

Abstract
Autotransporters (ATs) are the largest group of proteins secreted by Gram-negative bacteria and include many virulence factors from human pathogens. ATs are synthesized as large precursors with a C-terminal domain that is inserted in the outer membrane (OM) and is essential for the translocation of an N-terminal passenger domain to the extracellular milieu. Several mechanisms have been proposed for AT secretion. Self-translocation models suggest transport across a hydrophilic channel formed by an internal pore of the β-barrel or by the oligomerization of C-terminal domains. Alternatively, an assisted-translocation model suggests that transport employs a conserved machinery of the bacterial OM such as the Bam complex. In this work we have investigated AT secretion by carrying out a comparative study to analyze the conserved biochemical and functional features of different C-terminal domains selected from ATs of gammaproteobacteria, betaproteobacteria, alphaproteobacteria, and epsilonproteobacteria. Our results indicate that C-terminal domains having an N-terminal α-helix and a β-barrel constitute functional transport units for the translocation of peptides and immunoglobulin domains with disulfide bonds. In vivo and in vitro analyses show that multimerization is not a conserved feature in AT C-terminal domains. Furthermore, we demonstrate that the deletion of the conserved α-helix severely impairs β-barrel folding and OM insertion and thereby blocks passenger domain secretion. These observations suggest that the AT β-barrel without its α-helix cannot form a stable hydrophilic channel in the OM for protein translocation. The implications of our data for an understanding of AT secretion are discussed.
The classical autotransporter (AT) family, also known as the type Va protein secretion system, represents the largest group of proteins secreted by Gram-negative bacteria and includes many virulence factors from important human pathogens (10, 17). Bacteria produce AT proteins as large polypeptide precursors, with their virulence activity (e.g., cytotoxins, adhesins, and proteases, etc.) present in a passenger domain flanked by an N-terminal signal peptide (sp) for Sec-dependent translocation across the bacterial inner membrane (IM) and a C-terminal domain of ∼30 to 40 kDa for insertion into the bacterial outer membrane (OM) (see Fig. 2A). A self-translocation model was originally proposed to explain the secretion mechanism of AT proteins across the OM, based mostly on data obtained with the IgA protease (IgAP) from Neisseria gonorrhoeae (43). In this model the C-terminal domain of ATs was supposed to fold in the OM as a β-barrel protein with an internal hydrophilic pore that could be used for the translocation of the passenger domain. The finding that the B subunit of cholera toxin (CtxB) should not have disulfide bonds for its secretion when fused as a heterologous passenger to the C-terminal domain of IgAP (30, 31) indirectly suggests passenger translocation in an unfolded conformation through a narrow channel expected for a β-barrel. Similar observations with the C-terminal domains of IcsA from Shigella flexneri (56) and AIDA-I from Escherichia coli (36) supported this model.
