

Abstract
It has been reported that complement is activated on the surface of activated platelets, despite the presence of multiple regulators of complement activation. To reinvestigate the mechanisms by which activated platelets bind to complement components, the presence of complement proteins on the surfaces of nonactivated and thrombin receptor-activating peptide-activated platelets was analyzed by flow cytometry and Western blot analyses. C1q, C4, C3, and C9 were found to bind to thrombin receptor-activating peptide-activated platelets in lepirudin-anticoagulated platelet-rich plasma (PRP) and whole blood. However, inhibiting complement activation at the C1q or C3 level did not block the binding of C3 to activated platelets. Diluting PRP and chelating divalent cations also had no effect, further indicating that the deposition of complement components was independent of complement activation. Furthermore, washed, activated platelets bound added C1q and C3 to the same extent as platelets in PRP. The use of mAbs against different forms of C3 demonstrated that the bound C3 consisted of C3(H2O). Furthermore, exogenously added soluble complement receptor 1 was shown to bind to this form of platelet-bound C3. These observations indicate that there is no complement activation on the surface of platelets under physiological conditions. This situation is in direct contrast to a number of pathological conditions in which regulators of complement activation are lacking and thrombocytopenia and thrombotic disease are the ultimate result. However, the generation of C3(H2O) represents nonproteolytic activation of C3 and after factor I cleavage may act as a ligand for receptor binding.
Aberrations in the complement system are associated with an increased risk of pathological thrombotic processes, such as myocardial infarction and stroke. Support for direct involvement of complement in these conditions has come from reports of the generation of complement activation fragments in myocardial infarction and unstable angina (1–4). Complement activation also is associated with a wide range of other clinical conditions that involve platelet activation, including sepsis (5) and systemic lupus erythematosis (6).
We have shown recently that complement activation is triggered by the chondroitin sulfate (CS) that is released by activated platelets (7). Activation by thrombin receptor-activating peptide (TRAP)6 was found to induce fluid-phase complement activation, as reflected by the generation of the complement activation products C3a and sC5b-9. C1q was identified as the recognition molecule involved, because it bound directly to CS and CS-triggered complement activation could be restored in C1q-depleted serum by adding purified C1q. TRAP activation of whole blood increased the expression of CD11b on leukocytes and the generation of leukocyte/platelet complexes. These leukocyte functions were dependent on C3 activation and signaling via C5a, because they could be inhibited by complement inhibition and blockade of C5aR (7).
This reaction adds an additional mechanism to the previously reported mechanisms of complement activation under physiological conditions, which have been confined to the platelet surface. Del Conde et al. (8) and Peerschke et al. (9) have reported that complement is activated by the alternative and the classical pathways, respectively, on the surfaces of activated platelets. However, there is a lack of congruity between the observation that complement activation takes place on activated platelets and the previously reported expression of complement regulatory proteins on the platelet surfaces: decay-accelerating factor, MCP, and CD59 have been reported to be present on platelets (10–12).
The protective capacity of these regulators is in line with the complement activation reported in several pathological conditions in which complement regulators are missing or are dysfunctional, such as paroxysmal nocturnal hemoglobulinuria and atypical hemolytic-uremic syndrome (aHUS). Under these conditions, complement attack leads to platelet activation and thrombotic conditions. Supporting the notion that complement activation is carefully regulated on the platelet surface is a series of papers by Sims and coworkers (13), who have demonstrated that directed complement attack is needed to trigger complement activation and that this complement-mediated attack leads to platelet activation and the generation of microparticles via C5b-9. Thus, the mechanisms by which complement activation might be triggered by activated platelets under physiological conditions still need to be established.
In the current study, we have reinvestigated the previously reported binding of complement proteins to activated platelets. We found that many of the important components of the complement cascade bound to the surfaces of activated platelets, including C1q, C4, C3, and C9. However, we saw no evidence of any major proteolytically triggered complement activation, suggesting that no regular complement activation occurs when platelets are activated. Thus, our findings suggest that under physiological conditions complement activation is regulated carefully on the surfaces of activated platelets. Furthermore, we found that C3 present on activated platelets may act as a ligand for binding to complement receptor 1 (CR1).
Materials and Methods
Purified complement proteins
Human C1q was prepared from human serum as described by Tenner et al. (14), and C3 and factor I were prepared from plasma according to Hammer et al. (15) and Fearon (16), respectively. Factor H was prepared from human serum essentially according to Hammer et al. (15) except that the first step consisted of euglobulin precipitation (17). C3(H2O) was produced by repeated freeze-thawing of C3 (18); C3b was produced by incubating C3 with trypsin, followed by gel filtration to remove C3a; and iC3b was produced by incubating C3b with factor I, using factor H as a cofactor. Soluble CR1 (sCR1) BRL55730 was a kind gift from Dr. Henry Marsh (GlaxoSmithKline, Research Triangle Park, NC). The α-2–macroglobulin M6159 was purchased from Sigma-Aldrich (St. Louis).
Blood samples
Blood samples were obtained from healthy volunteers who had received no medication for at least 10 d prior to donation. Blood was drawn into 7-ml vacutainer tubes containing 100 μl of 5 mg/ml lepirudin (Schering AG, Saksa, Germany). To obtain platelet-rich plasma (PRP), blood was centrifuged at 150 × g for 15 min at room temperature (RT) within 30 min of collection.
Platelet activation
Platelets were activated in lepirudin-anticoagulated whole blood or PRP at 37°C by adding TRAP6 (Invitrogen, Carlsbad, CA) to a final concentration of 25 μg/ml (33.5 μM). Activation was stopped by the addition of 10 mM EDTA (final concentration). In other experiments, PRP or whole blood was activated in the presence of 10 μM Compstatin (a potent C3 inhibitor, Ac-ICV (1MeW)QDWGAHRCT-NH2) (19), 10 mM EDTA, or 10 mM EGTA with 2.5 mM Mg2+. In separate experiments, PRP was diluted 1:10 in Veronal-buffered saline (pH 7.4) containing 0.15 mM Ca2+ and 0.5 mM Mg2+ (VBS2+) to attenuate the alternative pathway (20), and incubated with 25 μg/ml C1q-inhibiting mAb (anti–C1q-85) (21, 22) for 10 min at RT. The PRP was centrifuged at 1100 × g for 15 min at RT, and the plasma was removed.
