

Photoactivatable fluorescent proteins have become an important addition to the set of molecular probes used to understand cellular function. Known as molecular highlighters, their fluorescence is switched on by irradiation, thereby enabling non-invasive tracking of protein trafficking and dynamics.[1] They are also the basis for the imaging technique PhotoActivated Light Microscopy (PALM)[2] in which multiple emitted photons are observed from individual active fluorophores which are sequentially activated from a large pool of inactive proteins and then photobleached. By locating the center of the point spread function for these emitted photons, it is possible to determine the location of the active fluorophore at better resolution than the theoretical diffraction limit. Previous GFP mutants exhibiting photomodulatory behaviour have been reported including Kaede[3] and KikGR[4] whose emission wavelength change irreversibly, Dronpa[5] whose fluorescence can be reversibly activated with light, and photoactivatable GFP (paGFP)[1], whose excitation wavelength can be irreversibly changed with light. For Kaede, KikGR, and Dronpa, the precise three dimensional structure of the fluorophore must be preserved to maintain photoswitching behavior[4, 6]. In paGFP, the shift in excitation wavelength is mediated by a light dependent decarboxylation of Glu222.[1, 7] The loss of this carboxyl group is believed to cause reorientation of an internal hydrogen bond network, which changes the protonation state of the fluorophore and leads to an irreversible shift in the excitation maximum from 397 nm to 475 nm. This mechanism of GFP photoactivation requires the preservation of active site residues His203, His148, Ser205 and Glu222. Unfortunately the sequence restrictions necessary to maintain photomodulatory behavior are not always compatible with the mutations necessary to produce other fluorescent protein variants including YFP[8, 9] and RFP[10]. In addition, Kaede, KikGR and paGFP exist in two forms with differing excitation and emission wavelengths, precluding the simultaneous use of these wavelengths for other purposes. To address these problems, we have developed a general strategy for photoactivating GFP based upon unnatural amino acid mutagenesis[11] with the photocaged tyrosine analog o-nitrobenzyl-O-tyrosine (ONBY).[12] Replacing the fluorophore tyrosine 66 with ONBY yields a GFP molecule that is nonfluorescent as observed for other o-nitrobenzyl appended fluorophores including fluoresceine,[13] texas red,[14] and quantum dots.[15] An earlier unnatural variant of GFP was produced using a similar strategy, however, the high preirradiation background fluorescence and low protein yield made this unsuitable for use as a molecular marker.[16] Fast, time resolved UV-Vis spectroscopy measurements indicate that the fluorescence quenching likely occurs through Photo-induced Electron Transfer (PET). Irradiation at 365 nm is sufficient to remove the o-nitrobenzyl group and restore fluorescence to GFP Tyr66→ONBY (GFP66ONBY).