Previous work done by our group challenged the original self-translocation model, since a 45-kDa C-terminal fragment of IgAP was shown to form oligomeric ring-shaped complexes with a central hydrophilic pore of ∼2 nm (63). In addition, this C-terminal fragment of IgAP was found to translocate folded immunoglobulin (Ig) domains with disulfide bonds to the bacterial surface, indicating that at least a ∼2-nm pore was being used for passenger secretion (61, 62). These data led us to propose a “multimeric” version of the self-translocation model in which the secretion of the passenger may occur through the central channel assembled by the oligomerization of the C-terminal domains in the OM. Studies with IcsA from S. flexneri (7, 46, 47, 64) and EspP from E. coli (53) also provided evidence indicating that native and heterologous passengers adopt folded or at least partially folded conformations in the periplasm before OM translocation. Conversely, a limited capacity for the translocation of folded native passengers with engineered disulfide bonds has been reported by studies with Hbp from E. coli (23) and pertactin from Bordetella pertussis (24). Crystallographic structures of the C-terminal domains of NalP from Neisseria meningitidis (41) and EspP from E. coli (2) revealed distinct β-barrel folding with 12 amphipathic β-strands and one N-terminal α-helix filling the central hydrophilic pore of the β-barrel. No indication of oligomerization was obtained with the crystallographic data. In addition, the putative protein-conducting channels of the EspP and NalP β-barrels (of ∼1 nm in diameter) were found to be closed due to the presence of the internal α-helix, which would impede the transport of passenger polypeptides (either folded or unfolded) through the reported structures. Thus, an alternative model was proposed for the assisted translocation of ATs (3, 41), in which the protein-conducting channel for secretion across the OM would be provided by the conserved Bam complex. The Bam complex is required for the insertion of β-barrel proteins (32), and the depletion of its essential component BamA (formerly YaeT in E. coli and Omp85 in Neisseria) prevents the insertion of several ATs in the OM (i.e., IcsA and SepA from S. flexneri, AIDA-I and Hbp from E. coli, and BrkA from B. pertussis) (21, 50). BamA was reported to form hydrophilic pores in lipid membranes in vitro (54) and to cross-link in vivo with the passenger domain of a slow-secretion mutant of EspP (19), which supports a role for BamA in translocation.
Despite the above-described progress made in our understanding of ATs, their actual molecular mechanism of secretion remains uncertain. This is partially because the reported information is based on studies with different model AT proteins and nonhomogenous experimental approaches used by different laboratories, which sometimes produce data that are difficult to compare or may be conflicting. Here, we report a comparative study to determine conserved biochemical and functional properties found in AT C-terminal domains. Following a uniform experimental approach for six AT C-terminal domains selected from the gammaproteobacteria, betaproteobacteria, alphaproteobacteria, and epsilonproteobacteria, we have investigated their capacities for the secretion of peptides and globular domains, their pore formation and oligomerization properties, and their requirement for an N-terminal α-helix for AT function and C-terminal domain stability. Our results shed light on the secretion mechanism of ATs from the conserved structural features found in their C-terminal domains.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The E. coli strains used in the experiments described in this work are listed in Table 1. Bacteria carrying plasmids were grown at 30°C on Luria-Bertani (LB) agar plates containing 2% (wt/vol) glucose for the repression of the Plac promoter and appropriate antibiotics for plasmid selection. Antibiotics were used at the following concentrations: chloramphenicol (Cm) at 40 μg/ml and ampicillin (Ap) at 100 μg/ml. Unless otherwise indicated, for expression, E. coli cells were grown in LB medium containing 2% (wt/vol) glucose and appropriate antibiotics overnight at 30°C without agitation. The next day, the cultures grown overnight were diluted to a final optical density at 600 nm (OD600) of 0.5 in fresh LB medium containing isopropyl-thio-β-d-galactoside (IPTG) (0.05 mM) and appropriate antibiotics, and the cultures were grown for 2 h at 30°C with gentle agitation (160 rpm). E. coli XL1-Blue was used for cloning and plasmid preparation.
TABLE 1.
