

Summary
Recently, the hypothesis that protein motions are involved in enzymatic turnover has gained significant attention. We review cases where there is evidence that protein motions are rate-limiting in the overall catalytic cycle and examine experimental and theoretical evidence for how such motions enhance the probability of sampling the transition state configurations relative to the ground state. The impact of tunneling, the possible role of vibrational coupling and the value of conformational chemical landscapes are also scrutinized.
Introduction
The enormous rate accelerations of enzyme reactions relative to their solution counterparts range in one recent tabulation from 1010–10 20 fold [1]. The source of this acceleration has been hypothesized, named, and debated in the numerous articles, reviews and books that have enzymology as their subject.
Currently, the most seductive rationale connects the enzyme’s dynamics to its catalytic prowess. The issue is far from settled, however, and this topic has become the focus of intense, often contentious debate. It is outside the scope of this article to capture the nuanced details offered in various papers to bolster a particular viewpoint, so we will attempt to report accurately the gist of the key arguments and to identify areas where there might be a consensus. Please also keep in mind that although this review is restricted to recent publications, many of the concepts debated are not novel but have been informed by recent experimental and computational methods.
To begin, an incisive summary of available protein motions as well as the experimental methods to detect each time scale serves as a needed reference base (Figure 1) [2]. The great majority of enzyme catalyzed reactions exhibit turnover cycles that are limited by rates in the millisecond to second range. Consequently, the method of choice to document motions in specific regions of the enzyme has been a variety of NMR spectroscopic methods, particularly relaxation dispersion experiments that have provided both structural and kinetic dynamic information across the above time scale in solution under reaction conditions. X-ray diffraction, likewise, has provided time-resolved high-resolution structures but is limited to the crystalline state. Advances in computational methods have also provided deeper insights.
Figure 1. Timescales of molecular motions and means of their detection.

Figure reproduced with permission from ref. [2].
Relationship of motions to catalysis
A more liberal definition describes this relationship in terms of a series of correlated conformational changes that occur throughout the enzyme and act along the reaction coordinate to facilitate the chemical reaction. Such facilitation may be viewed as active-site reorganization providing stabilizing interactions as well as increased sampling of chemically productive substrate/active-site configurations [3]. There is as yet no evidence, experimental or theoretical, nor is it meant, that such conformational motions are coupled directly to vibrations within the transition state of the chemical reaction.
The more conservative definition of this relationship posits a direct linkage between the transition state and motions within the protein. That linkage however, must be established by evidence for a large deviation from transition theory manifested in a small transmission coefficient, κ, and non-Boltzmann coherent motions and distributions [4]. As yet such data, experimental or theoretical, have not been described for any enzyme.
Moreover, there is a temptation to blur the distinction between these two definitions, and further confusion in the literature arises from imprecise usage of the terms dynamics and catalysis. Rigorously, dynamics is defined as nonequilibrium barrier recrossing effects, and catalysis is defined as rate enhancement relative to a model reaction in solution. Often the term dynamics is assumed to be synonymous with the above liberal definition of motions, and catalysis is not related to an appropriate reference system.
Invoking the more liberal definition, we will present several case histories where experiments have implicated the rate of conformational movement of specific regions that appear to impact the turnover cycle. We will end with an abbreviated review of recent explanations that invoke motions influencing the rate of the chemical step in enzyme reactions.
1. Dihydrofolate reductase (DHFR)
DHFR catalyzes the reduction of 7,8-dihydrofolate (H2F) to 5,6,7,8-tetrahydrofolate (H4F) through a stereospecific transfer of the pro-R hydrogen from the cofactor nicotinamide adenine dinucleotide phosphate (NADPH; also abbreviated as NH) to the C6 atom of the pterin nucleus, with concomitant protonation at N5 (Figure 2).
Figure 2. (Top) The reaction catalyzed dihydrofolate reductase (DHFR) and a model of E. coli).

. The reactant substrate is dihydrofolate (H2F), the product is tetrahydrofolate (H4F) and the cofactor is reduced nicotinamide adenine dinucleotide phosphate (NADPH), which is converted to the oxidized form NADP+. R = p-aminobenzoylglutamate, R' = adenosine-diphosphoribose. (Middle) The reaction catalyzed by Cyclophilin A (CypA) and a model of Human CypA. Model of CypA showing the bound peptide (in wire-frame), residues in the coupled network (sticks) and Ser99 (spheres). (Bottom) The reaction catalyzed by Triosephosphate isomerase (TIM) and and model of chicken TIM. Model of chicken TIM with loop 6 in green sticks and loop7 residues in yellow sticks. DHAP = dihydroxyacetone phosphate; G3P = glyceraldehyde-3-phosphate.
X-ray structures have been reported for E. coli DHFR complexed with a wide variety of substrate, product and cofactor analogs. E. coli DHFR is a small (~18 kD) protein with an α/β structure consisting of a central 8-stranded β-sheet (comprised of β-strands A–H) and four flanking α-helices (designated αB, αC, αE, and αF) (Figure 2) [5,6]. The active-site cleft divides the protein into two structural subdomains: the adenosine binding subdomain and the loop subdomain. The adenosine binding subdomain (residues 38–88 in E. coli DHFR) is the smaller of the two subdomains and provides the binding site for the adenosine part of the cofactor. The loop subdomain consists of ~100 residues and contains a set of three loops (Met20 [residues 9–24], βF-βG [116–132], and βG-βH [142–150]) close to the active site.
