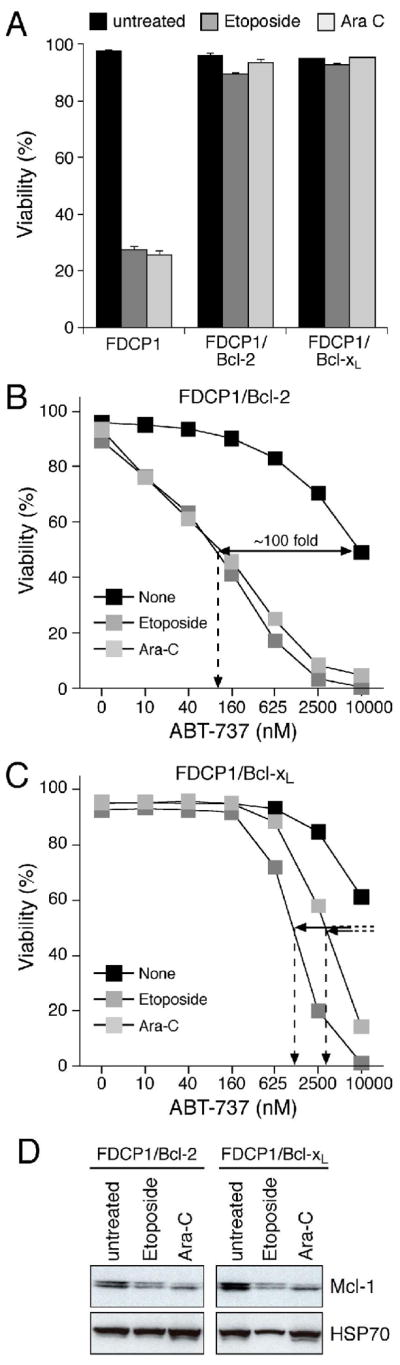
ABT-737 potently sensitizes cells overexpressing Bcl-2 to genotoxic agents
A: Bcl-2 or Bcl-xL overexpression renders FDC-P1 cells resistant to genotoxic agents. FDC-P1 cells or FDC-P1 cells overexpressing Bcl-2 or Bcl-xL were treated with Etoposide (25 μM) or Cytosine Arabinoside (25 μM) for 24 h and viability determined by PI exclusion.
B, C: FDC-P1 cells overexpressing Bcl-2 (B) or Bcl-xL (C) were treated with ABT-737 (0–10 μM), and Etoposide (25μM) or Cytosine Arabinoside (Ara-C; 25 μM) or no other drug (none) for 24 h and the viability determined by PI exclusion. Filled lines - fold increase in killing efficacy; hatched lines - EC50 values.
D: Cytotoxic agents trigger Mcl-1 degradation. Equivalent amounts of lysates prepared from cells overexpressing Bcl-2 or Bcl-xL that were left untreated or after 24 h exposure to Etoposide (25 μM) or Ara-C (25 μM) were probed for Mcl-1 or HSP70 (loading control).
Data in A–C represent means ± SD from a representative experiment.
Removing cytokine support sensitizes cells overexpressing Bcl-2 or Bcl-xL to ABT-737
Since sensitizing cells to ABT-737 with genotoxic agents (Fig. 7) may be less effective in the many tumors where p53 mutations blunt genotoxic responses, we considered alternative strategies to counter Mcl-1. As Mcl-1 expression is usually maintained by cytokines in hematopoietic cells (Kozopas et al., 1993), we reasoned that eliminating cytokine support might well sensitize such cells to ABT-737, even if Bcl-2 were overexpressed. We therefore tested FDC-P1 cells overexpressing Bcl-2 or Bcl-xL, which tolerate prolonged IL-3 deprivation (Vaux et al., 1988). Upon IL-3 withdrawal, the Mcl-1 level dropped significantly and that of the BH3-only protein Bim rose (Fig. 8A), but the overexpressed Bcl-2 or Bcl-xL prevented apoptosis. Nevertheless, the IL-3-deprived Bcl-2-overexpressing cells were now readily killed by ABT-737, their sensitivity rising by ~three orders of magnitude (Fig. 8B). The starved FDC-P1 cells overexpressing Bcl-xL were also sensitized to ABT-737, albeit to a much lesser degree (Fig. 8B).
Figure 8.

Alternative ways to target Mcl-1 and sensitize cells to ABT-737
A: IL-3 withdrawal triggers Mcl-1 degradation and Bim accumulation in FDC-P1 cells. Lysates prepared from Bcl-2-overexpressing FDC-P1 cells grown for 0–24 h in the absence of its essential growth factor IL-3 were blotted for Mcl-1, Bim or HSP70 (loading control).