Platelet preparation
After activation and centrifugation, pellets were washed three times with Tyrode's medium I (pH 6.5), containing 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 0.36 mM NaH2PO4, 12 mM NaHCO3, 2 mM CaCl2, 5.5 mM glucose, 3.5 mg/ml BSA (Sigma-Aldrich), 1 μM PGE1 (Sigma-Aldrich), and 2 IU/ml heparin (Bioiberica, Barcelona, Spain). Each wash cycle consisted of suspension of the pellet in Tyrode's medium and incubation at 37°C for 10 min, followed by centrifugation at 1100 × g at RT for 10 min.
In some experiments, the platelets were washed prior to activation. Platelets were pelleted from PRP by centrifugation at 1100 × g for 10 min and washed three times as described above to remove plasma proteins. After being washed, the platelets were pelleted and resuspended in Tyrode's medium I without the addition of BSA, heparin, or PGE1. The platelets then were activated by the addition of TRAP and incubated as described above. After activation, they were washed once and resuspended in Tyrode's medium I with BSA, heparin, and PGE1. Activated and nonactivated platelets were diluted to a concentration of 200 × 109 platelets/l, and purified C1q (80 μg/ml), α-2–macroglobulin (250 μg/ml), or C3 (250 μg/ml) was added to the platelets and incubated for 30 min at RT. In experiments where platelets were incubated with purified C3, the protease inhibitor PMSF (1 mM) was added to the washing buffer to inhibit putative proteases that could cleave C3. In a few experiments, C1q was preincubated with soluble CS (50 mg/ml; Sigma-Aldrich) or low m.w. heparin (Fragmin) 50 mg/ml (Pfizer, New York, NY) on a shaker for 30 min at RT prior to being added to activated and nonactivated platelets. Platelets then were washed and labeled with Abs as described below.
In addition, supernatants from washed and TRAP-activated platelets were collected and analyzed for C3a (23).
To study whether C3 bound to activated platelets could act as a ligand for CR1, 100 μg/ml biotinylated sCR1 was added to platelets that had been incubated with or without C3 (500 μg/ml) and incubated for 30 min at RT. After being washed, platelet binding was detected with streptavidin-FITC (Amersham Biosciences, Little Chalfont, U.K.).
Flow cytometric analysis of platelet-bound complement proteins
Flow cytometry was used to monitor the activation of platelets and demonstrate the binding of complement proteins to nonactivated platelets or platelets activated with 25 μg/ml (33.5 μM) TRAP. Samples (100 μl) containing 50 × 109 platelets/l were incubated for 60 min at RT with each of the following Abs: mouse anti-human P-selectin–R-phycoerythrin (DakoCytomation, Carpinteria, CA), rabbit anti-human C3c (DakoCytomation), mAb 4SD17.3 against C3a (24), mAb C3–9 (25), rabbit anti-human C4 (DakoCytomation), sheep anti-human C9 (The Binding Site, Birmingham, U.K.), neo anti-human C9 mAb aE11 (Diatec, Oslo, Norway) (26) or sheep anti-human C1q (The Binding Site), anti-Mannan–binding lectin mAb (Antibody Shop A/S, Gentofte, Denmark), rabbit anti-human C-reactive protein (DakoCytomation), and rabbit anti-human α-2–macroglobulin (DakoCytomation). All of the Abs were labeled using an Alexa Fluor 488 kit (Invitrogen). Exposure of CS on TRAP-activated platelets was monitored with biotinylated IgM monoclonal anti–CS-A (Seikagaku Kogyo, Tokyo, Japan) and detected with streptavidin-FITC (Amersham Biosciences). The following were used as negative controls at a final concentration of 10 μg/ml: sheep anti-mouse Ig-FITC (The Binding Site), rabbit anti-mouse Ig-FITC (DakoCytomation), mouse Ig-G1-FITC (DakoCytomation), and mouse IgG1-biotin and goat anti-rabbit Ig-biotin, both detected using streptavidin-FITC (Amersham Biosciences). In each experiment the activation status of the platelets (control and experimental) was ascertained by monitoring the expression of P-selectin.
After being stained, the platelets were washed and fixed with 0.2% paraformaldehyde. The bound fluorochrome-labeled Abs were monitored with a FACSCalibur (BD Biosciences, San Jose, CA) and CellQuest Pro software. For each sample, ~30,000 cells were analyzed.
Biotinylation of CS
CS-A sodium salt (Sigma-Aldrich) was biotinylated via a primary amine in the protein core. CS (20 mg) was dissolved in 0.1 M phosphate buffer (pH 7.6) (2 ml), and 1.7 mg biotinamidohexanoic acid N-hydroxysuccinimide ester (Sigma-Aldrich) was added to the solution and incubated at 4°C for 10 h. The unconjugated biotin and reaction byproduct were removed with a PD-10 desalting column (GE Healthcare, Uppsala, Sweden).
Preparation of CS-coated microtiter plates
Microtiter plates (Nunc Maxisorb Immunoplates; Nunc, Naperville, IL) were coated with 100 μl streptavidin (5 μg/ml; Sigma-Aldrich). The wells were then blocked with 0.05% Tween 20 (Sigma-Aldrich) in PBS for 30 min at RT. Biotinylated CS (100 μl, 5 μg/ml) was subsequently bound to the streptavidin layer by incubation for 60 min at RT.
Binding of C1q and C3 fragments to CS
Purified C1q (70 μg/ml) was incubated with soluble CS (0–20 mg/ml) for 30 min at RT. Thereafter, the samples were transferred to CS-coated microtiter plates, and bound C1q was detected by sheep anti-human C1q (The Binding Site) followed by HRP-conjugated rabbit anti-sheep IgG (Dako-Cytomation).