NMR studies
A recent comprehensive dynamic study of the participation of excited-state conformations, detected by NMR relaxation dispersion measurements, showed that the structural and dynamic changes that occurred in the protein in response to the binding and dissociation of substrates, cofactors and products were an integral part of the mechanism of this enzyme [7]. Moreover, the relaxation rates measured for the adjacent excited state conformations associated with the key reaction cycle species (E · NH · H2F → E · N+ · H4F → E · H4F → E · NH · H4F) closely approximated the rates in the kinetic scheme measured by presteady state methods [8]. For example, the excited state detected in E · NH · H4F resembled the ground state of the E · NH complex and decayed at the rate of 12–18 s−1 that compared favorably with the 12 s−1 obtained from the kinetics. In brief, the kinetic rate constants have their counterparts in the NMR measurements of the rate of structural conformational changes consistent with temporal gating on the timescale of the reaction coordinate. Although, the overall conformation of the E · H2F, E · F and E · H4F complexes was the same, there were significant differences in the μs-ms fluctuations in the active site and the loop regions. Similar differences were observed in the E · NH · H4F and E · NH · F ternary complexes. This was interpreted in terms of highly orchestrated sampling of conformational motions, which are integral to the protein fold, to funnel the enzyme through intermediates along the catalytic cycle [9]. Another recent NMR study on a mutant DHFR supported this general picture [10].
Theoretical studies
Classical molecular dynamics (MD) simulations on various DHFR complexes provided residue-based maps of correlated motions indicating strongly correlated and anti-correlated motions involving spatially distinct regions [11]. When combined with the genomic sequence conservation data, a conserved set of residues emerged that may act to facilitate hydride transfer [12]. Hybrid quantum-classical MD simulations of the hydride transfer reaction catalyzed by DHFR have provided further information about enzyme motion in DHFR [12,13]. These hybrid simulations provide evidence for a network of coupled motions extending throughout the protein and ligands. Fast, stochastic thermal motions lead to slower conformational changes along the collective reaction coordinate (i.e., reorganization of the environment) that facilitate the chemical reaction. These equilibrium molecular motions are not dynamically coupled to the chemical transformation of the substrate and cofactor, but rather give rise to conformations of the ternary complex that are conducive to hydride transfer because of short transfer distances, suitable orientation of substrate and cofactor, and a favorable electrostatic environment for charge transfer. Another computational study on DHFR using different computational methods generated qualitatively similar results [14].
Hybrid quantum-classical MD simulations of DHFR enzymes with distal mutations are consistent with experimental rate measurements and suggest that the mutations may modify the network of coupled motions through structural perturbations [15,16]. These hybrid simulations suggest that the mutations alter the conformational sampling of the enzyme and ligands, thereby lowering the probability of sampling configurations conducive to hydride transfer, increasing the free energy barrier, and decreasing the reaction rate. Additional computation-based studies have emphasized the role of hydrogen bonding between the Met20 loop and the cofactor as strongly contributing to the modulation of the reactivity of the E · NH · H2F ternary complex by distal mutations [17].
Solvent effects
In a recent study on Escherichia coli and Thermotoga maritima DHFR, Allemann and colleagues have questioned the coupling of long range protein motions to the hydride transfer step [18,19]. Using solvents of different composition, they showed that changing the solvent viscosity did not influence the hydride transfer rate for both E. coli and T. maritima enzymes; however, the rates were altered by changes in the dielectric constant of the medium. Since the changes in dielectric constant influenced the protein flexibility, short range motions and conformational substates were implicated to contribute to catalysis. Although this makes for an important observation, changes in the bulk solvent viscosity are not expected to change the internal protein motions thought to facilitate the hydride transfer [3]. The large scale protein motions, but not the internal protein motions, have been shown to be slaved to the bulk solvent and controlled by the solvent viscosity [20,21]. Thus, these viscosity experiments do not provide information about the impact of internal protein motions on hydride transfer.
2. Cyclophilin A
Cyclophilin A (CypA) catalyzes the reversible isomerization (cis-trans) of prolyl peptide bonds (Figure 2) [22]. It acts on a wide variety of proteins and peptide substrates and plays a role in protein folding, intracellular protein transport and signaling. Human CypA is a 165 amino acid long polypeptide with eight anti-parallel beta strands, three helices and several flexible loops that cradle the active site (Figure 2). The enzyme was originally identified as the target of cyclosporin A, an immunosuppressant [23].
The dynamics of the isomerase have been characterized in the substrate-free state and during catalysis by NMR relaxation methods. As with DHFR the characteristic enzyme motions detected during catalysis are preserved in the free enzyme with frequencies corresponding to the catalytic turnover rates. Specifically, the enzyme interconverts between at least three states during catalysis, free enzyme and two substrate complexes with substrate bound in the cis and trans configurations. The exchange values from the dispersion profiles of 30 amides approximates the trans/cis substrate isomerization [kex = 2700 (dispersion NMR); kex = 2500 (cis ↔ trans)]. Site-specific mutations were shown by chemical shift changes to be propagated across the protein. Based on these studies, the authors proposed that protein motions are “harvested by the enzyme” for substrate turnover [24].