To produce photoactivatable o-nitrobenzyl photocaged GFP (cGFP), we started with the stabilized GFP variant ‘10+1’[17], which should be capable of folding in spite of the added bulk from introducing the o-nitrobenzyl group into the fluorophore pocket. GFP ‘10+1’ (referred to hereafter as WT) was placed into a pET vector, and a C-terminal hexahistidine tag was added. Finally, fluorophore tyrosine 66, encoding the chromophore tyrosine residue was mutated to ONBY. This was accomplished by converting the codon for Tyr66 to the amber stop codon, TAG, through site directed mutagenesis to afford pET GFP66TAG. This plasmid was cotransformed into E. coli BL21(DE3) cells with plasmid pSUPAR-ONBY-A10,[18] containing the aminoacyl tRNA synthetase (aaRS) evolved to aminoacylate ONBY and an amber suppressor tRNA, both derived from the Methanococcus jannaschi tyrosyl tRNA/aaRS pair. Cells were grown to OD600= 0.7 in Terrific Broth and ONBY was added to a final concentration of 0.2 mM, and protein production was induced by the addition of 1 mM IPTG. After 20 hours, the cells were harvested by centrifugation at 5000 × G, lysed and GFP was purified using Ni-NTA affinity chromatography. For WT GFP, protein yields were 62 mg/L; GFP66ONBY was produced at 38 mg/L. To verify the identity of each protein, both were subjected to liquid chromatography coupled electrospray ionization mass spectrometry. For WT GFP, the expected mass was 26675 and ob served mass 26675, while cGFP had a calculated mass of 26809, with observed mass 26810 (figure S1). In figure 1a and c, it can be seen that fully caged GFP66ONBY protein is practically non-fluorescent with excitation at either 387 or 486 nm (fluorescence quantum yield 0.006; for details see Supporting Information). However, irradiation with 365 nm light is capable of restoring fluorescence. In figure 1b, the absorption spectrum of photolyzed GFP66ONBY exhibits a typical for GFP. The ε486 for cGFP increases at least 100 times upon photolysis. Thus the relative brightness at 486 nm, which is defined as ε486Χϕf486, increases at least 4 orders of magnitude during photoactivation. The actual change in relative brightness is probably greater than this because the measured OD486 of cGFP is greater than the real absorbance due to trace amounts of activated cGFP produced during purification, and experimental manipulation. In addition there is an isosbestic point at 417 nm, which demonstrates that activated GFP is stable to the 365 nm radiation necessary for photoactivation. The uncaged cGFP shows nearly identical photophysical properties compared to WT GFP. Both have the same fluorescence spectra and the fluorescence lifetimes are similar (2.4 ns and 2.7 ns, respectively).
Figure 1.
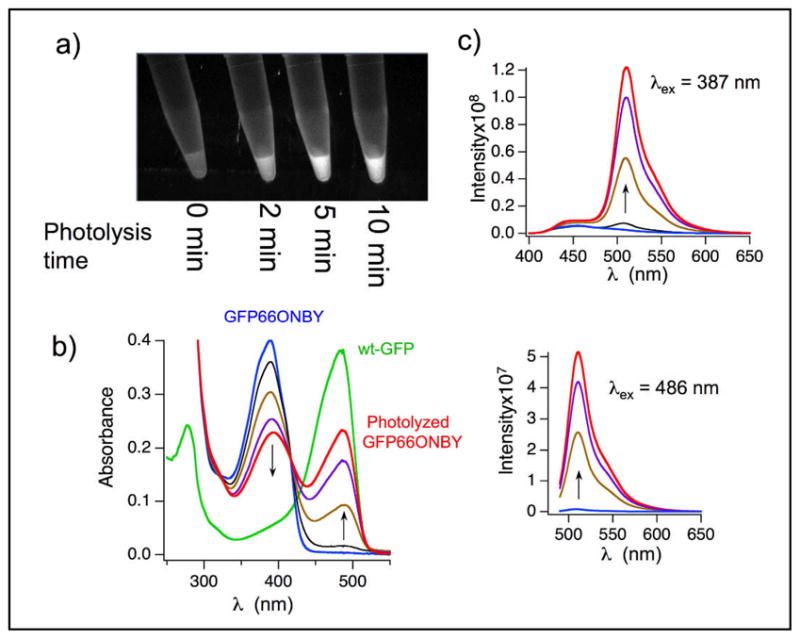
GFP66ONBY is non-fluorescent until irradiation with 365 nm light. a) GFP66ONBY purified using Ni-NTA was irradiated using a Spectra Physics 500 watt lamp with a 365 nm filter for the indicated lengths of time and then visualized using a UV transilluminator. b) UVVisible absorbance spectra of GFP66ONBY upon photolysis. c) Emission spectra of photolyzed GFP66ONBY excited at 387 nm (top) and 486 nm (bottom). For b) and c), samples were photolyzed at 365 nm for 3 (black), 23 (brown), 43 (purple), and 60 min (red) using a 4 watt UVP lamp.