Serum, diluted 1:10 in VBS2+ in the presence or absence of the inhibitory mAb anti–C1q-85 (21) (0–50 μg/ml), was incubated in the wells of microtiter plates coated with CS at 37°C for 60 min. After being washed, bound C1q was detected as above and C3 fragments (resulting from complement activation) were detected using HRP-conjugated rabbit anti-human C3c (DakoCytomation). Control experiments using serum diluted 1:10 in VBS supplemented with 10 mM EGTA and 2.5 mM Mg2+ were performed in parallel. Data are presented with background values (i.e., EGTA-Mg2+ control) subtracted. This set of experiments was performed to test the inhibitory effect of the anti–C1q-85 mAb on surface-induced complement activation by the classical pathway.
Western blot analyses
Western blot analysis was performed to characterize the platelet-associated C3. One milliliter of activated or nonactivated PRP was centrifuged at 1100×g for 10 min at RT, and the pellet was washed three times as described earlier. After being washed, the platelets were incubated for 10 min at 37°C with PBS containing 0.05% Tween 20 and 1 mM PMSF to remove surface-bound proteins. The samples then were centrifuged, and the supernatants were boiled with 10% SDS reducing sample buffer and subjected to 10% SDS-PAGE. The separated proteins were electrophoretically transferred onto a polyvinylidene difluoride membrane and blocked with PBS containing 1% BSA. HRP-conjugated polyclonal anti-C3c (DakoCytomation) or anti-C3a (24) Abs were used to detect the blotted proteins. Purified C3, C3b, iC3b, C3 (H2O), and iC3(H2O) were used as controls together with zymosan-activated normal human serum (containing C3a in addition to nonactivated intact C3). In initial experiments, platelets were first eluted as described above to remove surface-bound proteins, then lysed in the presence of 1 mM PMSF and subjected to Western blot analysis. Because no C3 fragments were detected in the lysate, subsequent experiments were performed on the platelet eluate only.
In other experiments, washed, TRAP-activated platelets prepared as described above were incubated with C3 (500 μg/ml) and factor I (10 μg/ml) in the presence and absence of factor H (80 μg/ml). After incubation, the platelets were washed twice and then incubated for 10 min at 37°C with PBS containing 0.05% Tween 20 and 1 mM PMSF or with PBS containing 0.05% Tween 20, 10 mM EDTA, 50 μg/ml soybean trypsin inhibitor, 1 mM theophylline, 20 mM benzamidine, and 0.5 μg/ml PGE1 (final concentrations). The samples then were centrifuged, and the supernatants were boiled with 10% SDS reducing sample buffer and subjected to 10% SDS-PAGE as described above. HRP-conjugated anti-C3c was used for detection.
Statistical analysis
Results are presented as means ± SEM. Differences between means were evaluated statistically using the Wilcoxon paired nonparametric test or repeated measures ANOVA followed by the Tukey-Kramer multiple comparison test for calculating differences in data containing more than two variables. For statistical analysis, Prism 4 for Macintosh software (Graph-Pad, San Diego, CA) was used. In each case, the n values refer to independent experiments using platelets or plasma from different blood donors.
Results
Binding of complement proteins to activated platelets in PRP and whole blood
Deposition of complement components on the surfaces of activated platelets in PRP and whole blood was investigated by flow cytometry. Platelets in lepirudin-anticoagulated PRP and whole blood were activated with 33.5 μM TRAP. These experiments clearly indicated that C1q (p = 0.0011), C4 (p =0.0002), C3 (p = 0.0001), and C9 (p = 0.0039) bound to TRAP-activated platelets in PRP (Fig. 1) to the same extent as in whole blood (data not shown). The mAb aE11 specific for a neoepitope in activated C9 did not bind to platelets under these conditions (data not shown). No binding of Mannan-binding lectin, C-reactive protein, or α-2–macroglobulin to activated platelets was detected.
FIGURE 1.

Binding of complement components C1q (n = 12) (A, B), C4 (n = 12) (C, D), C3 (n = 13) (E, F), and C9 (n = 9) (G, H) to nonactivated (black line) and TRAP-activated platelets (gray line) in lepirudin PRP, as detected by flow cytometry. Dotted lines indicate binding of species-matched negative control Abs to activated platelets: sheep anti-mouse Ig (A, G) and rabbit anti-mouse Ig (C, E). The difference in binding between the nonactivated and activated samples was statistically significant. **p < 0.01; ***p < 0.001.
Inhibition of C1q binding to immobilized CS by preincubation with soluble CS
Purified C1q bound to CS immobilized on the surfaces of microtiter plates. The binding of C1q decreased in a dose-dependent fashion when C1q had been preincubated with soluble CS and demonstrated that the binding was specific. Fifty percent inhibition was seen at ~4 mg/ml CS (Fig. 2A). By extrapolation of the dose curve, it was estimated that total inhibition of binding of C1q required ~50 mg/ml soluble CS. Therefore, this concentration of CS was employed in subsequent experiments using flow cytometry.
FIGURE 2.

Binding of purified C1q (A) (which had been preincubated with solublea CS at the indicated concentrations) to CS immobilized on ELISA plates. Data are presented as means ± SEM (n = 3). Binding of purified C1q (B–E) (n = 6) to nonactivated (black lines) and TRAP-activated (gray lines) platelets, which had been incubated in the absence (thick lines) or the presence (thin lines) of 50 mg/ml soluble CS (B, C) or 50 mg/ml low m.w. heparin (D, E) prior to being added to the platelets. The curves indicating binding of species-matched negative control Abs (sheep anti-mouse Ig) to activated platelets coincide with the curves for nonactivated platelets using specific Abs. The difference in binding between the nonactivated and activated samples was statistically significant. **p < 0.01; ***p < 0.001.
Binding of purified C1q and C3 to washed, activated platelets
To demonstrate that C1q and C3 bind to activated platelets in the absence of plasma complement components, washed platelets were TRAP-activated and incubated with purified C1q (Fig. 2B, 2D). Binding of C1q to washed, TRAP-activated platelets was slightly higher when purified C1q was used compared with binding of C1q to platelets activated in PRP, which is believed to be due to the noncompetitive environment created when platelets are washed and incubated with purified proteins. The binding of C1q could be inhibited substantially by preincubation with both CS (Fig. 2B, 2C) and low m.w. heparin (Fig. 2D, 2E).
Likewise, purified C3 that was added to washed, TRAP-activated platelets in PRP (Fig. 3A, 3B) had a binding pattern similar to those of C3 bound to platelets activated in PRP (Fig. 1E) and in whole blood (data not shown), suggesting that binding of C3 to activated platelets occurred independent of complement activation.