In a recent study by Alber, Kern and coworkers, the observations from the NMR studies were corroborated using ambient temperature crystallography [25]. From the X-ray data analysis by the Ringer method they indeed could identify within the crystal, substates observed in solution. Furthermore, introduction of a mutation (Ser99Thr, located >14 Å away from the active site) effected reversal of the substates. The Ser99Thr mutation also decreased the rate of inter-conversion of the conformational substates that had been deduced by NMR-relaxation dispersion experiments. This reduced rate of inter-conversion coincides well with the drop in the turnover number of the mutant relative to the wild-type enzyme.
3. Triose phosphate isomerase (TIM)
Triose phosphate isomerase catalyzes the reversible isomerization of dihydroxyacetone phosphate (DHAP) to D-glyceraldehyde 3- phosphate (G3P), the fifth step in the glycolysis pathway (Figure 2)[26]. TIM is a homodimer, 53 kDa in size and has the α/β-barrel structural motif (TIM-barrel). Structural analysis of TIM showed that it exists in two major conformations, an open and a closed form, where the active-site loop (denoted loop 6) modulates the substrate entry and product release [27,28]. This 11 residue (166–176) loop undergoes a 7Å movement as it transitions between the open and closed forms. Loop 6 residues interact with loop 7, which is another evolutionarily conserved motif in TIM. The hydrogen bonding interaction between loop 6 and loop 7 residues is known to stabilize the closed complex. The catalytic conversion of DHAP to G3P occurs in the closed conformation, and the loop 6 opening is partially rate-limiting during the turnover. Also, several distant residues have been implicated in assisting the motions of the protein that mediate the transition of the TIM between the closed and open forms.
Loria and coworkers have studied conformational changes in the loop 6 mutants by NMR relaxation as they relate to the opening and closing of the loop [29]. Based upon the NMR studies, the timescale of the transition between the open and closed complex (103 to 104 s−1) correlates with the rate of substrate turnover (~104 s−1). When residue Tyr208 in loop 7 was mutated to Phe, there was a significant change in equilibrium between the open and the closed conformations that led to more than 1000-fold reduction in the kcat of the enzyme. Interestingly, the loop motion at the C-terminal part of loop 6 was severely affected by the mutation explaining the altered rate of isomerization. In another temperature dependent NMR study that involved replacement of the loop 7 residues from the chicken TIM with those from an archaeal homologue, a similar shift in the equilibrium in favor of the open complex was noted. This dramatic effect of loop mutations highlights the significance of the conformational transitions in the loop for TIM catalysis [29].
4. Overview of case studies
Based on these examples, several unifying features can be noted. There are now a handful of cases where the substrate-free form of the enzyme samples conformational states along the reaction cycle [7,25,29–31]. In the case of DHFR and Cyclophilin A, the intermediates have been shown to make excursions into the conformations resembling the adjacent species along the reaction cycle. Mutations in the regions involved in conformational changes severely affect the rate of the reaction, and in the characterized instances, there is a concomitant change in the rate of conformational changes. The functional role of these motions is corroborated by multiple methods, and the system follows pathways encoded by the fold to optimize a catalytically competent enzyme-substrate complex [30]. While these steps are a necessary prelude to an active enzyme, their coupling to the chemical step is problematic.
Explanations for catalysis
Tunneling
Since the first clean demonstration [32] of nuclear tunneling in an enzymatic reaction, continuing studies of kinetic isotope effects have furnished solid evidence for nuclear tunneling in a variety of enzyme-catalyzed reactions [33–36]. The various results and their interpretation have been succinctly reviewed [37]. Hydrogen tunneling depends strongly on the hydrogen donor-acceptor distance and is particularly subject to motions that impact this distance. Particularly germane to the interpretation of the meaning of the magnitude of the kinetic isotope effect (KIE) and its temperature dependence has been the parallel development of theory [35,38–42]. Recent theories include the quantum mechanical effects of the transferring hydrogen nucleus and the proton donor-acceptor motion, as well as the reorganization of the protein and solvent environment [40,43].
A recent example is the temperature dependence of the KIE for distal mutants of soybean lipoxygenase, where comparison of the x-ray structures ruled out a change in structure of the enzyme’s ground state as responsible [44]. Theoretical modeling of these mutants [44,45] indicated that the distal mutation decreased the frequency of the proton donor-acceptor motion, enhancing the proton donor-acceptor fluctuations and thereby increasing the temperature dependence of the KIE. The interpretation of this finding, as well as those from other examples, where generally a mutation in the wild-type enzyme led to a temperature dependent kinetic isotope effect, was that flexibility had been introduced into the active site that enhanced the temperature-dependent proton donor-acceptor motion. Consequently, the temperature dependence of the KIE has been directly linked to the active-site conformational sampling alluded to above. Note that the catalytic effect of hydrogen tunneling and the associated heavy-atom motions is still under debate since the same effects also exist in solution [4,39,46–52].