B: IL-3 deprivation sensitizes FDC-P1 cells overexpressing Bcl-2 (squares) or Bcl-xL (circles) to ABT-737. Viability was determined for the cells, cultured with (filled symbols) or without (unfilled symbols) IL-3 and exposed to ABT-737 (0–10 μM) for 24 h.
C: The protein synthesis inhibitor cycloheximide (CHX) and the CDK inhibitor Seliciclib both reduce Mcl-1 expression. HeLa cells were treated with 50 μg/mL cycloheximide or 30 μM Seliciclib (R-roscovitine/CYC202) for 12 h and Mcl-1 expression measured by immunoblotting (HSP-70, loading control).
D: HeLa cells were left untreated, treated with 2.5 μM ABT-737, 50 μg/mL cycloheximide or 30 μM Seliciclib (R-roscovitine/CYC202), or combinations of ABT-737 with cycloheximide or Seliciclib, for 14 h. Statistical analyses were performed using two-tailed unpaired Student’s t-test.
Data in B and D represent means ± SD from 3 independent experiments.
These results suggest that combining ABT-737 with selected cytokine antagonists, to reduce Mcl-1 levels, might be an effective strategy to eliminate Bcl-2-overexpressing malignancies in vivo.
Inhibitors of Mcl-1 production also sensitize cells to ABT-737
Since both mcl-1 mRNA and Mcl-1 protein have very short half-lives (Craig, 2002), strategies that reduce synthesis at either level may render cells sensitive to ABT-737. Notably, the cyclin-dependent kinase inhibitor Seliciclib (R-roscovitine/CYC202), now in Phase II clinical trials, has recently been shown to act by blocking production of mcl-1 mRNA (MacCallum et al., 2005; Raje et al., 2005). Indeed, we found that both Seliciclib and the protein synthesis inhibitor cycloheximide (CHX) reduced Mcl-1 levels (Fig. 8C) and markedly boosted the action of ABT-737 in HeLa carcinoma cells (Fig. 8D) and modestly augmented it in MEFs (data not shown). Thus, strategies exploiting the lability of Mcl-1 have promise.
Discussion
A critical but challenging task with any new therapeutic agent, such as a BH3 mimetic, is determining its biological mechanism of action. We reasoned that any agents mimicking the BH3-only proteins must act through their essential downstream effectors Bax and Bak (Cheng et al., 2001; Lindsten et al., 2000; Zong et al., 2001). Hence, we compared the ability of putative BH3 mimetics to kill wild-type cells and equivalent cells deficient for Bax and Bak. Six of the seven BH3 mimetic compounds tested at doses previously reported to be efficacious, caused non-specific toxicity, as they killed cells independently of Bax/Bak (Fig. 1). Although these compounds bind Bcl-2-like proteins with low affinities, their predominant cytotoxic activity thus seems to be mediated through pathway(s) other than those regulated by Bcl-2. This non-specific activity presumably would limit their therapeutic efficacy and potentially provoke undesirable side effects. Nevertheless, some of them could well be useful leads for developing higher affinity derivatives that kill as true BH3 mimetics.
Of the compounds tested, only ABT-737, developed by structure-based design and greatly improved by medicinal chemistry (Oltersdorf et al., 2005), acted like an authentic BH3 mimetic. Its highly specific action makes it a good candidate for clinical trials, as its selectivity for its targets should limit undesirable toxicity. Consistent with the absence of non-specific effects in vitro observed here, ABT-737 appears to cause minimal adverse effects in mice (Oltersdorf et al., 2005) (AHW, KDM, AWR and DCSH, unpublished observations). As ABT-737 effectively targets Bcl-2, Bcl-xL and Bcl-w (Fig. 2; Oltersdorf et al., 2005), the compound might have been expected to induce toxic effects in vivo related to some of the developmental defects in mice lacking each of those proteins (Cory et al., 2003; Ranger et al., 2001). However, it seems likely that the transient, and probably partial, neutralization of these proteins in adult tissues, in contrast to their constitutive absence in the developing tissues of knock-out animals, limits co-lateral damage. Nevertheless, more detailed in vivo studies will be required to preclude all adverse side effects.