The mechanism of uncaging involves photoexcitation of the o-nitrobenzyl group with UV-light (365 nm) and cleavage of the o-nitrobenzyl group from the protein (Scheme 1).[19] Fluorescence activation occurs on a timescale faster than 1 second, comparable to rates of other photoactivatable GFP variants.[1] The photoactivation quantum yield, ϕPA is around 0.0003. This quantum yield, which is lower than for other photoactivatable GFPs, is probably due to other competing chromophores and because the photochemistry of this reaction requires excitation of the o-nitrobenzyl group rather than excitation of the benzylidene imidazolinone, typical for other photoreactive fluorescent proteins. To compensate for the lower quantum yield, more intense 365 light or longer irradiation times can be used.
Scheme 1.

Uncaging of the non-fluorescent GFP66ONBY by UV photolysis to generate fluorescent protein
Unlike most earlier GFP mutants with unnatural fluorophores, this protein exhibited practically no fluorescence (fluorescence quantum yield 0.006). The most likely causes for this are either a lack of fluorophore maturation in the photocaged protein, which is fully matured only after photolysis, or quenching of the singlet state due by the o-nitrobenzyl group. To determine which of these mechanisms is correct, we determined whether GFP66ONBY was capable of forming a mature fluorophore by x-ray crystallography. To produce protein sufficiently pure for crystal growth, Ni-NTA purified protein was further purified using a Pharmacia 10/10 mono column in 50 mM Tris buffer, pH 8.0, and then eluted in a gradient from 0 to 1 M NaCl over 5 column volumes. Fractions judged to be completely pure by SDS-PAGE analysis were combined and concentrated to 10 mg/ml. Crystallization of cGFP was accomplished by the hanging-drop vapor diffusion method. Hanging drops containing 2 μl of protein solution at 10 mg/ml and 2 μl of buffer (0.1 M HEPES, pH 7.0, 20 % PEG 6k) were equilibrated at 20 °C against 1 ml of the same buffer in the reservoir. Rodlike crystals formed after 2 weeks which diffracted to 2.1 Å. Data collection and Refinement statistics can be see in table S1.
From the crystal structure (Figure 2a), it is apparent that both the o-nitrobenzyl group and the benzylidene imidazolinone are both present. Thus, GFP66ONBY is still capable of the backbone cyclization necessary for maturation of all fluorescent proteins. Alignment of the cGFP structure with the most closely related GFP shows near perfect overlap except for β-strand 7. A four amino acid stretch from Asn145 to Asn 148 from this strand does not have well resolved electron density because the tight packing of this β-stand against the fluorophore in WT GFP is disrupted by the added bulk of the o-nitrobenzyl group in cGFP. Although the actual trace of residue 145–148 is not clear, it seems that a rearrangement of residues in this region is necessary to accommodate the o-nitrobenzyl group.
Figure 2.
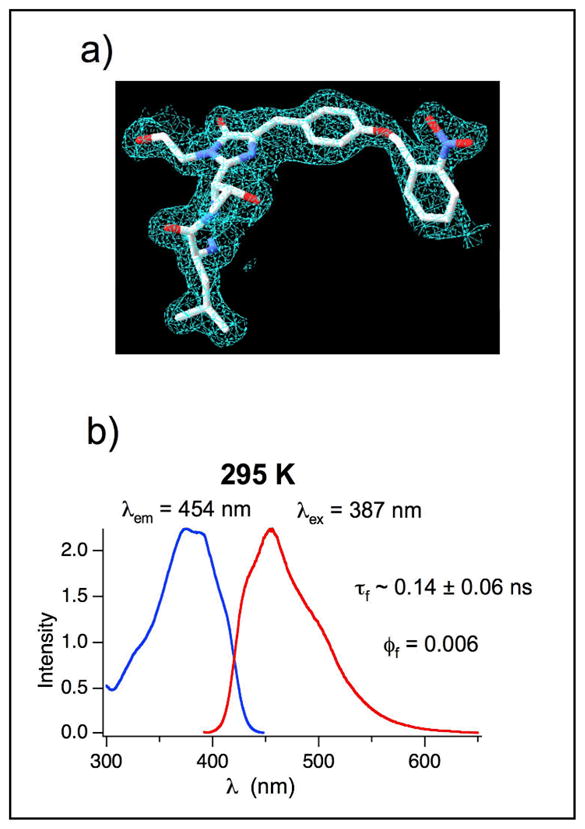
GFP66ONBY fluorescence is likely quenched through photon induced electron transfer to the o-nitrobenzyl group a)Histagged GFP66ONBY was purified with affinity chromatography and crystallized to yield crystals that diffracted to 2.1 Å. Electron density from the X-ray structure clearly shows the presence of the o-nitrobenzyl group and cyclized backbone. b) Fluorescence excitation (blue) and emission (red) spectra of caged GFP66ONBY in tris buffer (pH = 8) at room temperature; caged GFP66ONBY is practically non-fluorescent (fluorescence quantum yield = 0.006) and shows only a short fluorescence lifetime (0.14 ± 0.06 ns).