Fast vibrational modes dynamically coupled to transition state
Another proposal is that fast femtosecond to picosecond dynamic motions at the transition state contribute significantly to catalysis [53]. Studies on purine nucleoside phosphorylase (PNP) showed that replacement of two residues more than 25Å from the active site yielded an enzyme with largely unchanged catalytic properties but revealed KIEs and transition state analog binding differing from the parent [54,55]. Information as to the importance of the specific vibrational distortion [54,55] was furnished by transition-path sampling calculations [56]. To reach the PNP transition state the calculations implicated a promoting vibration of His257 that correlated with compression of three oxygens (O5′ and O4′ of the ribose and oxygen of the phosphate) and expulsion of the leaving guanine (Figure 3). The transition state lifetime was estimated at ~10 fs with a reaction time ~60 fs. From this perspective, dynamics directly couples a promoting vibration to a transition state reactive bond. As stated in Ref. [53]: “In the few femtoseconds of the transition state’s existence, the transition-state ensemble cannot equilibrate with surroundings and cannot be described on the basis of thermodynamic parameters. In this interpretation of enzymatic catalysis, the transition-state barrier is probabilistic in nature and is determined by the formation of rare promoting vibrations at the catalytic site that lower the barrier for bond change.”
Figure 3. Intermediate in Purine nucleotide phosphorylase (PNP) catalysis.
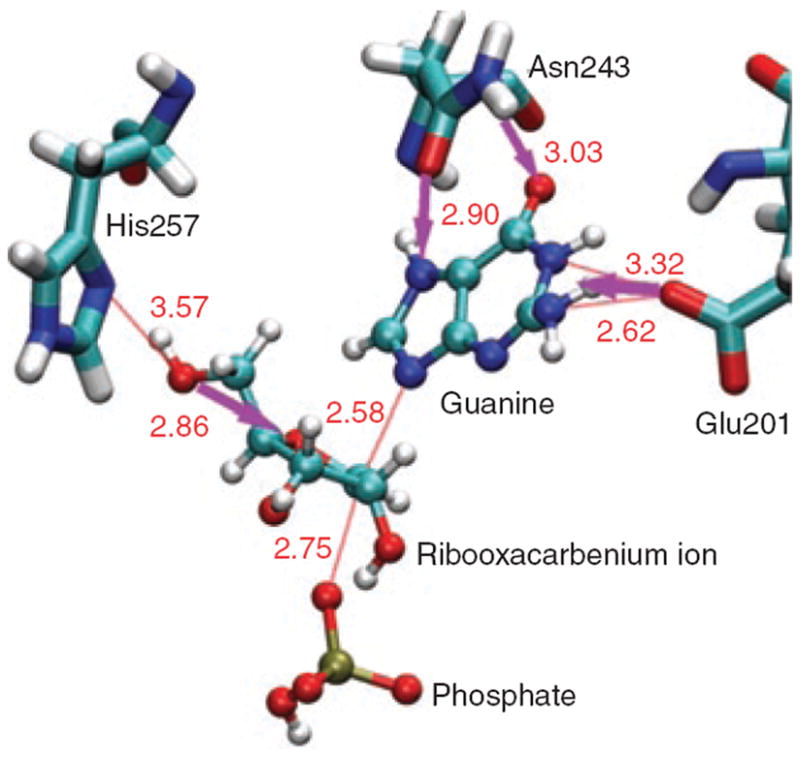
Compression of oxygens by His-257 and expulsion of guanine. Figure reproduced with permission from ref. [53].
There appears to be consensus that fast thermal fluctuations of the enzyme and ligands play an important role in enabling the stochastic search through the multidimensional conformational phase space for the chemical step [57]. These stochastic motions lead to the overall conformational changes, also denoted the reorganization of the environment, enabling the chemical reaction. By definition, the free energy barrier for the chemical step is related to the relative probability of sampling transition state configurations relative to reactant configurations, so clearly the free energy barrier is determined by the probability of sampling the relatively rare configurations conducive to the chemical reaction [3]. From this perspective, the experimentally measured rate for the chemical step is determined by the stochastic sampling of multidimensional conformational phase space to obtain the configurations conducive to the chemical reaction (i.e., the “transition state configurations” at the top of the barrier), and the chemical reaction itself (i.e., the bond breaking and forming) is very fast relative to this slower timescale. It is reasonable that specific modes in the protein will impact the very fast barrier crossing of the dividing surface at the top of the barrier [53]. The issue that is still under debate is whether the direct coupling of specific modes to this fast (femtosecond) barrier crossing is catalytically relevant in the sense of influencing the overall rate of the enzyme-catalyzed chemical reaction (on the millisecond timescale).
Preorganization
Another perspective is that preorganization forms the sole basis of the catalytic effect [4]. In this perspective, the active site of the enzyme is preorganized so that the reorganization of the environment required to evolve from the reactant to the transition state is lower in the enzyme than in solution. As stated in Ref. [4], “The real difference [between solution and enzyme] is the amplitude of the change in the [environmental] coordinates during the reaction, which determines the reorganization energy and generally is smaller in the enzyme because of the preorganization of the active site.” This perspective is consistent with the concept that stochastic conformational sampling determines the free energy barrier, which in turn strongly impacts the chemical reaction rate [3]. The reorganization of the environment from the reactant to the transition state (i.e., surmounting the free energy barrier) requires motions within the enzyme, even when the enzyme active site is preorganized. Altering the coupled motions associated with conformational sampling by distal mutation has been shown to impact the free energy barrier and therefore the catalytic rate [16]. In addition, achieving the preorganized active site may require rate-limiting conformational changes within the turnover cycle.