E. coli strains and plasmids
| Strain or plasmid | Genotype and/or relevant properties | Reference |
|---|---|---|
| E. coli strains | ||
| UT5600 | K-12 (F− λ−) Δ(ompT-fepC)266 | 16 |
| BL21(DE3) omp8 | F′ ompT hsdSB (rB− mB−) gal dcm (DE3) ΔlamB ompF::Tn5 ΔompA ΔompC | 45 |
| XL-1 Blue | recA1 gyrA96 relA1 endA1 hsdR17 supE44 thi1 lac [F′ proAB lacIqlacZΔM15 Tn10] Tcr | 8 |
| Plasmids | ||
| pAK-Not | Cmr; lacIq-Plac promoter; pBR322-ori | 61 |
| pOmpG | Apr; OmpG with 6×His tag at C terminus; pUC-ori | 55 |
| pFosβ | pAK derivative; Fosβ fusion (pelB-FosLZ-E tag-IgAP1120-1532) | 60 |
| pHEA | pAK derivative; HEA fusion (pelB-6×His-E tag-EhaA989-1327) | This study |
| pHES | pAK derivative; HES fusion (pelB-6×His-E tag-ShdA1720-2039) | This study |
| pHEI | pAK derivative; HEI fusion (pelB-6×His-E tag-IgAP1226-1532) | This study |
| pHEN | pAK derivative; HEN fusion (pelB-6×His-E tag-NalP777-1084) | This study |
| pHEBA | pAK derivative; HEBA fusion (pelB-6×His-E tag-BruA3085-3422) | This study |
| pHEV | pAK derivative; HEV fusion (pelB-6×His-E tag-VacA915-1287) | This study |
| pJunA | pAK derivative; JunA fusion (pelB-JunLZ-E tag-EhaA989-1327) | This study |
| pJunS | pAK derivative; JunS fusion (pelB-JunLZ-E tag-ShdA1720-2039) | This study |
| pJunI | pAK derivative; JunI fusion (pelB-JunLZ-E tag-IgAP1226-1532) | This study |
| pJunN | pAK derivative; JunN fusion (pelB-JunLZ-E tag-NalP777-1084) | This study |
| pJunBA | pAK derivative; JunBA fusion (pelB-JunLZ-E tag-BruA3085-3422) | This study |
| pJundA | pAK derivative; JundA fusion (pelB-JunLZ-E tag-EhaA1034-1327) | This study |
| pJundS | pAK derivative; JundS fusion (pelB-JunLZ-E tag-ShdA1752-2039) | This study |
| pJundI | pAK derivative; JundI fusion (pelB-JunLZ-E tag-IgAP1279-1532) | This study |
| pJundN | pAK derivative; JundN fusion (pelB-JunLZ-E tag-NalP815-1084) | This study |
| pJundBA | pAK derivative; JundBA fusion (pelB-JunLZ-E tag-BruA3124-3422) | This study |
| pJunAI | pAK derivative; JunAI fusion (pelB-JunLZ-E tag-EhaA1007-1046-IgAP1285-152) | This study |
| pJunNI | pAK derivative; JunNI fusion (pelB-JunLZ-E tag-NalP786-818-IgAP1285-1532) | This study |
| pVHH-A | pAK derivative; VHH-A fusion (pelB-VHH-E tag-EhaA989-1327) | This study |
| pVHH-S | pAK-derivative; VHH-S fusion (pelB-VHH-E tag-ShdA1720-2039) | This study |
| pVHH-I | pAK derivative; VHH-I fusion (pelB-VHH-E tag-IgAP1226-1532) | This study |
| pVHH-N | pAK derivative; VHH-N fusion (pelB-VHH-E tag-NalP777-1084) | This study |
| pVHH-BA | pAK derivative; VHH-BA fusion (pelB-VHH-E tag-BruA3085-3422) | This study |
Plasmids, DNA constructs, and oligonucleotides.