FIGURE 3.
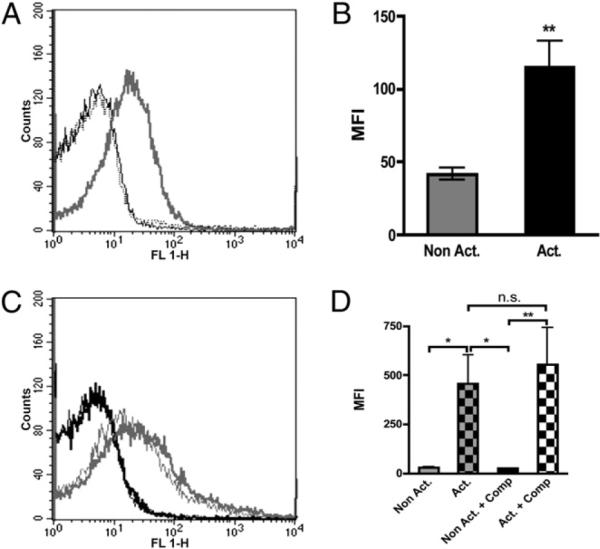
Binding of C3 to (A, B) washed, nonactivated (black line) and TRAP-activated (gray line) platelets (n = 9), as detected by flow cytometry. Binding of C3 to (C, D) nonactivated (black line) and TRAP-activated (gray line) platelets in lepirudin PRP in the absence (thick lines) or presence of 10 μM Compstatin (thin lines) (n = 8). The curves for the negative control Abs (rabbit anti-mouse Ig) to activated platelets coincide with the curves for nonactivated platelets using specific Abs. The difference in binding between nonactivated and activated samples was statistically significant. *p < 0.05; **p < 0.01.
Neither C1q nor C3 was detected on washed, activated platelets that had not received exogenously added components. In addition, no C3a was detected in the supernatants from washed, TRAP-activated platelets.
Attempts to block the binding of C3 to activated platelets by inhibiting complement activation
The addition of Compstatin, which blocks the activation of complement at the C3 level, did not affect the binding of either C3 (Fig. 3C, 3D) or C9 (data not shown) to the surfaces of activated platelets in PRP or whole blood, as detected by flow cytometry.
A 10-fold dilution of PRP, which attenuates the activation of the alternative pathway (20), also did not significantly affect the binding of C3 to platelets (Fig. 4A versus Fig. 1E, p = 1.00 for activated platelets). In addition, the presence of the inhibitory anti–C1q-85 mAb (25 μg/ml) did not inhibit the binding of C3 under the same conditions (Fig. 4A, 4B). This Ab binds to the globular heads of C1q, blocking the binding of C1q (21, 22) and thereby attenuating the activation of the classical pathway. In control experiments, serum diluted 1:10 was incubated in CS-coated microtiter plates in the presence or absence of anti–C1q-85. The Ab was effective in inhibiting the binding of C1q (Fig. 4C) and C3 fragments (the result of complement activation), even at low concentrations, with total inhibition at an Ab concentration of 25 μg/ml (Fig. 4D). Consequently, this concentration of anti–C1q-85 was chosen for subsequent experiments using flow cytometry.
FIGURE 4.
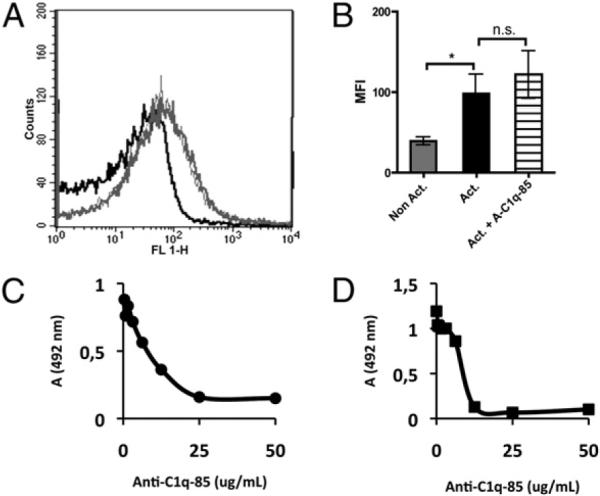
Binding of C3 (A, B) to nonactivated platelets (black line) and TRAP-activated platelets (gray line) in PRP, diluted 1:10 in VBS2+ (to inhibit complement activation by the alternative pathway), in the absence (thick lines) or presence (thin lines) of the inhibitory anti–C1q-85 mAb at a concentration of 25 μg/ml. The curve for the negative control (rabbit anti-mouse Ig) to activated platelets coincides with that for nonactivated platelets using specific Abs (n = 4). The difference in binding between nonactivated and activated samples was statistically significant. *p < 0.05. C, Blocking of C1q binding from serum diluted 1:10 in VBS2+ to CS immobilized on ELISA plates by preincubating the diluted serum with anti–C1q-85 mAb. D, Complement activation induced by incubating serum diluted 1:10 in VBS2+ in microtiter plates with immobilized CS was detected as binding of C3 fragments. The activation was inhibited when the serum was preincubated with anti–C1q-85 mAb. Data are shown as means ± SEM (n = 3). The SE lies within the symbols.
Consistent with these results was our observation that the binding of C1q, C4, and C3 was not attenuated when PRP was activated by TRAP in the presence of EDTA (which totally inhibits complement activation) or EGTA (which inhibits classical pathway activation; data not shown).
Characterization of the bound C3 on activated platelets
To further characterize the bound C3 on activated platelets, we made use of two anti-C3 mAbs (Fig. 5), both of which bind to neoepitopes of activated and conformationally changed C3 products but not to native C3. Anti–C3–9 is specific for the C-terminal α-chain–derived polypeptide of C3c (α4) that is exposed in C3b, iC3b, C3c, and C3(H2O) but not in native C3 and 4SD17.3 that binds to C3a (α1) in both C3(H2O) and C3a but not in native C3 (presented in Fig. 6D). Both Abs bound to TRAP-activated platelets in PRP (Fig. 5) with binding profiles similar to that of polyclonal anti-C3c (Fig. 1E), indicating that the bound C3 was nonproteolytically activated and still contained the C3a fragment, consistent with the binding of C3(H2O).