Landscapes
There is a consensus that enzymatic reactions can be effectively represented in two dimensions that combine a catalytic and conformational landscape (Figure 4) [7,57–60]. The existence of multiple conformations that in series or in parallel are able to traverse several transition states is embodied in models that posit searching of the landscape is associated with catalysis [37,57]. This landscape will be significantly different in the enzyme and in solution and hence will impact the conformational sampling that determines the relative free energy barriers. Whether landscapes account for the rate acceleration attributed to enzymes, however, is still under debate.
Figure 4. Schematic representation of the standard free energy landscape for an enzyme reaction.
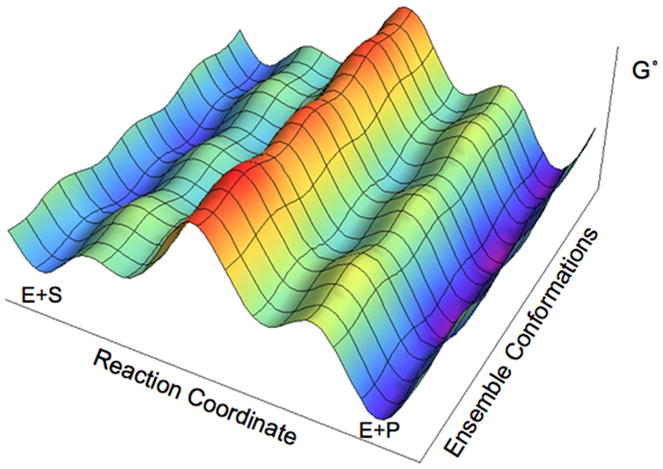
Conformational changes occur along both axes. The conformational changes occurring along the Reaction Coordinate axis correspond to the environmental reorganization that facilitates the chemical reaction, and the conformational changes occurring along the Ensemble Conformations axis represent the ensembles of configurations existing at all stages along the reaction coordinate. Figure from Ref. [57].
Conclusions
Overall, it is clear that protein conformational changes are manifest during different steps along the turnover cycle of enzymes. Motions on different timescales are important for binding the substrate, chaperoning the substrate along the reaction coordinate, and ultimately releasing the product. There is strong evidence that the turnover cycle of several enzymes is limited by conformational changes on the ms-sec timescale that preorganize the active site and facilitate its conformational sampling of transition state conformations. However, there is no evidence, to our knowledge, as to whether fast fs-ps timescale motions are directly linked to the bond breaking and forming process of the transition state.
Although, due to space limitations, we provided only three specific examples where conformational changes have been shown to play a functional role in enzyme turnover, there are other systems where there is now substantial evidence supporting a similar claim. Therefore, it appears that such conformational changes are universal and are programmed in the protein fold to facilitate specific functions. There is much to be learned about the origins of the catalytic power of enzymes. However, one can be assured that modern experimental and computational tools will lead to deeper understanding of enzyme catalysis in specific and broader contexts [61].
Acknowledgments
This work was supported by grants NIH GM56207 (SHS) and NIH 1R01GM092946 (SJB)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Vishal C. Nashine, Email: vcn1@psu.edu, Department of Chemistry, The Pennsylvania State University, 414 Wartik Laboratory, University Park, Pennsylvania, 16802. Tel 814-865-9508
Sharon Hammes-Schiffer, Email: shs@chem.psu.edu, Department of Chemistry, The Pennsylvania State University, 104 Chemistry Building, University Park, Pennsylvania, 16802. Tel 814-865-6442.
Stephen J. Benkovic, Email: sjb1@psu.edu, Department of Chemistry, The Pennsylvania State University, 414 Wartik Laboratory, University Park, Pennsylvania, 16802. Tel 814-865-2882, Fax: 814-865-2973
References and recommended reading
Papers of particular interest, published within the period of interest, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Wolfenden R, Snider MJ. The depth of chemical time and the power of enzymes as catalysts. Acc Chem Res. 2001;34:938–945. doi: 10.1021/ar000058i. [DOI] [PubMed] [Google Scholar]
- 2.Henzler-Wildman K, Kern D. Dynamic personalities of proteins. Nature. 2007;450:964–972. doi: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- 3.Hammes-Schiffer S, Benkovic SJ. Relating protein motion to catalysis. Annu Rev Biochem. 2006;75:519–541. doi: 10.1146/annurev.biochem.75.103004.142800. [DOI] [PubMed] [Google Scholar]
- •4.Kamerlin SC, Warshel A. At the dawn of the 21st century: Is dynamics the missing link for understanding enzyme catalysis? Proteins. 2010;78:1339–1375. doi: 10.1002/prot.22654. A comprehensive review on enzyme dynamics and its contribution to enzyme catalysis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bolin JT, Filman DJ, Matthews DA, Hamlin RC, Kraut J. Crystal structures of Escherichia coli and Lactobacillus casei dihydrofolate reductase refined at 1.7 A resolution. I. General features and binding of methotrexate. J Biol Chem. 1982;257:13650–13662. [PubMed] [Google Scholar]