How might ABT-737 be used in the clinic? Our results suggest that ABT-737 is likely to be most efficacious as a single agent in those tumors where Mcl-1 is low, absent or inactivated. Overexpression of A1, which ABT-737 also fails to bind, can also limit its action but to a lesser extent (Fig. 5B). ABT-737 has shown single-agent efficacy in many cases of follicular lymphoma, chronic lymphocytic leukemia and small cell lung carcinoma (Oltersdorf et al., 2005). Significantly, the expression of mcl-1 and a1 mRNA is very low in most malignancies of those types (see Gene Expression Omnibus repository at http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=geo). On the other hand, in those tumors where Mcl-1 is the predominant survival protein, such as multiple myeloma (Zhang et al., 2002), ABT-737 is unlikely to be effective as a single agent. Thus, the expression levels of pro-survival proteins, particularly Mcl-1 and A1, in individual tumors should be valuable prognostic markers for responses to ABT-737. In small cell lung cancer cell lines, resistance to ABT-737 correlates with elevated Mcl-1 expression (C. Tse, S. K. Tahir, S. Fesik, S. Rosenberg, S. Elmore; personal communication). Our results also predict that tumors initially sensitive to ABT-737 may eventually become resistant by Mcl-1 up-regulation. Indeed, the efficacy of ABT-737 to prolong survival of mice transplanted with a lymphoma is severely compromised if Mcl-1 is overexpressed (Fig. 6).
ABT-737 is likely to be effective (Figs. 5–8, S3, S4) even in the presence of the very high levels of Bcl-2 or Bcl-xL found in many tumors (Cory et al., 2003). It has previously been shown to be highly cytotoxic to most follicular lymphoma cells (Oltersdorf et al., 2005), in which Bcl-2 is overexpressed due to translocation of the gene (Cory et al., 2003). We found that the drug could override overexpression of either Bcl-2 or Bcl-xL in various scenarios. A striking but consistent finding was that ABT-737 sensitized cells overexpressing Bcl-2 to a much greater extent than those overexpressing Bcl-xL (Figs. 5, 7, 8, S4), even though the affinity of ABT-737 for Bcl-2 and Bcl-xL is comparable (Fig. S1A and Oltersdorf et al., 2005). This may reflect as yet unexplored differences in the biological action or regulation of these two proteins.
Although with many cells ABT-737 is not a potent cytotoxic agent when used alone, we found that most cells could be readily sensitized by eliminating Mcl-1, such as by overexpressing Noxa, or by down-regulating Mcl-1 using RNA interference (Figs. 2–4). We also identified more clinically amenable ways to reduce Mcl-1 expression. Firstly, Mcl-1 degradation can be induced by DNA damage (Cuconati et al., 2003; Nijhawan et al., 2003), and we showed that genotoxic agents synergize with ABT-737, even in cells overexpressing pro-survival Bcl-2 proteins. The potent sensitization observed here (Figs. 7, S4) and by others (Oltersdorf et al., 2005), suggests that combination therapy with ABT-737 should render genotoxic agents more effective at lower doses, potentially reducing undesirable co-lateral damage or ensuring more stable remissions with conventional doses. This approach could be particularly effective in overcoming the chemoresistance imparted by overexpression of Bcl-2 or Bcl-xL (Fig. 7). Nevertheless, how well normal tissues will tolerate ABT-737 in combination with a standard cytotoxic agent needs further evaluation and may require optimization of treatment protocols.
Secondly, the observations that Mcl-1 is a labile protein (Nijhawan et al., 2003), maintained in many cell types by cytokine signaling (Kozopas et al., 1993), prompted us to test whether cytokine deprivation could sensitize cells to ABT-737. Indeed, striking synergy was obtained, even when Bcl-2 was overexpressed (Fig. 8). Hence, antagonists of certain growth factors may well sensitize tumor cells to ABT-737. For example, antagonists of IL-6 or VEGF signalling may sensitize multiple myeloma, CLL, and perhaps other tumor types (eg. Huang et al., 2000; Jourdan et al., 2003; Le Gouill et al., 2004) to ABT-737.
Thirdly, the rapid turnover of mcl-1 mRNA and protein raised the interesting prospect of targeting intracellular signalling pathways that control its transcription and translation. The well tolerated cyclin-dependent kinase inhibitor Seliciclib (R-roscovitine/CYC202), currently in Phase II clinical trials for non-small cell lung cancer and breast tumors, is now thought to function by impairing RNA synthesis by RNA polymerase II, with mcl-1 mRNA being a key target because of its rapid turnover (MacCallum et al., 2005; Raje et al., 2005). Seliciclib showed notable synergy with ABT-737 in HeLa cells (Fig. 8D). We also found that interference with protein synthesis, using cycloheximide, enhanced ABT-737 action, presumably at least in part by reducing Mcl-1 production (Fig. 8D). In accord with this notion, recent results indicate that the multi-kinase inhibitor BAY 43-9006, now under Phase II/III clinical evaluation, acts predominantly by inhibiting Mcl-1 translation (Rahmani et al., 2005; Yu et al., 2005). Although this drug and cycloheximide inhibit translation by different mechanisms (Rahmani et al., 2005), both these and other agents such as flavopirodol (Kitada et al., 2000) preferentially affect short-lived proteins like Mcl-1. Thus, the lability of Mcl-1 renders it vulnerable to inhibition in multiple ways.