FIGURE 5.
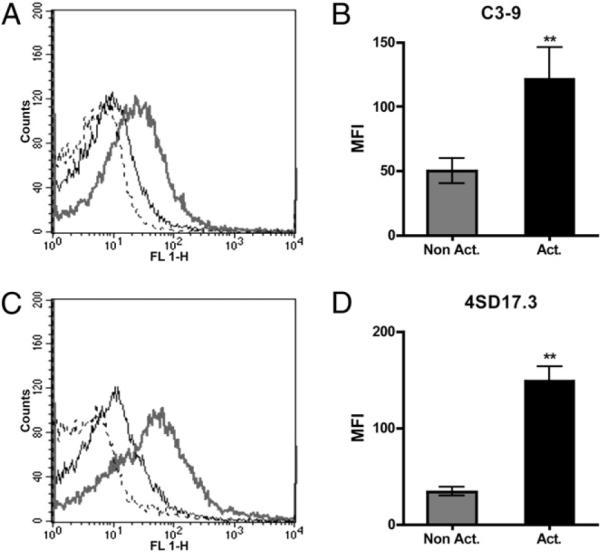
Binding of C3 to platelets, as detected with C3–9 mAb (A, B) specific for neoepitopes in the C-terminal α-chain-derived polypeptide of C3c (α4; Fig. 6D) exposed in conformationally changed C3 fragments or anti-C3a mAb 4SD17.3 specific for neoepitopes exposed in C3a or conformationally changed C3 (C, D), to nonactivated (black line) or TRAP-activated platelets (gray line). Dotted lines indicate the binding of species-matched negative control Abs (mouse IgG1) to activated platelets (n = 6). The difference in binding between nonactivated and activated samples was statistically significant. **p < 0.01.
FIGURE 6.

Platelet-associated C3, as detected by Western blot analysis using polyclonal anti-C3c Abs (A, B) or anti-C3a mAb 7SD17.3 (C). A, Platelets were activated by TRAP in PRP for 0, 15, or 30 min. Lanes 1–3 show membrane-bound C3 at the three time points, and lanes 4–6 show control C3, C3b, and iC3b, respectively, at 3 μg each. B, Binding and cleavage of purified C3 after incubation with washed, activated platelets in the absence (lane 1) or presence of factor I (lane 2) or with both factor I and factor H (lane 3). Lane 4 shows a reference comprising a mixture of C3b and iC3b. C, Membrane-bound C3 from nonactivated (lane 1) or TRAP-activated platelets (lane 2). Lane 3 shows control zymosan-activated serum (containing C3a in addition to nonactivated C3), and lane 4 shows control C3(H2O) incubated with factor I and factor H producing iC3(H2O). D, Diagram of the structure of C3 and its various proteolytic activation fragments.
Western blot analyses
The conclusion from our binding experiments that C3(H2O) was formed on the platelet surface was supported further by the results of our Western blot analyses. Using a polyclonal anti-C3c Ab specific for the C-terminal end of the α-chain (α4) and the β-chain, we were able to detect the α-chain, the β-chain, and the α4 fragment (Fig. 6A, 6B); with a polyclonal anti-C3a Ab, we detected an 80-kDa polypeptide (α1–3) (Fig. 6C). Both of these patterns are consistent with the pattern obtained when C3(H2O) is cleaved by factor I and factor H (Fig. 6D). Thus, in our system, C3 was activated in a nonproteolytic way and cleaved to some extent by factor I.
Binding of sCR1 to platelet-bound C3
The binding of sCR1 to platelets was investigated by flow cytometry. Washed, nonactivated and activated platelets were incubated with purified C3, and after being washed, sCR1 was added to the platelets. sCR1 was shown to bind to TRAP-activated platelets that had been exposed to native C3, whereas on nonactivated platelets only background levels of sCR1 were detected (p < 0.05) (Fig. 7). Washed, nonactivated and TRAP-activated platelets that had not been incubated with purified C3 were used as controls and showed only background levels of sCR1 (data not shown).
FIGURE 7.

Binding of sCR1 to nonactivated and TRAP-activated platelets exposed to native C3. The background level of sCR1 binding to platelets without the presence of C3 has been subtracted from the levels obtained in the presence of C3 for each individual experiment. The data are shown as means ± SEM (n = 7; *p < 0.05).
Discussion
In the current study, we have shown that activation of platelets elicits binding of complement components to the membranes of these cells. Our results indicated that this binding is not the result of complement activation but rather of the direct binding of the components to the activated platelets.
Binding of complement components to activated platelets has been demonstrated in a number of studies (e.g., Refs. 27–29). In the current study, we demonstrated that the complement components C1q, C4, C3, and C9 are present on the activated platelet surfaces. These components represent the whole range of complement components, from the beginning of the classical pathway to the end of the cascade (formation of the membrane attack complex). The general view has been that the deposition of complement components is a result of complement activation (8, 9). Both the classical and the alternative pathways have been proposed as the activation pathway. However, there remains a need to clarify the exact mechanism by which platelets bind complement proteins.
The presence of CS-A as the main glycosaminoglycan in platelets has been well established by both biochemical and histologic techniques (30, 31). The rapid release of CS from platelets has been demonstrated to occur in response to a variety of agonists, including ADP, collagen, adrenalin, and thrombin, resulting in an increase in plasma CS by up to 2 μg/ml within 3 min after activation (32). CS also has been reported to be exposed on the surfaces of platelets after activation (30), and we have shown previously that CS released from activated platelets can activate the complement system in the fluid phase (7). In this reaction, C1q was identified as the recognition molecule. We believe that the binding of C1q to platelets also is mediated partially by CS that is expressed on the activated platelets. Consistent with this hypothesis, we were able to substantially inhibit the binding of C1q to platelets by incubating C1q with soluble CS and low m.w. heparin.