- 6.Sawaya MR, Kraut J. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: crystallographic evidence. Biochemistry. 1997;36:586–603. doi: 10.1021/bi962337c. [DOI] [PubMed] [Google Scholar]
- 7.Boehr DD, McElheny D, Dyson HJ, Wright PE. The dynamic energy landscape of dihydrofolate reductase catalysis. Science. 2006;313:1638–1642. doi: 10.1126/science.1130258. [DOI] [PubMed] [Google Scholar]
- 8.Fierke CA, Johnson KA, Benkovic SJ. Construction and evaluation of the kinetic scheme associated with dihydrofolate reductase from Escherichia coli. Biochemistry. 1987;26:4085–4092. doi: 10.1021/bi00387a052. [DOI] [PubMed] [Google Scholar]
- ••9.Boehr DD, McElheny D, Dyson HJ, Wright PE. Millisecond timescale fluctuations in dihydrofolate reductase are exquisitely sensitive to the bound ligands. Proc Natl Acad Sci U S A. 2010;107:1373–1378. doi: 10.1073/pnas.0914163107. The dispersion NMR studies demonstrate the exquisite sensitivity of the protein motions to the oxidation state of the folate ligand used. This elegant study also demonstrates sampling of higher energy conformational states by DHFR as it cycles through intermediates in the turnover cycle. The higher energy states resemble the intermediates adjacent in the turnover cycle. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mauldin RV, Lee AL. Nuclear magnetic resonance study of the role of M42 in the solution dynamics of Escherichia coli dihydrofolate reductase. Biochemistry. 2010;49:1606–1615. doi: 10.1021/bi901798g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Radkiewicz JL, Brooks CL. Protein dynamics in enzymatic catalysis: Exploration of dihydrofolate reductase. J Am Chem Soc. 2000;122:225–231. [Google Scholar]
- 12.Agarwal PB, SR, Rajagopalan PTR, Benkovic SJ, Hammes-Schiffer S. Network of coupled promoting motions in enzyme catalysis. Proc Natl Acad Sci U S A. 2002;99:2794–2799. doi: 10.1073/pnas.052005999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Agarwal PB, SR, Hammes-Schiffer S. Nuclear quantum effects and enzyme dynamics in dihydrofolate reductase catalysis. J Phys Chem B. 2002;106:3283–3293. [Google Scholar]
- 14.Garcia-Viloca M, Truhlar DG, Gao J. Reaction-path energetics and kinetics of the hydride transfer reaction catalyzed by dihydrofolate reductase. Biochemistry. 2003;42:13558–13575. doi: 10.1021/bi034824f. [DOI] [PubMed] [Google Scholar]
- 15.Watney JB, Agarwal PK, Hammes-Schiffer S. Effect of mutation on enzyme motion in dihydrofolate reductase. J Am Chem Soc. 2003;125:3745–3750. doi: 10.1021/ja028487u. [DOI] [PubMed] [Google Scholar]
- 16.Wong KF, Selzer T, Benkovic SJ, Hammes-Schiffer S. Impact of distal mutations on the network of coupled motions correlated to hydride transfer in dihydrofolate reductase. Proc Natl Acad Sci U S A. 2005;102:6807–6812. doi: 10.1073/pnas.0408343102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rod TR, JL, Brooks CL. Correlated motion and the effect of distal mutations in dihydrofolate reductase. Proc Natl Acad Sci U S A. 2003;100:6980–6985. doi: 10.1073/pnas.1230801100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loveridge EJ, Evans RM, Allemann RK. Solvent effects on environmentally coupled hydrogen tunnelling during catalysis by dihydrofolate reductase from Thermotoga maritima. Chemistry. 2008;14:10782–10788. doi: 10.1002/chem.200801804. [DOI] [PubMed] [Google Scholar]
- 19.Loveridge EJ, Tey LH, Allemann RK. Solvent effects on catalysis by Escherichia coli dihydrofolate reductase. J Am Chem Soc. 2010;132:1137–1143. doi: 10.1021/ja909353c. [DOI] [PubMed] [Google Scholar]
- 20.Fenimore PW, Frauenfelder H, McMahon BH, Young RD. Bulk-solvent and hydration-shell fluctuations, similar to alpha- and beta-fluctuations in glasses, control protein motions and functions. Proc Natl Acad Sci U S A. 2004;101:14408–14413. doi: 10.1073/pnas.0405573101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •21.Frauenfelder H, Chen G, Berendzen J, Fenimore PW, Jansson H, McMahon BH, Stroe IR, Swenson J, Young RD. A unified model of protein dynamics. Proc Natl Acad Sci U S A. 2009;106:5129–5134. doi: 10.1073/pnas.0900336106. This study demonstrates the presence of two distinct hydration levels of proteins and their influence on the protein motions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao Y, Ke H. Crystal structure implies that cyclophilin predominantly catalyzes the trans to cis isomerization. Biochemistry. 1996;35:7356–7361. doi: 10.1021/bi9602775. [DOI] [PubMed] [Google Scholar]
- 23.Takahashi N, Hayano T, Suzuki M. Peptidyl-prolyl cis-trans isomerase is the cyclosporin A-binding protein cyclophilin. Nature. 1989;337:473–475. doi: 10.1038/337473a0. [DOI] [PubMed] [Google Scholar]