Strategies like these, which combine ABT-737 with another available therapeutic modality, may well provide substantial clinical benefit. Indeed, eventually it may prove feasible to enhance Mcl-1 degradation by augmenting the activity of the ubiquitin E3 ligase Mule (also known as ARF-BP1, Lasu, HectH9), which bears a BH3 domain targeting it to Mcl-1 (Zhong et al., 2005). Furthermore, because we have identified a Noxa BH3 domain that acts selectively on Mcl-1 (Fig. 5A), it should be feasible to develop a BH3 mimetic drug that specifically neutralizes Mcl-1 (and/or A1). Thus, Mcl-1 appears to be an attractive novel target for pharmacological intervention, if concerns about the consequences of compromising its essential physiological roles can be addressed (Opferman et al., 2005; Rinkenberger et al., 2000).
Why is Mcl-1 down-regulation so important for killing by ABT-737 or Bad? Firstly, the rapid degradation of Mcl-1 following certain cytotoxic stimuli (Cuconati et al., 2003; Nijhawan et al., 2003; Willis et al., 2005) (Figs. 7, 8) may help to ensure irreversible commitment to apoptosis. Secondly, since Mcl-1 and Bcl-xL are the only pro-survival proteins that guard Bak (Willis et al., 2005), Mcl-1 is the only barrier to Bak-mediated apoptosis when ABT-737 engages Bcl-xL.
Although the activation of Bax and Bak has been proposed to require their direct binding by certain ‘activator’ BH3-only proteins, notably Bim and truncated Bid (Kuwana et al., 2005; Letai et al., 2002), we have proposed that Bak, which is anchored in the mitochondrial outer membrane, is instead activated simply by its displacement from Mcl-1 and Bcl-xL by BH3-only proteins (Willis et al., 2005). In accord with that model, ABT-737 promoted release of cytochrome c from a mitochondrial fraction if the lysate derived from cells expressing Noxa (to neutralize Mcl-1), but not cells expressing Bad (Fig. 3C). The simplest interpretation of this result is that ABT-737 neutralized the remaining protective pro-survival proteins (eg. Bcl-xL).
In conclusion, the present studies validate the feasibility of targeting Bcl-2-like proteins using BH3 mimetics such as ABT-737 to induce apoptosis (Oltersdorf et al., 2005). The mechanistic insights provided here suggest ways in which ABT-737 might be used efficaciously as a single agent and in combination therapy. They also identify Mcl-1 and A1 as likely prognostic markers for clinical responses and suggest that Mcl-1 up-regulation or stabilization may well emerge as a mechanism of resistance to the drug. The development of ABT-737 (Oltersdorf et al., 2005), together with the recent demonstration of selectivity in the action of BH3-only proteins (Chen et al., 2005) and their pro-survival targets (Willis et al., 2005), suggest that the Bcl-2-regulated gateway to apoptosis is ripe for further therapeutic manipulation.
Experimental procedures
Expression, retroviral and RNAi constructs
FLAG-tagged mammalian expression vectors (in pEF PGKpuro or pEF PGKhygro) for Bcl-2 or Bcl-xL, and HA-tagged Bax or Bak have been described (Huang et al., 1997b; O’Connor et al., 1998; Willis et al., 2005), as have retroviral expression constructs expressing BimS, BimS 4E or BimL, and HA-tagged Bad, Noxa or Noxa 3E (Chen et al., 2005). Constructs for HA-tagged tBid (amino acids 60–195 of mouse Bid), and FLAG-tagged human Bcl-2, Bcl-xL, Mcl-1 or A1 (Bfl-1) were made by sub-cloning into the same pMIG retroviral vector. The retroviral constructs that target Mcl-1 and/or A1 (Fig. 5) replaced residues 51–76 of human BimS with residues 68–93 of mouse Noxa BH3 B (Oda et al., 2000) or a mutation of it (E74F). In pMIH retroviral constructs, the GFP (green fluorescent protein) cassette of pMIG is replaced by a hygromycin B resistance gene to link expression of human Noxa or Noxa 3E, and FLAG-tagged human Bcl-2, Bcl-xL, Mcl-1 or A1, to that of the selectable marker. All cDNAs used are of human origin except for mouse Bad, Bid and Mcl-1 (in addition to the human gene).