The plasmids used in this study are summarized in Table 1. Oligonucleotides were synthesized by Sigma Genosys and are listed in Table 2. PCRs were performed with Vent DNA polymerase (New England Biolabs), and DNA constructs were sequenced (Secugen). Details of plasmid constructions are described below. For plasmids pHEA and pJunA, a DNA fragment corresponding to EhaA (amino acids 989 to 1327) was PCR amplified from genomic DNA of E. coli O157:H7 EDL933 stx (14, 42) with oligonucleotides H3-ALOH and SacII-ALOH (Table 2), digested with HindIII and SacII, and ligated into the same sites of the vector backbones pHEβ (63) and pJunβ (60) to give pHEA and pJunA, respectively. For plasmid pJundA, a DNA fragment corresponding to EhaA (amino acids 1034 to 1327) was PCR amplified from pHEA with oligonucleotides ALOH-D1 and pAK-direct, digested with HindIII and SacII, and ligated into same sites of the pJunA vector backbone. For plasmids pHES and pJunS, a DNA fragment corresponding to ShdA (amino acids 1720 to 2039) was PCR amplified from genomic DNA of Salmonella enterica serovar Typhimurium LT2 with oligonucleotides Sac2tromShdA and H3ShdA, digested with HindIII and SacII, and ligated into the same sites of the pHEβ or pJunβ vector backbone to give pHES and pJunS, respectively. For plasmid pJundS, a DNA fragment corresponding to ShdA (amino acids 1752 to 2039) was PCR amplified from pHES with oligonucleotides ShdA-D1 and H3ShdA, digested with HindIII and SacII, and ligated into same sites of the pJunA vector backbone. For plasmids pHEI and pJunI, a DNA fragment corresponding to IgAP (amino acids 1226 to 1532) was PCR amplified from pHEβ with oligonucleotides Sac2tromIgAP and pAK-direct, digested with HindIII and SacII, and ligated into the same sites of the pHEβ or pJunβ vector backbone to give pHEI and pJunI, respectively. For plasmid pJundI, a DNA fragment corresponding to IgAP (amino acids 1279 to 1532) was PCR amplified from pHEI with oligonucleotides IgAP-D2 and pAK-direct, digested with HindIII and SacII, and ligated into the same sites of the pJunA vector backbone. For plasmids pHEN and pJunN, a DNA fragment corresponding to NalP (amino acids 777 to 1084) of N. meningitidis MC58 was PCR amplified from pC-AspA (57) with oligonucleotides Sac2tromNP and H3NP, digested with HindIII and SacII, and ligated into the same sites of the pHEβ or pJunβ vector backbone to give pHEN and pJunN, respectively. The AspA and NalP C-terminal sequences are identical. For plasmid pJundN, a DNA fragment corresponding to NalP (amino acids 815 to 1084) was PCR amplified from pHEN with oligonucleotides NalP-D1 and H3NP, digested with HindIII and SacII, and ligated into same sites of the pJunA vector backbone. For plasmids pHEBA and pJunBA, a DNA fragment corresponding to BruA (amino acids 3085 to 3422) was PCR amplified from the genomic DNA of Brucella abortus strain 2308 (9) with oligonucleotides H3BA and SacII-tromBA, digested with HindIII and SacII, and ligated into the same sites of the pHEβ or pJunβ vector backbone. For plasmid pJundBA, a DNA fragment corresponding to BruA (amino acids 3124 to 3422) was PCR amplified from pHEBA with oligonucleotides BruA-D1 and pAK-direct, digested with HindIII and SacII, and ligated into the same sites of the pJunA vector backbone. For plasmid pHEV, a DNA fragment corresponding to VacA (amino acids 915 to 1287) was PCR amplified from pT1-VacA (40) with oligonucleotides Sac2tromVacA and VacA-NotI, digested with SacII and NotI, and ligated into the XbaI and NotI sites of the vector backbone pMTb (58) along with a third DNA fragment obtained from SacII- and XbaI-digested pHEβ that contains the ribosome binding site (RBS), E tag, and His tag. For plasmid pJunAI, two DNA fragments, one corresponding to the EhaA α-helix (amino acids 1007 to 1046), PCR amplified from pHEA with oligonucleotides ALOH-IgAP1 and ALOH-IgAP2, and a second one corresponding to the IgAP β-barrel (amino acids 1285 to 1532), PCR amplified from pHEI with oligonucleotides ALOH-IgAP3 and H3-IgAP, were assembled by fusion PCR. The assembled DNA fragment was subsequently PCR amplified with oligonucleotides ALOH-IgAP1 and H3-IgAP, digested with HindIII and SacII, and ligated into the same sites of the pJunA vector backbone. For plasmid pJunNI, two DNA fragments, one corresponding to the NalP α-helix (amino acids 786 to 818), PCR amplified from pHEN with oligonucleotides NalP-IgAP1 and NalP-IgAP2, and a second one corresponding to the IgAP β-barrel (amino acids 1285 to 1532), PCR amplified from pHEI with oligonucleotides NalP-IgAP3 and pAK-direct, were assembled by fusion PCR. The assembled DNA fragment was subsequently PCR amplified with oligonucleotides NalP-IgAP1 and pAK-direct, digested with HindIII and SacII, and ligated into the same sites of the pJunA vector backbone. For plasmids pVHH-A, pVHH-S, pVHH-I, pVHH-N, and pVHH-BA, a DNA fragment corresponding to a variable domain of heavy-chain-only (VHH) nanobody against human fibrinogen selected by phage display (our unpublished data) was digested with SfiI and NotI from the phagemid vector and cloned into the same sites of the pHEA, pHES, pHEI, pHEN, and pHEBA vectors, replacing their 6×His tag sequences.