Despite the fact that C1q binds complement components and activates the complement classical pathway in serum and plasma, its binding to activated platelets does not appear to activate complement. This situation was evident when potential complement activation was inhibited at the C1q (mAb C1q-85) and C3 levels (Compstatin) and the binding of C3 and C9 to activated platelets was studied by flow cytometry. Using polyclonal anti-C3c, we saw no difference in C3 binding, indicating that the inhibition of complement activation at the C1q and C3 levels had no effect on its binding. Furthermore, we found that purified C3 bound equally well to washed platelets in the absence of other complement components. Dilution of PRP, which attenuates the alternative pathway, and the presence of EDTA, which totally inhibits complement activation, had no effect on C3 binding. Western blot analyses demonstrated that the bound form of C3 had not been cleaved by convertases. Taken together, these experiments indicate that the binding of complement proteins to the surfaces of activated platelets is not a result of regular complement activation.
To clarify the nature of bound C3, we analyzed the activated platelets by flow cytometry using two mAbs specific for amino acid sequences present in C3c and C3a, respectively. Unlike the anti-C3c polyclonal Ab, the anti-C3c mAb (C3–9) does not bind to native C3. The binding of this mAb to activated platelets demonstrated that C3 was bound in an activated form. Likewise, the second mAb (4SD.17.3), which is specific for a neoepitope in C3a, does not bind to native C3, but it also detects C3(H2O). The results of this analysis suggested that C3(H2O) had bound to the activated platelets. In a complementary set of experiments, Western blot analyses of extracts from activated and nonactivated platelets revealed a high m.w. 80-kDa band that could be detected with anti-C3a. This size of this polypeptide band corresponds exactly to that of the N-terminal end of the factor I-cleaved C3(H2O) α-chain.
We also demonstrated that purified C3 bound to washed platelets could be cleaved by factor I. The fact that C3 was cleaved by factor I demonstrates that the thiol ester was disrupted, as is the case for C3(H2O). The cleavage occurred in the absence of exogenously added factor H, supporting the presence of factor H on the platelets (33, 34). Considered together, our findings indicate that the disruption of the thiol ester of native C3 that was observed here is consistent with the formation of C3(H2O).
Although our data indicate that C3 deposited on the platelets was activated by the disruption of the thiol ester in C3 in the absence of proteolytic cleavage, the mechanism responsible for this reaction is still unclear. Studies to elucidate this mechanism are currently underway. Thus far, we have employed the blocking polyclonal Ab against P-selectin that was used in the original report by Del Conde (8) to determine whether P-selectin is involved in binding of C3 in our experimental system; the results have indicated that the P-selectin–specific Ab has no effect on the binding of C3 in our system (data not shown).
Our results support the concept that complement is not proteolytically activated on the platelet surface under physiological conditions. This lack of surface-associated complement activation may be explained by the large number of membrane-bound (e.g., decay-accelerating factor, MCP, and CD59) and soluble (e.g., factor H) complement regulators found on the activated platelet surface that probably serve to protect the platelets from complement attack (10–12). Consistent with these observations, Ståhl et al. (29) have shown recently that aHUS patients with mutated factor H have a higher level of deposition of C3 and C9 and complement activation onto their platelets than control individuals; unlike the situation in normal individuals, this increased level is observed for both nonactivated and activated platelets. Combining aHUS patient sera containing mutated factor H and normal platelets resulted in complement activation and the activation and aggregation of platelets. The complement deposition and platelet activation were abrogated when platelets were preincubated with normal factor H or when normal serum was used (29). Platelet activation and a decrease in platelet counts also have been reported in paroxysmal nocturnal hemoglobulinuria. Insertion of C5b-9 complexes into the platelet membrane as a result of diminished surface expression of decay-accelerating factor and CD59 results in complement activation (35). In both of these conditions, the absence of complement regulatory function on the platelets contributes to the deposition of complement activation products, platelet aggregation, and thrombocytopenia. In normal individuals, platelets are more resistant to activation because of their surface expression of a normal repertoire of complement inhibitors.
It should be emphasized that the generation of C3(H2O) represents nonproteolytic activation of C3 into functionally active forms. Generation of C3(H2O) is pivotal for the initiation of the alternative pathway according to the tick-over theory reflecting that this form of C3 can form a C3 convertase (36). However, as shown in this work, the platelet-bound C3(H2O) is cleaved by factor I and inactivated with respect to convertase formation into iC3(H2O). It is also well established that C3(H2O) and iC3(H2O) interact with C3 receptors, such as CR1(CD35) (23), CR2 (CD21) (37), and a CR3 (CD11b/CD18)-like molecule from Candida albicans (38). Interestingly, we found that sCR1 bound to washed and activated platelets that had been exposed to purified native C3 (39, 40), which further links the transition of native C3 into C3 (H2O)/ iC3(H2O) upon binding to activated platelets to a biological function. We have demonstrated previously complex formation between platelets and monocytes/polymorphonuclear neutrophils, which was dependent on platelet-mediated C5a generation (7). The binding of sCR1 to platelets exposed to exogenously added native C3 suggests that the bound C3(H2O) may act as a receptor ligand and be involved in complex formation (41).
Taken together with the results of our previous study, the findings presented here indicate that complement activation occurs in the fluid phase in the vicinity of activated platelets, but under physiological conditions, no complement activation occurs on the surfaces of the platelets. This seems logical, because such activation would damage the platelets and elicit thrombotic reactions. This normal scenario is in direct contrast to that in a number of pathological conditions, such as paroxysmal nocturnal hemoglobulinuria and aHUS, in which regulators of complement activation are lacking and complement attack contributes to thrombocytopenia and thrombotic disease. In such conditions, complement activation is detrimental to the platelets and leads to platelet activation and thrombotic complications.
Acknowledgments
This work was supported by National Institutes of Health Grants AI068730, EB003968, GM62134, and GM069736; Swedish Research Council Grants 2009-4462, 2009-4675, and 15244; and grants from the Natural Sciences Faculty, University of Kalmar.
Abbreviations used in this paper
- aHUS
atypical hemolytic-uremic syndrome
- CR1
complement receptor 1
- CS
chondroitin sulfate
- PRP
platelet-rich plasma
- RT
room temperature
- sCR1
soluble CR1
- TRAP
thrombin receptor-activating peptide
- VBS
Veronal-buffered saline
Footnotes
Disclosures The authors have no financial conflicts of interest.