Retroviral vectors for RNA interference were constructed by ligating annealed oligonucleotides encoding short hairpin sequences into the pRetroSuper vector (Brummelkamp et al., 2002). The human Mcl-1 short hairpin targets the sequence 5′-GCAAGAGGATTATGGCTAA. The hairpin oligonucleotides are:
Mcl-1 Sense:
5′-GATCCCCGCAAGAGGATTATGGCTAATTCAAGAGATTAGCCATAATCCTCTTGCTTTTTGGAAA
Mcl-1 anti-sense:
5′-AGCTTTTCCAAAAAGCAAGAGGATTATGGCTAATCTCTTGAATTAGCCATAATCCTCTTGCGGG
The control short hairpin targets the mouse caspase-12 sequence
5′-GGCCACATTGCCAATTCCCA.
All constructs were verified by sequencing and details of all oligonucleotides and constructs are available from the authors.
Mouse lymphoma model
Eμ-myc/bcl-2 bi-transgenic mice on a C57BL/6 genetic background develop disseminated lymphoid tumors with primitive markers at about 6 wks of age (Strasser et al., 1990). Tumors from two such mice (#9 and #16) were expanded by injecting 106 cells intravenously into syngeneic wild-type (non-transgenic) recipient males (6–8 wks old). Once these mice developed tumors, lymphomatous masses harvested from their mesenteric lymph nodes were made into a single cell suspension and infected with the indicated retroviruses by spin infection (Schmitt et al., 2000). Twenty-four h later, the infected (GFP+ve) cells were further expanded in recipient mice and their tumor mass pooled for use in the lymphoma study.
Cohorts of 6–8 wk old mice (n = 3) were inoculated (iv) with 106 lymphoma cells infected with the control virus or ones overexpressing Bcl-2 or Mcl-1. Four days later, a 14-day course of daily ip injections of ABT-737 (75 mg/kg) (Oltersdorf et al., 2005), or vehicle alone, was initiated. The mice were culled when deemed unwell (lethargy, tremor, hind leg paralysis, > 5% weight loss, palpable tumor masses) by the animal husbandry staff, who are blinded to the experiment.
All mouse experiments were performed in accordance with guidelines administered by the Melbourne Health Research Directorate Animal Ethics Committee.
Tissue culture, retroviral infections, cell death induction and apoptosis assays
Immunoblotting
Affinity measurements and solution competition assays
Flow cytometric analysis
In vitro cytochrome c release assays
Supplementary Material
Acknowledgments
We thank Abbott Laboratories (S Fesik, S Rosenberg, S Elmore, A Shoemaker and colleagues) for providing ABT-737; P Colman, J Fletcher, A Strasser, D Vaux for discussions; H Ierino, B Kolevski, L Parma, T Pham for excellent technical assistance; D Coopper, K Pioch, G Siciliano for animal care; R Anderson, J Borst, G Dewson, P Ekert, S Korsmeyer, J Silke, M Suzuki, C Thompson, K Watson, W Welch for reagents. Our work is supported by grants from the Australian NHMRC (Program Grant 257502), US NCI (CA80188, CA43540), Leukemia and Lymphoma Society (SCOR 7015-02), Australian Cancer Research Foundation, Marsden Fund (NZ); a Melbourne International Research Scholarship (MFvD); and fellowships from Leukaemia Foundation of Victoria (AHW), Leukemia and Lymphoma Society (CLS), Cancer Council of Victoria (KDM, SNW, AWR), Sylvia and Charles Viertel Charitable Foundation (DCSH) and NHMRC (CLS, JMA, AWR, DCSH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baell JB, Huang DCS. Prospects for targeting the Bcl-2 family of proteins to develop novel cytotoxic drugs. Biochem Pharmacol. 2002;64:851–863. doi: 10.1016/s0006-2952(02)01148-6. [DOI] [PubMed] [Google Scholar]
- Brummelkamp T, Bernards R, Agami R. Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell. 2002;2:243–247. doi: 10.1016/s1535-6108(02)00122-8. [DOI] [PubMed] [Google Scholar]
- Chan SL, Lee MC, Tan KO, Yang LK, Lee AS, Flotow H, Fu NY, Butler MS, Soejarto DD, Buss AD, Yu VC. Identification of chelerythrine as an inhibitor of Bcl-xL function. J Biol Chem. 2003;278:20453–20456. doi: 10.1074/jbc.C300138200. [DOI] [PubMed] [Google Scholar]
- Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DCS. Differential targeting of pro-survival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, Korsmeyer SJ. Bcl-2, Bcl-xL sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell. 2001;8:705–711. doi: 10.1016/s1097-2765(01)00320-3. [DOI] [PubMed] [Google Scholar]
- Cochran AG. Protein-protein interfaces:mimics and inhibitors. Curr Opin Chem Biol. 2001;5:654–659. doi: 10.1016/s1367-5931(01)00262-9. [DOI] [PubMed] [Google Scholar]
- Cory S, Huang DCS, Adams JM. The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene. 2003;22:8590–8607. doi: 10.1038/sj.onc.1207102. [DOI] [PubMed] [Google Scholar]
- Craig RW. Mcl-1 provides a window on the role of the Bcl-2 family in cell proliferation, differentiation and tumorigenesis. Leukemia. 2002;16:444–454. doi: 10.1038/sj.leu.2402416. [DOI] [PubMed] [Google Scholar]
- Cuconati A, Mukherjee C, Perez D, White E. DNA damage response and Mcl-1 destruction initiate apoptosis in adenovirus-infected cells. Genes Dev. 2003;17:2922–2932. doi: 10.1101/gad.1156903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A, Lugovskoy A, Cardone M, Mulley B, Wagner G, Mitchison T, Yuan J. Identification of small-molecule inhibitors of interaction between the BH3 domain and Bcl-xL. Nat Cell Biol. 2001;3:173–182. doi: 10.1038/35055085. [DOI] [PubMed] [Google Scholar]
- Ekert PG, Read SH, Silke J, Marsden VS, Kaufmann H, Hawkins CJ, Gerl R, Kumar S, Vaux DL. Apaf-1 and caspase-9 accelerate apoptosis, but do not determine whether factor-deprived or drug-treated cells die. J Cell Biol. 2004;165:835–842. doi: 10.1083/jcb.200312031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enyedy IJ, Ling Y, Nacro K, Tomita Y, Wu X, Cao Y, Guo R, Li B, Zhu X, Huang Y, et al. Discovery of small-molecule inhibitors of Bcl-2 through structure-based computer screening. J Med Chem. 2001;44:4313–4324. doi: 10.1021/jm010016f. [DOI] [PubMed] [Google Scholar]
- Fesik SW. Promoting apoptosis as a strategy for cancer drug discovery. Nat Rev Cancer. 2005;5:876–885. doi: 10.1038/nrc1736. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Huang DCS, Cory S, Strasser A. Bcl-2, Bcl-xL and adenovirus protein E1B19kD are functionally equivalent in their ability to inhibit cell death. Oncogene. 1997a;14:405–414. doi: 10.1038/sj.onc.1200848. [DOI] [PubMed] [Google Scholar]
- Huang DCS, O’Reilly LA, Strasser A, Cory S. The anti-apoptosis function of Bcl-2 can be genetically separated from its inhibitory effect on cell cycle entry. EMBO J. 1997b;16:4628–4638. doi: 10.1093/emboj/16.15.4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DCS, Strasser A. BH3-only proteins – essential initiators of apoptotic cell death. Cell. 2000;103:839–842. doi: 10.1016/s0092-8674(00)00187-2. [DOI] [PubMed] [Google Scholar]
- Huang HM, Huang CJ, Yen JJ. Mcl-1 is a common target of stem cell factor and interleukin-5 for apoptosis prevention activity via MEK/MAPK and PI-3K/Akt pathways. Blood. 2000;96:1764–1771. [PubMed] [Google Scholar]
- Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4:321–328. doi: 10.1016/s1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108:153–164. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- Jourdan M, Veyrune JL, Vos JD, Redal N, Couderc G, Klein B. A major role for Mcl-1 anti-apoptotic protein in the IL-6-induced survival of human myeloma cells. Oncogene. 2003;22:2950–2959. doi: 10.1038/sj.onc.1206423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann SH, Vaux DL. Alterations in the apoptotic machinery and their potential role in anticancer drug resistance. Oncogene. 2003;22:7414–7430. doi: 10.1038/sj.onc.1206945. [DOI] [PubMed] [Google Scholar]
- Kitada S, Leone M, Sareth S, Zhai D, Reed J, Pellechia M. Discovery, characterisation, and structure-activity relationships studies of pro-apoptotic polyphenols targeting B-cell lymphocyte/leukemia-2 proteins. J Med Chem. 2003;46:4259–4264. doi: 10.1021/jm030190z. [DOI] [PubMed] [Google Scholar]
- Kitada S, Zapata JM, Andreeff M, Reed JC. Protein kinase inhibitors flavopiridol and 7-hydroxy-staurosporine down-regulate anti-apoptosis proteins in B-cell chronic lymphocytic leukemia. Blood. 2000;96:393–397. [PubMed] [Google Scholar]