- 24.Eisenmesser EZ, Millet O, Labeikovsky W, Korzhnev DM, Wolf-Watz M, Bosco DA, Skalicky JJ, Kay LE, Kern D. Intrinsic dynamics of an enzyme underlies catalysis. Nature. 2005;438:117–121. doi: 10.1038/nature04105. [DOI] [PubMed] [Google Scholar]
- •25.Fraser JS, Clarkson MW, Degnan SC, Erion R, Kern D, Alber T. Hidden alternative structures of proline isomerase essential for catalysis. Nature. 2009;462:669–673. doi: 10.1038/nature08615. An elegant example of structural approach employed to identify minor conformational substates in enzymes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rieder SV, Rose IA. The mechanism of the triosephosphate isomerase reaction. J Biol Chem. 1959:234. [PubMed] [Google Scholar]
- 27.Pompliano DL, Peyman A, Knowles JR. Stabilization of a reaction intermediate as a catalytic device: Definition of the functional role of the flexible loop in triosephosphate isomerase. Biochemistry. 1990;29:3186–3194. doi: 10.1021/bi00465a005. [DOI] [PubMed] [Google Scholar]
- 28.Kempf JG, Jung JY, Ragain C, Sampson NS, Loria JP. Dynamic requirements for a functional protein hinge. J Mol Biol. 2007;368:131–149. doi: 10.1016/j.jmb.2007.01.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berlow RB, Igumenova TI, Loria JP. Value of a hydrogen bond in triosephosphate isomerase loop motion. Biochemistry. 2007;46:6001–6010. doi: 10.1021/bi700344v. [DOI] [PubMed] [Google Scholar]
- 30.Henzler-Wildman KA, Thai V, Lei M, Ott M, Wolf-Watz M, Fenn T, Pozharski E, Wilson MA, Petsko GA, Karplus M, et al. Intrinsic motions along an enzymatic reaction trajectory. Nature. 2007;450:838–844. doi: 10.1038/nature06410. [DOI] [PubMed] [Google Scholar]
- 31.Kale S, Ulas G, Song J, Brudvig GW, Furey W, Jordan F. Efficient coupling of catalysis and dynamics in the E1 component of Escherichia coli pyruvate dehydrogenase multienzyme complex. Proc Natl Acad Sci U S A. 2008;105:1158–1163. doi: 10.1073/pnas.0709328105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cha Y, Murray CJ, Klinman JP. Hydrogen tunneling in enzyme reactions. Science. 1989;243:1325–1330. doi: 10.1126/science.2646716. [DOI] [PubMed] [Google Scholar]
- 33.Billeter SR, Webb SP, Agarwal PK, Iordanov T, Hammes-Schiffer S. Hydride transfer in liver alcohol dehydrogenase: quantum dynamics, kinetic isotope effects, and role of enzyme motion. J Am Chem Soc. 2001;123:11262–11272. doi: 10.1021/ja011384b. [DOI] [PubMed] [Google Scholar]
- 34.Kohen A, Cannio R, Bartolucci S, Klinman JP. Enzyme dynamics and hydrogen tunnelling in a thermophilic alcohol dehydrogenase. Nature. 1999;399:496–499. doi: 10.1038/20981. [DOI] [PubMed] [Google Scholar]
- 35.Hammes-Schiffer S. Hydrogen tunneling and protein motion in enzyme reactions. Acc Chem Res. 2006;39:93–100. doi: 10.1021/ar040199a. [DOI] [PubMed] [Google Scholar]
- 36.Nagel ZD, Klinman JP. Tunneling and dynamics in enzymatic hydride transfer. Chem Rev. 2006;106:3095–3118. doi: 10.1021/cr050301x. [DOI] [PubMed] [Google Scholar]
- •37.Nagel ZD, Klinman JP. A 21st century revisionist's view at a turning point in enzymology. Nat Chem Biol. 2009;5:543–550. doi: 10.1038/nchembio.204. A concise review on protein motions in catalysis which also provides a description of the tunneling model as it relates to the kinetic isotope effect and their temperature dependence in enzymes. [DOI] [PubMed] [Google Scholar]
- 38.Olsson MH, Siegbahn PE, Warshel A. Simulations of the large kinetic isotope effect and the temperature dependence of the hydrogen atom transfer in lipoxygenase. J Am Chem Soc. 2004;126:2820–2828. doi: 10.1021/ja037233l. [DOI] [PubMed] [Google Scholar]
- 39.Olsson MH, Parson WW, Warshel A. Dynamical contributions to enzyme catalysis: critical tests of a popular hypothesis. Chem Rev. 2006;106:1737–1756. doi: 10.1021/cr040427e. [DOI] [PubMed] [Google Scholar]
- 40.Knapp MJ, Rickert K, Klinman JP. Temperature-dependent isotope effects in soybean lipoxygenase-1: correlating hydrogen tunneling with protein dynamics. J Am Chem Soc. 2002;124:3865–3874. doi: 10.1021/ja012205t. [DOI] [PubMed] [Google Scholar]
- 41.Hay S, Pang J, Monaghan P, Wang X, Evans R, Sutcliffe M, Allemann R, Scrutton N. Secondary kinetic isotope effects as probes of environmentally-coupled enzymatic hydrogen tunneling reactions. ChemPhysChem. 2008;9:1875–1881. doi: 10.1002/cphc.200800291. [DOI] [PubMed] [Google Scholar]
- 42.Pu J, Gao J, Truhlar DG. Multidimensional tunneling, recrossing, and the transmission coefficient for enzymatic reactions. Chem Rev. 2006;106:3140–3169. doi: 10.1021/cr050308e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hatcher E, Soudackov AV, Hammes-Schiffer S. Proton-coupled electron transfer in soybean lipoxygenase: dynamical behavior and temperature dependence of kinetic isotope effects. J Am Chem Soc. 2007;129:187–196. doi: 10.1021/ja0667211. [DOI] [PubMed] [Google Scholar]