References
- 1.Iltumur K, Karabulut A, Toprak G, Toprak N. Complement activation in acute coronary syndromes. APMIS. 2005;113:167–174. doi: 10.1111/j.1600-0463.2005.apm1130303.x. [DOI] [PubMed] [Google Scholar]
- 2.Langlois PF, Gawryl MS. Detection of the terminal complement complex in patient plasma following acute myocardial infarction. Atherosclerosis. 1988;70:95–105. doi: 10.1016/0021-9150(88)90103-7. [DOI] [PubMed] [Google Scholar]
- 3.Yasuda M, Kawarabayashi T, Akioka K, Teragaki M, Oku H, Kanayama Y, Takeuchi K, Takeda T, Kawase Y, Ikuno Y. The complement system in the acute phase of myocardial infarction. Jpn. Circ. J. 1989;53:1017–1022. doi: 10.1253/jcj.53.1017. [DOI] [PubMed] [Google Scholar]
- 4.Yasuda M, Takeuchi K, Hiruma M, Iida H, Tahara A, Itagane H, Toda I, Akioka K, Teragaki M, Oku H, et al. The complement system in ischemic heart disease. Circulation. 1990;81:156–163. doi: 10.1161/01.cir.81.1.156. [DOI] [PubMed] [Google Scholar]
- 5.Levi M. Platelets in sepsis. Hematology. 2005;10(Suppl. 1):129–131. doi: 10.1080/10245330512331390177. [DOI] [PubMed] [Google Scholar]
- 6.Ekdahl KN, Bengtsson AA, Andersson J, Elgue G, Rönnblom L, Sturfelt G, Nilsson B. Thrombotic disease in systemic lupus erythematosus is associated with a maintained systemic platelet activation. Br. J. Haematol. 2004;125:74–78. doi: 10.1111/j.1365-2141.2004.04858.x. [DOI] [PubMed] [Google Scholar]
- 7.Hamad OA, Ekdahl KN, Nilsson PH, Andersson J, Magotti P, Lambris JD, Nilsson B. Complement activation triggered by chondroitin sulfate released by thrombin receptor-activated platelets. J. Thromb. Haemost. 2008;6:1413–1421. doi: 10.1111/j.1538-7836.2008.03034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Del Conde I, Crúz MA, Zhang H, López JA, Afshar-Kharghan V. Platelet activation leads to activation and propagation of the complement system. J. Exp. Med. 2005;201:871–879. doi: 10.1084/jem.20041497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peerschke EI, Yin W, Grigg SE, Ghebrehiwet B. Blood platelets activate the classical pathway of human complement. J. Thromb. Haemost. 2006;4:2035–2042. doi: 10.1111/j.1538-7836.2006.02065.x. [DOI] [PubMed] [Google Scholar]
- 10.Nicholson-Weller A, Spicer DB, Austen KF. Deficiency of the complement regulatory protein, “decay-accelerating factor,” on membranes of granulocytes, monocytes, and platelets in paroxysmal nocturnal hemoglobinuria. N. Engl. J. Med. 1985;312:1091–1097. doi: 10.1056/NEJM198504253121704. [DOI] [PubMed] [Google Scholar]
- 11.Yu GH, Holers VM, Seya T, Ballard L, Atkinson JP. Identification of a third component of complement-binding glycoprotein of human platelets. J. Clin. Invest. 1986;78:494–501. doi: 10.1172/JCI112601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morgan BP. Isolation and characterization of the complement-inhibiting protein CD59 antigen from platelet membranes. Biochem. J. 1992;282:409–413. doi: 10.1042/bj2820409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sims PJ, Wiedmer T. The response of human platelets to activated components of the complement system. Immunol. Today. 1991;12:338–342. doi: 10.1016/0167-5699(91)90012-I. [DOI] [PubMed] [Google Scholar]
- 14.Tenner AJ, Lesavre PH, Cooper NR. Purification and radio-labeling of human C1q. J. Immunol. 1981;127:648–653. [PubMed] [Google Scholar]
- 15.Hammer CH, Wirtz GH, Renfer L, Gresham HD, Tack BF. Large scale isolation of functionally active components of the human complement system. J. Biol. Chem. 1981;256:3995–4006. [PubMed] [Google Scholar]
- 16.Fearon DT. Purification of C3b inactivator and demonstration of its two polypeptide chain structure. J. Immunol. 1977;119:1248–1252. [PubMed] [Google Scholar]
- 17.Nilsson UR, Mueller-Eberhard HJ. Isolation of βIF-globulin from human serum and its characterization as the fifth component of complement. J. Exp. Med. 1965;122:277–298. doi: 10.1084/jem.122.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Isenman DE, Kells DI, Cooper NR, Müller-Eberhard HJ, Pangburn MK. Nucleophilic modification of human complement protein C3: correlation of conformational changes with acquisition of C3b-like functional properties. Biochemistry. 1981;20:4458–4467. doi: 10.1021/bi00518a034. [DOI] [PubMed] [Google Scholar]
- 19.Katragadda M, Magotti P, Sfyroera G, Lambris JD. Hydrophobic effect and hydrogen bonds account for the improved activity of a complement inhibitor, compstatin. J. Med. Chem. 2006;49:4616–4622. doi: 10.1021/jm0603419. [DOI] [PubMed] [Google Scholar]
- 20.Nilsson UR, Storm KE, Elwing H, Nilsson B. Conformational epitopes of C3 reflecting its mode of binding to an artificial polymer surface. Mol. Immunol. 1993;30:211–219. doi: 10.1016/0161-5890(93)90050-l. [DOI] [PubMed] [Google Scholar]
- 21.Wouters D, Wiessenberg HD, Hart M, Bruins P, Voskuyl A, Daha MR, Hack CE. Complexes between C1q and C3 or C4: novel and specific markers for classical complement pathway activation. J. Immunol. Methods. 2005;298:35–45. doi: 10.1016/j.jim.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 22.McGrath FD, Brouwer MC, Arlaud GJ, Daha MR, Hack CE, Roos A. Evidence that complement protein C1q interacts with C-reactive protein through its globular head region. J. Immunol. 2006;176:2950–2957. doi: 10.4049/jimmunol.176.5.2950. [DOI] [PubMed] [Google Scholar]