- Kozopas KM, Yang T, Buchan HL, Zhou P, Craig RW. mcl-1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to bcl-2. Proc Natl Acad Sci U S A. 1993;90:3516–3520. doi: 10.1073/pnas.90.8.3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, Newmeyer DD. BH3 Domains of BH3-Only Proteins Differentially Regulate Bax-Mediated Mitochondrial Membrane Permeabilization Both Directly and Indirectly. Mol Cell. 2005;17:525–535. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Le Gouill S, Podar K, Amiot M, Hideshima T, Chauhan D, Ishitsuka K, Kumar S, Raje N, Richardson PG, Harousseau JL, Anderson KC. VEGF induces Mcl-1 up-regulation and protects multiple myeloma cells against apoptosis. Blood. 2004;104:2886–2892. doi: 10.1182/blood-2004-05-1760. [DOI] [PubMed] [Google Scholar]
- Letai A, Bassik M, Walensky L, Sorcinelli M, Weiler S, Korsmeyer S. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–192. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- Lindsten T, Ross AJ, King A, Zong W, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, et al. The combined functions of pro-apoptotic Bcl-2 family members Bak and Bax are essential for normal development of multiple tissues. Mol Cell. 2000;6:1389–1399. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Dai S, Zhu Y, Marrack P, Kappler JW. The structure of a Bcl-xL/Bim fragment complex: Implications for Bim function. Immunity. 2003;19:341–352. doi: 10.1016/s1074-7613(03)00234-6. [DOI] [PubMed] [Google Scholar]
- MacCallum DE, Melville J, Frame S, Watt K, Anderson S, Gianella-Borradori A, Lane DP, Green SR. Seliciclib (CYC202, R-roscovitine) induces cell death in multiple myeloma cells by inhibition of RNA polymerase II-dependent transcription and down-regulation of Mcl-1. Cancer Res. 2005;65:5399–5407. doi: 10.1158/0008-5472.CAN-05-0233. [DOI] [PubMed] [Google Scholar]
- Nijhawan D, Fang M, Traer E, Zhong Q, Gao W, Du F, Wang X. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 2003;17:1475–1486. doi: 10.1101/gad.1093903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor L, Strasser A, O’Reilly LA, Hausmann G, Adams JM, Cory S, Huang DCS. Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 1998;17:384–395. doi: 10.1093/emboj/17.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N. Noxa, a BH3-only member of the bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288:1053–1058. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- Opferman J, Iwasaki H, Ong CC, Suh H, Mizuno S, Akashi K, Korsmeyer SJ. Obligate role of anti-apoptotic Mcl-1 in the survival of hematopoietic stem cells. Science. 2005;307:1101–1104. doi: 10.1126/science.1106114. [DOI] [PubMed] [Google Scholar]
- Petros AM, Nettseheim DG, Wang Y, Olejniczak ET, Meadows RP, Mack J, Swift K, Matayoshi ED, Zhang H, Thompson CB, Fesik SW. Rationale for Bcl-xL/Bad peptide complex formation from structure, mutagenesis, and biophysical studies. Protein Sci. 2000;9:2528–2534. doi: 10.1110/ps.9.12.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmani M, Davis EM, Bauer C, Dent P, Grant S. Apoptosis induced by the kinase inhibitor BAY 43-9006 in human leukemia cells involves down-regulation of Mcl-1 through inhibition of translation. J Biol Chem. 2005;280:35217–32227. doi: 10.1074/jbc.M506551200. [DOI] [PubMed] [Google Scholar]
- Raje N, Kumar S, Hideshima T, Roccaro A, Ishitsuka K, Yasui H, Shiraishi N, Chauhan D, Munshi NC, Green SR, Anderson KC. Seliciclib (CYC202 or R-roscovitine), a small-molecule cyclin-dependent kinase inhibitor, mediates activity via down-regulation of Mcl-1 in multiple myeloma. Blood. 2005;106:1042–1047. doi: 10.1182/blood-2005-01-0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranger AM, Malynn BA, Korsmeyer SJ. Mouse models of cell death. Nature Genetics. 2001;28:113–118. doi: 10.1038/88815. [DOI] [PubMed] [Google Scholar]
- Rinkenberger JL, Horning S, Klocke B, Roth K, Korsmeyer SJ. Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes Dev. 2000;14:23–27. [PMC free article] [PubMed] [Google Scholar]