TABLE 2.
List of oligonucleotides
| Oligonucleotide | Sequence |
|---|---|
| ALOH-D1 | 5′-TCCCCGCGGACCGACATGGTGACGGGTGAG-3′ |
| ALOH-IgAP1 | 5′-TCCCCGCGGGGTAGCTACATTGCGAACCTTGC-3′ |
| ALOH-IgAP2 | 5′-TGACATCCAAACACTGTTCATAGTGGTTTGTTTCTGCTCACC-3′ |
| ALOH-IgAP3 | 5′-AACAGTGTTTGGATGTCAAAC-3′ |
| BruA-D1 | 5′-GATTCCCCGCGGGTCTGGGCCAAGCACCCTGTGCCTG-3′ |
| H3-ALOH | 5′-GTCAAGCTTTCAGAATTGCCACTTAATGCC-3′ |
| H3-IgAP | 5′-GTCAAGCTTGAGTTCAGACGGCATTTTATT-3′ |
| H3BA | 5′-CGGGCAGGAAGCTTCTACCAACGTACGCGCAGACCCACA-3′ |
| H3NP | 5′-CGGGCAGGAAGCTTTCAGAACCGGTAGCCTACGCCGACT-3′ |
| H3-ShdA | 5′-CGGGCAGGAAGCTTCCGCCCGGTTTTGTCTAAC-3′ |
| IgAP-D2 | 5′-TCCCCGCGGCGCGCTGATGCCGAAAAAAAC-3′ |
| NalP-D1 | 5′-TCCCCGCGGAACGGCACGGGTCTGCGCGTC-3′ |
| NalP-IgAP1 | 5′-TCCCCGCGGGCCGCTACCGTCTATGCCGAC-3′ |
| NalP-IgAP2 | 5′-GGTGTTTGACATCCAAACACTGTTACCCGTGCCGTTGTGGTCCAAC-3′ |
| NalP-IgAP3 | 5′-AACAGTGTTTGGATGTCAAACACC-3′ |
| pAK-direct | 5′-GTGACGCAGTAGCGGTAAACGGCAGAC-3′ |
| SacII-ALOH | 5′-GTTCCCCGCGGACACCTACGCCGGGTCCGGAT-3′ |
| Sac2tromBA | 5′-GTTCCCCGCGGCTGGTTCCGCGTGGTTCCCCGACGGACCCCGAAACACCGCTTTA-3′ |
| Sac2tromIgAP | 5′-GTTCCCCGCGGCTGGTTCCGCGTGGTTCCTTACAACCAAGAGCCGCGCAG-3′ |
| Sac2tromNP | 5′-GTTCCCCGCGGCTGGTTCCGCGTGGTTCCGACGGTGTACGCATCTTCAACAGTCT-3′ |
| Sac2tromShdA | 5′-GTTCCCCGCGGCTGGTTCCGCGTGGTTCCGGCGGCGATGTCGCACCGCAGTACCG-3′ |
| Sac2tromVacA | 5′-GTTCCCCGCGGCTGGTTCCGCGTGGTTCCAGCCATGATGCGGGTTATGCC-3′ |
| ShdA-D1 | 5′-TCCCCGCGGCGCGAGGGCAGCCAGTATCGT-3′ |
| VacA-NotI | 5′-GAGTCATTCTGCGGCCGCTTAGAAACTATACCTCATTCCTAAATTGG-3′ |
SDS-PAGE and Western blots.