- 23.Nilsson Ekdahl K, Nilsson B, Pekna M, Nilsson UR. Generation of iC3 at the interface between blood and gas. Scand. J. Immunol. 1992;35:85–91. doi: 10.1111/j.1365-3083.1992.tb02837.x. [DOI] [PubMed] [Google Scholar]
- 24.Nilsson B, Svensson KE, Inganaäs M, Nilsson UR. A simplified assay for the detection of C3a in human plasma employing a monoclonal antibody raised against denatured C3. J. Immunol. Methods. 1988;107:281–287. doi: 10.1016/0022-1759(88)90229-3. [DOI] [PubMed] [Google Scholar]
- 25.Hack CE, Paardekooper J, Smeenk RJ, Abbink J, Eerenberg AJ, Nuijens JH. Disruption of the internal thioester bond in the third component of complement (C3) results in the exposure of neodeterminants also present on activation products of C3. An analysis with monoclonal antibodies. J. Immunol. 1988;141:1602–1609. [PubMed] [Google Scholar]
- 26.Mollnes TE, Lea T, Frøland SS, Harboe M. Quantification of the terminal complement complex in human plasma by an enzyme-linked immunosorbent assay based on monoclonal antibodies against a neoantigen of the complex. Scand. J. Immunol. 1985;22:197–202. doi: 10.1111/j.1365-3083.1985.tb01871.x. [DOI] [PubMed] [Google Scholar]
- 27.Polley MJ, Nachman R. The human complement system in thrombin-mediated platelet function. J. Exp. Med. 1978;147:1713–1726. doi: 10.1084/jem.147.6.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sandvik T, Endresen GK, Førre O. Studies on the binding of complement factor C4 in human platelets. Complement activation by means of cold agglutinins. Int. Arch. Allergy Appl. Immunol. 1984;74:152–157. doi: 10.1159/000233536. [DOI] [PubMed] [Google Scholar]
- 29.Ståhl AL, Vaziri-Sani F, Heinen S, Kristoffersson AC, Gydell KH, Raafat R, Gutierrez A, Beringer O, Zipfel PF, Karpman D. Factor H dysfunction in patients with atypical hemolytic uremic syndrome contributes to complement deposition on platelets and their activation. Blood. 2008;111:5307–5315. doi: 10.1182/blood-2007-08-106153. [DOI] [PubMed] [Google Scholar]
- 30.Ward JV, Packham MA. Characterization of the sulfated glycosaminoglycan on the surface and in the storage granules of rabbit platelets. Biochim. Biophys. Acta. 1979;583:196–207. doi: 10.1016/0304-4165(79)90427-6. [DOI] [PubMed] [Google Scholar]
- 31.Okayama M, Oguri K, Fujiwara Y, Nakanishi H, Yonekura H, Kondo T, Ui N. Purification and characterization of human platelet proteoglycan. Biochem. J. 1986;233:73–81. doi: 10.1042/bj2330073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Donato JL, Nogueira MD, Marcondes S, Antunes E, Nader HB, Dietrich CP, de Nucci G. The kinetics of chondroitin 4-sulfate release from stimulated platelets and its relation to thromboxane A2 formation and granule secretion. Braz. J. Med. Biol. Res. 1994;27:2163–2167. [PubMed] [Google Scholar]
- 33.Vaziri-Sani F, Hellwage J, Zipfel PF, Sjöholm AG, Iancu R, Karpman D. Factor H binds to washed human platelets. J. Thromb. Haemost. 2005;3:154–162. doi: 10.1111/j.1538-7836.2004.01010.x. [DOI] [PubMed] [Google Scholar]
- 34.Mnjoyan Z, Li J, Afshar-Kharghan V. Factor H binds to platelet integrin αIIbβ3. Platelets. 2008;19:512–519. doi: 10.1080/09537100802238494. [DOI] [PubMed] [Google Scholar]
- 35.Wiedmer T, Hall SE, Ortel TL, Kane WH, Rosse WF, Sims PJ. Complement-induced vesiculation and exposure of membrane prothrombinase sites in platelets of paroxysmal nocturnal hemoglobinuria. Blood. 1993;82:1192–1196. [PubMed] [Google Scholar]
- 36.Bexborn F, Andersson PO, Chen H, Nilsson B, Ekdahl KN. The tick-over theory revisited: formation and regulation of the soluble alternative complement C3 convertase (C3(H2O)Bb) Mol. Immunol. 2008;45:2370–2379. doi: 10.1016/j.molimm.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schwendinger MG, Spruth M, Schoch J, Dierich MP, Prodinger WM. A novel mechanism of alternative pathway complement activation accounts for the deposition of C3 fragments on CR2-expressing homologous cells. J. Immunol. 1997;158:5455–5463. [PubMed] [Google Scholar]
- 38.Alaei S, Larcher C, Ebenbichler C, Prodinger WM, Janatova J, Dierich MP. Isolation and biochemical characterization of the iC3b receptor of Candida albicans. Infect. Immun. 1993;61:1395–1399. doi: 10.1128/iai.61.4.1395-1399.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andersson J, Ekdahl KN, Larsson R, Nilsson UR, Nilsson B. C3 adsorbed to a polymer surface can form an initiating alternative pathway convertase. J. Immunol. 2002;168:5786–5791. doi: 10.4049/jimmunol.168.11.5786. [DOI] [PubMed] [Google Scholar]
- 40.Janssen BJC, Huizinga EG, Raaijmakers HCA, Roos A, Daha MR, Nilsson-Ekdahl K, Nilsson B, Gros P. Structures of complement component C3 provide insights into the function and evolution of immunity. Nature. 2005;437:505–511. doi: 10.1038/nature04005. [DOI] [PubMed] [Google Scholar]
- 41.Evangelista V, Manarini S, Sideri R, Rotondo S, Martelli N, Piccoli A, Totani L, Piccardoni P, Vestweber D, de Gaetano G, Cerletti C. Platelet/polymorphonuclear leukocyte interaction: P-selectin triggers protein-tyrosine phosphorylation-dependent CD11b/CD18 adhesion: role of PSGL-1 as a signaling molecule. Blood. 1999;93:876–885. [PubMed] [Google Scholar]