- Rutledge SE, Chin JW, Schepartz A. A view to a kill: ligands for Bcl-2 family proteins. Curr Opin Chem Biol. 2002;6:479–485. doi: 10.1016/s1367-5931(02)00352-6. [DOI] [PubMed] [Google Scholar]
- Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB, Chang BS, Minn AJ, et al. Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science. 1997;275:983–986. doi: 10.1126/science.275.5302.983. [DOI] [PubMed] [Google Scholar]
- Schmitt CA, Rosenthal CT, Lowe SW. Genetic analysis of chemoresistance in primary murine lymphomas. Nature Medicine. 2000;6:1029–1035. doi: 10.1038/79542. [DOI] [PubMed] [Google Scholar]
- Shibue T, Takeda K, Oda E, Tanaka H, Murasawa H, Takaoka A, Morishita Y, Akira S, Taniguchi T, Tanaka N. Integral role of Noxa in p53-mediated apoptotic response. Genes Dev. 2003;17:2233–2238. doi: 10.1101/gad.1103603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser A, Harris AW, Bath ML, Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature. 1990;348:331–333. doi: 10.1038/348331a0. [DOI] [PubMed] [Google Scholar]
- Su AI, Cooke MP, Ching KA, Hakak Y, Walker JR, Wiltshire T, Orth AP, Vega RG, Sapinoso LM, Moqrich A, et al. Large-scale analysis of the human and mouse transcriptomes. Proc Natl Acad Sci U S A. 2002;99:4465–4470. doi: 10.1073/pnas.012025199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto Y, Cossman J, Jaffe E, Croce CM. Involvement of the bcl-2 gene in human follicular lymphoma. Science. 1985;228:1440–1443. doi: 10.1126/science.3874430. [DOI] [PubMed] [Google Scholar]
- Tzung SP, Kim KM, Basanez G, Giedt CD, Simon J, Zimmerberg J, Zhang KY, Hockenbery DM. Antimycin A mimics a cell-death-inducing Bcl-2 homology domain 3. Nat Cell Biol. 2001;3:183–191. doi: 10.1038/35055095. [DOI] [PubMed] [Google Scholar]
- Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- Villunger A, Michalak EM, Coultas L, Müllauer F, Böck G, Ausserlechner MJ, Adams JM, Strasser A. p53- and drug-induced apoptotic responses mediated by BH3-only proteins Puma and Noxa. Science. 2003;302:1036–1038. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. Activation of Apoptosis in vivo by a Hydrocarbon-Stapled BH3 Helix. Science. 2004;305:1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JL, Liu D, Zhang ZJ, Shan S, Han X, Srinivasula SM, Croce CM, Alnemri ES, Huang Z. Structure-based discovery of an organic compound that binds Bcl-2 protein and induces apoptosis of tumor cells. Proc Natl Acad Sci U S A. 2000;97:7124–7129. doi: 10.1073/pnas.97.13.7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse NJ, Steel R, Kluck R, Trapani JA. Assaying cytochrome c translocation during apoptosis. Methods Mol Biol. 2004;284:307–313. doi: 10.1385/1-59259-816-1:307. [DOI] [PubMed] [Google Scholar]
- Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, Adams JM, Huang DC. Pro-apoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19:1294–1305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C, Bruzek LM, Meng XW, Gores GJ, Carter CA, Kaufmann SH, Adjei AA. The role of Mcl-1 down-regulation in the pro-apoptotic activity of the multikinase inhibitor BAY 43-9006. Oncogene. 2005;24:6861–6869. doi: 10.1038/sj.onc.1208841. [DOI] [PubMed] [Google Scholar]
- Zhai D, Jin C, Satterthwait AC, Reed JC. Comparison of chemical inhibitors of anti-apoptotic Bcl-2-family proteins. Cell Death Differ. 2006;13:1419–21. doi: 10.1038/sj.cdd.4401937. [DOI] [PubMed] [Google Scholar]
- Zhang B, Gojo I, Fenton RG. Myeloid cell factor-1 is a critical survival factor for multiple myeloma. Blood. 2002;99:1885–1893. doi: 10.1182/blood.v99.6.1885. [DOI] [PubMed] [Google Scholar]
- Zhong Q, Gao W, Du F, Wang X. Mule/ARF-BP1, a BH3-Only E3 Ubiquitin Ligase, Catalyzes the Polyubiquitination of Mcl-1 and Regulates Apoptosis. Cell. 2005;121:1085–1095. doi: 10.1016/j.cell.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev. 2001;15:1481–1486. doi: 10.1101/gad.897601. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.