Whole-cell protein extracts were prepared by mixing 100 μl of a suspension of induced bacteria (OD600 of 15.0) in 10 mM Tris-HCl (pH 8.0) with the same volume of SDS sample buffer (2×). The SDS sample buffer (1×) consists of 60 mM Tris-HCl (pH 6.8), 1% (wt/vol) SDS, 5% (vol/vol) glycerol, 0.005% (wt/vol) bromophenol blue, and 1% (vol/vol) 2-mercaptoethanol (2-ME). For immunoblotting, the proteins were separated by SDS-PAGE using the Miniprotean III electrophoresis system (Bio-Rad) and transferred onto a polyvinylidene difluoride (PVDF) membrane (Immobilon-P; Millipore) using a semidry electrophoresis transfer apparatus (Bio-Rad). For immunodetection of the E-tagged proteins, blots were incubated for 1 h at room temperature with an anti-E tag monoclonal antibody (MAb) (0.5 μg/ml; Phadia), followed by an anti-mouse peroxidase (POD) conjugate (1:5,000; Sigma). Maltose binding protein (MBP) was detected with anti-MBP (1:5,000; New England Biolabs), and OmpA was detected with specific rabbit polyclonal serum (1:5,000; a gift from Hiroshi Nikaido). Bound rabbit antibodies were detected with a protein A-POD conjugate (1:5,000; Zymed). Membranes were blocked, washed, and developed as previously described (25, 62).
Protease accessibility assays.
Bacteria were harvested by centrifugation and resuspended in 100 μl of 10 mM Tris-HCl (pH 8.0) to a final OD600 of 15.0. This bacterial suspension was incubated with trypsin (Sigma) at 10 μg/ml for 20 min at 37°C. Next, the trypsin inhibitor (Sigma) was added at 5 μg/ml to stop further proteolysis. The cell suspension was centrifuged (4,000 × g for 3 min), and the cell pellet was resuspended in 100 μl of 10 mM Tris-HCl (pH 8.0) and mixed with the same volume of SDS sample buffer (2×). Finally, the sample was boiled for 10 min, briefly sonicated (5 s) (Labsonic B; Braun), and centrifuged (14,000 × g for 5 min) to remove insoluble material before loading onto an SDS-PAGE gel.
Flow cytometric analysis.
Bacterial colonies were grown in LB medium containing 0.05 mM IPTG and the corresponding antibiotics overnight at 30°C without agitation. The next day, bacterial cells (equivalent to a final OD600 of 1.0) were harvested from the cultures, washed with phosphate-buffered saline (PBS), resuspended in 1 ml of PBS containing 5% (vol/vol) goat serum, and incubated for 30 min at room temperature. After the blocking step, an anti-E tag MAb (10 μg/ml; Phadia) was added to the cell suspension and incubated for 60 min. Next, bacteria were washed three times with 1 ml of PBS and resuspended in 0.5 ml of PBS-5% goat serum containing Alexa 488-conjugated anti-mouse IgG (1:1,000; Molecular Probes). After an additional incubation for 30 min at room temperature in the dark, the cells were washed twice with PBS and resuspended in 1 ml of PBS. For each experiment, at least 10,000 cells were analyzed with a cytometer (Coulter Epics XL-MCL; Beckman Coulter) using 488 nm as the excitation wavelength.
ELISAs.