Given that the Y66ONBY mutant still forms the chromophore, the non-fluorescent nature of cGFP probably originates in efficient quenching of the singlet excited state. The o-nitrobenzyl group sterically interferes with the tight packing of the β-sheet, against the fluorophore. This intimate association is necessary to exclude water and O2, which are both capable of quenching the excited state fluorophore. However, the most likely cause for the low fluorescence quantum yield is efficient intramolecular quenching by the o-nitrobenzyl group. It is reported that o-nitrobenzyl groups quench excited fluorophores through a photo-induced electron transfer from the S1 excited fluorophore to the o-nitrobenzyl group.[20, 21] Because no transient absorptions in the spectral range from 350 to 800 nm were observed in laser flash photolysis experiments in the microsecond time scale, this intramolecular electron transfer probably reverses fast to the ground state of caged Y66ONBY (for details on laser flash photolysis experiments see Supporting Information).
In conclusion, we have developed a photoactivatable GFP appropriate for use as a molecular marker or in super resolution imaging using unnatural amino acid mutagenesis with o-nitrobenzyl tyrosine whose fluorescence is activated on the same timescale as proteins previously used for these applications.[1, 2] Recent advances make it possible to utilize this technology in both prokaryotic and eukaryotic cells.[22] The o-nitrobenzyl group likely deactivates the excited fluorophore through photon induced electron transfer. The mechanism of photoactivation does not require the conservation of any other residues, and so it should be useful for producing a variety of fluorescent proteins ranging from GFP to RFP. Finally, the dark form of cGFP is practically nonfluorescent at any wavelength, unlike previous photoactivatable proteins, freeing up the rest of the visible spectrum for other uses.
Experimental Section
Complete materials and methods are listed in supplemental material.
Supplementary Material
Acknowledgments
P.G.S. would like to thank the NIH for support through grants R01GM62159 and PN22EY018241. S. J. and N. J. T. thank the National Science Foundation of the USA for financial support (NSFCHE-07-17518).
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Dr. Dan Groff, Department of Chemistry and the Skaggs Institute for Chemical Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037(USA), fax: (+1)858-784-9440.
Dr. Feng Wang, Department of Chemistry and the Skaggs Institute for Chemical Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037(USA), fax: (+1)858-784-9440.
Dr. Steffen Jockusch, Department of Chemistry, Columbia University, New York, New York, 10027, USA.
Prof. Nicholas J. Turro, Department of Chemistry, Columbia University, New York, New York, 10027, USA
Prof. Peter G. Schultz, Email: Schultz@scripps.edu, Department of Chemistry and the Skaggs Institute for Chemical Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037(USA), fax: (+1)858-784-9440.