- 44.Meyer MP, Tomchick DR, Klinman JP. Enzyme structure and dynamics affect hydrogen tunneling: the impact of a remote side chain (I553) in soybean lipoxygenase-1. Proc Natl Acad Sci U S A. 2008;105:1146–1151. doi: 10.1073/pnas.0710643105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Edwards SJ, Soudackov AV, Hammes-Schiffer S. Impact of Distal Mutation on Hydrogen Transfer Interface and Substrate Conformation in Soybean Lipoxygenase. J Phys Chem B. 2010 doi: 10.1021/jp100133p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cui Q, Karplus M. Quantum mechanics/molecular mechanics studies of triosephosphate isomerase-catalyzed reactions: effect of geometry and tunneling on proton-transfer rate constants. J Am Chem Soc. 2002;124:3093–3124. doi: 10.1021/ja0118439. [DOI] [PubMed] [Google Scholar]
- 47.Knapp MJ, Klinman JP. Environmentally coupled hydrogen tunneling. Linking catalysis to dynamics. Eur J Biochem. 2002;269:3113–3121. doi: 10.1046/j.1432-1033.2002.03022.x. [DOI] [PubMed] [Google Scholar]
- 48.Liu H, Warshel A. Origin of the temperature dependence of isotope effects in enzymatic reactions: the case of dihydrofolate reductase. J Phys Chem B. 2007;111:7852–7861. doi: 10.1021/jp070938f. [DOI] [PubMed] [Google Scholar]
- 49.Major DT, Heroux A, Orville AM, Valley MP, Fitzpatrick PF, Gao J. Differential quantum tunneling contributions in nitroalkane oxidase catalyzed and the uncatalyzed proton transfer reaction. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0911416106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Doll KM, Bender BR, Finke RG. The first experimental test of the hypothesis that enzymes have evolved to enhance hydrogen tunneling. J Am Chem Soc. 2003;125:10877–10884. doi: 10.1021/ja030120h. [DOI] [PubMed] [Google Scholar]
- 51.Cui Q, Elstner M, Karplus M. A theoretical analysis of the proton and hydride transfer in liver alcohol dehydrogenase (LADH) J Phys Chem B. 2002;106:2721–2740. [Google Scholar]
- 52.Benkovic SJ, Hammes-Schiffer S. A perspective on enzyme catalysis. Science. 2003;301:1196–1202. doi: 10.1126/science.1085515. [DOI] [PubMed] [Google Scholar]
- •53.Schwartz SD, Schramm VL. Enzymatic transition states and dynamic motion in barrier crossing. Nat Chem Biol. 2009;5:551–558. doi: 10.1038/nchembio.202. This review details the role of femtosecond timescale protein motions in transition state barrier crossing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Luo M, Li L, Schramm VL. Remote mutations alter transition-state structure of human purine nucleoside phosphorylase. Biochemistry. 2008;47:2565–2576. doi: 10.1021/bi702133x. [DOI] [PubMed] [Google Scholar]
- 55.Saen-Oon S, Quaytman-Machleder S, Schramm VL, Schwartz SD. Atomic detail of chemical transformation at the transition state of an enzymatic reaction. Proc Natl Acad Sci U S A. 2008;105:16543–16548. doi: 10.1073/pnas.0808413105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bolhuis P, Chandler D, Dellago C, Geissler P. Transition path sampling: Throwing ropes over rough mountain passes, in the dark. Annu Rev Phys Chem. 2002;53:291–318. doi: 10.1146/annurev.physchem.53.082301.113146. [DOI] [PubMed] [Google Scholar]
- ••57.Benkovic SJ, Hammes GG, Hammes-Schiffer S. Free-energy landscape of enzyme catalysis. Biochemistry. 2008;47:3317–3321. doi: 10.1021/bi800049z. The concept of a free energy landscape of enzyme catalysis, in which the standard free energy is viewed as a two-dimensional surface in terms of the reaction coordinate and ensembles of conformations, is examined in the context of both experimental and theoretical evidence. [DOI] [PubMed] [Google Scholar]
- 58.Xiang Y, Goodman MF, Beard WA, Wilson SH, Warshel A. Exploring the role of large conformational changes in the fidelity of DNA polymerase beta. Proteins. 2008;70 :231–247. doi: 10.1002/prot.21668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brooks BR, Janezic D, Karplus M. Harmonic analysis of large systems. 1. Methodology. Journal of Computational Chemistry. 1995;16:1522–1542. [Google Scholar]
- 60.Min W, English BP, Luo GB, Cherayil BJ, Kou SC, Xie XS. Fluctuating enzymes: Lessons from single-molecule studies. Acc Chem Res. 2005;38:923–931. doi: 10.1021/ar040133f. [DOI] [PubMed] [Google Scholar]
- 61.Zalatan JG, Herschlag D. The far reaches of enzymology. Nat Chem Biol. 2009;5:516–520. doi: 10.1038/nchembio0809-516. [DOI] [PubMed] [Google Scholar]