Enzyme-linked immunosorbent assays (ELISAs) were done as described elsewhere previously (61). Briefly, to detect the binding of VHH to antigen, either human fibrinogen (25 mg/ml) or bovine serum albumin (BSA) (50 mg/ml; New England Biolabs) diluted in PBS was adsorbed onto ELISA plates (Maxisorb; Nunc). Next, plates were blocked for 1 h with PBS containing 3% (wt/vol) skimmed milk, and the bacterial suspension was added at an OD600 of 3.0 in PBS and incubated for an additional hour. The plates were then washed with PBS, and the presence of bound bacteria was detected with an anti-E tag MAb (1 μg/ml; Phadia), followed by an anti-mouse POD conjugate (1:4,000; Sigma). For assaying the accessibility of the peptidoglycan (PG) of E. coli in the ELISAs, the bacterial suspension was added to uncoated plates at an OD600 of 3.0 in PBS and incubated for 1 h at room temperature. The plates were blocked with PBS containing 3% (wt/vol) skimmed milk, and the peptidoglycan was detected with anti-E. coli peptidoglycan serum (1:1,000) (48) and protein A-POD conjugate (1:5,000; Zymed). The permeabilized bacterial control corresponds to E. coli cells carrying pAK-Not (empty vector) in PBS containing 10 mM EDTA and briefly sonicated with three pulses of 10 s (Labsonic U B Braun sonicator). ELISAs were developed by using o-phenylenediamine (OPD; Sigma), and results were read at 490 nm. The ELISA values reported were from at least two independent experiments with triplicates. Graphs of mean and standard deviation values were done with Prism v5.0 (GraphPad Software, CA).
Thiol-PEGylation assays.
Disulfide bond formation in VHH fusion proteins was determined by a PEGylation assay of free thiol groups performed with high-molecular-weight polyethylene glycol maleimide (mPEG-MAL-5000; Nektar Therapeutics) as described previously (6).
Antibiotic sensitivity.
IPTG-induced bacteria bearing the corresponding plasmids were plated onto LB agar plates containing 0.05 mM IPTG (to maintain the induction of the plasmids). Discs of Whatman paper (diameter of 5 mm) with the indicated antibiotic were placed on top of the LB agar plates and incubated at 30°C for 24 h. The antibiotic sensitivity was calculated by measuring the diameter of the growth inhibition halo. The experiment was done in triplicate using independent transformants of E. coli BL21(DE3) omp8.
Purification of AT C-terminal domains.
The purification of His and E (HE)-tagged C-terminal domains was performed basically as described previously for HEβ (63), with small modifications. E. coli UT5600 cells with the corresponding plasmids were grown in 2-liter cultures of 2× yeast extract-tryptone (YT) medium (16 g/liter Bacto tryptone, 10 g/liter yeast extract, 5 g/liter NaCl) at 30°C, and when the OD600 reached ∼0.5, they were induced with 0.1 mM IPTG for 4 h. Cells were subsequently harvested by centrifugation (4,000 × g for 10 min) and resuspended in 100 ml of TN buffer (20 mM Tris-HCl [pH 8.0], 10 mM NaCl) containing DNase I (0.1 mg/ml; Roche), pancreatic RNase A (0.1 mg/ml; Amresco), 0.1 mM phenylmethylsulfonyl fluoride (PMSF), and a cocktail of protease inhibitors (Complete EDTA-free; Roche). Here, all steps were carried out at 4°C. The suspension of cells was passed through a French press (Sim-Aminco; Electronic Instruments) at 1,000 lb/in2, followed by centrifugation (4,000 × g for 10 min) to discard nonlysed cells. The supernatant was centrifuged once more (100,000 × g for 1 h) in a 50.2 Ti rotor (Beckman). The pellet, containing the cellular envelope, was resuspended in 20 ml of TN buffer containing 1.5% (vol/vol) Triton X-100 (TX-100). After 30 min of incubation, this sample was centrifuged (100,000 × g for 1 h) in a 90 Ti rotor (Beckman), and the subsequent pellet, which corresponded to the OM fraction, was resuspended in 10 ml of TN buffer containing 1% (wt/vol) Zwittergent 3-14 (Calbiochem). After further incubation for 30 min, the mixture was centrifuged (100,000 × g for 1 h) in a 90 Ti rotor (Beckman), and the supernatant, containing the solubilized OM proteins (OMPs), was diluted with TN buffer to a final concentration of 0.1% (wt/vol) Zwittergent 3-14. A 10-ml aliquot of a cobalt-containing agarose resin (50%, vol/vol) (Talon; Clontech) equilibrated in TNZ buffer (TN buffer plus 0.1% [wt/vol] Zwittergent 3-14) was then added. The resulting suspension was incubated overnight with slow agitation on a gyratory wheel to allow efficient binding of the His-tagged transporter domain. The next day, this mixture was passed through a chromatography column (Econopac; Bio-Rad) containing an additional 3 ml of the Talon resin. This column was washed with 100 ml of TNZ buffer and 20 ml of the same buffer containing 5 mM imidazole. The His-tagged transporter domain was eluted in 1-ml fractions with the same buffer containing 120 mM imidazole.