References
- 1.Patterson GH, Lippincott-Schwartz J. Science. 2002;297:1873. doi: 10.1126/science.1074952. [DOI] [PubMed] [Google Scholar]
- 2.Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Science. 2006;313:1642. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 3.Ando R, Hama H, Yamamoto-Hino M, Mizuno H, Miyawaki A. Proc Natl Acad Sci U S A. 2002;99:12651. doi: 10.1073/pnas.202320599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsutsui H, Karasawa S, Shimizu H, Nukina N, Miyawaki A. EMBO Rep. 2005;6:233. doi: 10.1038/sj.embor.7400361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Habuchi S, Ando R, Dedecker P, Verheijen W, Mizuno H, Miyawaki A, Hofkens J. Proc Natl Acad Sci U S A. 2005;102:9511. doi: 10.1073/pnas.0500489102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mizuno H, Mal TK, Tong KI, Ando R, Furuta T, Ikura M, Miyawaki A. Mol Cell. 2003;12:1051. doi: 10.1016/s1097-2765(03)00393-9. [DOI] [PubMed] [Google Scholar]
- 7.Henderson JN, Gepshtein R, Heenan JR, Kallio K, Huppert D, Remington SJ. J Am Chem Soc. 2009;131:4176. doi: 10.1021/ja808851n. [DOI] [PubMed] [Google Scholar]
- 8.Ormo M, Cubitt AB, Kallio K, Gross LA, Tsien RY, Remington SJ. Science. 1996;273:1392. doi: 10.1126/science.273.5280.1392. [DOI] [PubMed] [Google Scholar]
- 9.Elsliger MA, Wachter RM, Hanson GT, Kallio K, Remington SJ. Biochemistry. 1999;38:5296. doi: 10.1021/bi9902182. [DOI] [PubMed] [Google Scholar]
- 10.Matz MV, Fradkov AF, Labas YA, Savitsky AP, Zaraisky AG, Markelov ML, Lukyanov SA. Nat Biotechnol. 1999;17:969. doi: 10.1038/13657. [DOI] [PubMed] [Google Scholar]
- 11.Wang L, Brock A, Herberich B, Schultz PG. Science. 2001;292:498. doi: 10.1126/science.1060077. [DOI] [PubMed] [Google Scholar]
- 12.Deiters A, Groff D, Ryu Y, Xie J, Schultz PG. Angew Chem Int Ed Engl. 2006;45:2728. doi: 10.1002/anie.200600264. [DOI] [PubMed] [Google Scholar]
- 13.Krafft GA, Sutton WR, Cummings RT. Journal of the American Chemical Society. 1988;110:301. [Google Scholar]
- 14.Kobayashi T, Urano Y, Kamiya M, Ueno T, Kojima H, Nagano T. J Am Chem Soc. 2007;129:6696. doi: 10.1021/ja070376d. [DOI] [PubMed] [Google Scholar]
- 15.Han G, Mokari T, Ajo-Franklin C, Cohen BE. Journal of the American Chemical Society. 2008;130:15811. doi: 10.1021/ja804948s. [DOI] [PubMed] [Google Scholar]
- 16.Wilkins BJ, Marionni S, Young DD, Liu J, Wang Y, Di Salvo ML, Deiters A, Cropp TA. Biochemistry. 49:1557. doi: 10.1021/bi100013s. [DOI] [PubMed] [Google Scholar]
- 17.Cabantous S, Waldo GS. Nat Methods. 2006;3:845. doi: 10.1038/nmeth932. [DOI] [PubMed] [Google Scholar]
- 18.Groff D, Thielges MC, Cellitti S, Schultz PG, Romesberg FE. Angew Chem Int Ed Engl. 2009;48:3478. doi: 10.1002/anie.200806239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Il’ichev YV, Schwörer MA, Wirz J. Journal of the American Chemical Society. 2004;126:4581. doi: 10.1021/ja039071z. [DOI] [PubMed] [Google Scholar]
- 20.Ueno T, Urano Y, Setsukinai K-i, Takakusa H, Kojima H, Kikuchi K, Ohkubo K, Fukuzumi S, Nagano T. Journal of the American Chemical Society. 2004;126:14079. doi: 10.1021/ja048241k. [DOI] [PubMed] [Google Scholar]
- 21.Kobayashi T, Urano Y, Kamiya M, Ueno T, Kojima H, Nagano T. Journal of the American Chemical Society. 2007;129:6696. doi: 10.1021/ja070376d. [DOI] [PubMed] [Google Scholar]
- 22.Thibodeaux GN, Liang X, Moncivais K, Umeda A, Singer O, Alfonta L, Zhang ZJ. PLoS ONE. 5(6):e11263. doi: 10.1371/journal.pone.0011263. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.