BN-PAGE.
Blue native PAGE (BN-PAGE) was performed as described previously (6), employing 4 to 15% polyacrylamide gradient gels in electrophoresis buffer (50 mM Bis-Tris-HCl [Fluka], 500 mM 6-aminocaproic acid [Sigma], 10% [vol/vol] glycerol [Merck] [pH 7.0]). Before gel loading, 20 μl of purified protein was mixed with 2.5 μl of 87% (vol/vol) glycerol and 2.5 μl Coomassie blue G-250 (5%, wt/vol; Bio-Rad) and kept on ice. The cathode buffer consisted of 50 mM Tricine (Fluka), 15 mM Bis-Tris-HCl (pH 7.0), and 0.002% (wt/vol) Coomassie blue G250. The anode buffer contained 50 mM Bis-Tris-HCl (pH 7.0) (Fluka). Protein standards of high molecular mass (66 to 669 kDa) for native electrophoresis (GE Healthcare) were resuspended at a 2.5-mg/ml final concentration in 50 mM Bis-Tris-HCl (pH 7.0) containing 750 mM 6-aminocaproic acid. A total of 5 to 10 μl of OMPs (or resuspended protein standards) was loaded per well, and electrophoresis was run with a Miniprotean III instrument (Bio-Rad) for 45 min at 100 V and for ca. 1 h at 500 V. Proteins were transferred onto PVDF membranes and incubated with anti-E tag MAb (0.5 μg/ml; Phadia) and anti-mouse POD conjugate (1:5,000; Sigma) for Western blotting.
Protein cross-linking.
Bacteria were induced with 0.1 mM IPTG for 3 h and resuspended in 1/10 of the original culture volume of PBS containing 250 mM dithiobis-succinimidyl propionate (DSP) (Pierce). Cross-linking was carried out for 30 min at room temperature and quenched with 50 mM Tris-HCl (pH 7.5) for 15 min. The cells were washed with 10 mM Tris-HCl (pH 7.5) and resuspended in the same buffer. One volume of SDS-PAGE sample buffer with or without 5% (vol/vol) 2-ME was added, and the samples were boiled for 10 min before gel loading. The absence of the reducing agent 2-ME in the SDS-PAGE sample buffer allowed the maintenance of a disulfide bridge in the cross-linker.
Bacterial aggregation assay.
Liquid cultures of IPTG-induced E. coli cells bearing pFosβ (Table 1) or the appropriate AT plasmid construct were adjusted to a final OD600 of 1.25. A volume of the bacterial culture expressing the AT was mixed with the same volume of E. coli cells bearing pFosβ and transferred into a 10-ml glass test tube. To calculate aggregation kinetics, 100-μl duplicate samples were withdrawn from the top of the tube, with intervals of 20 min, for measuring the OD600 across time. The τ parameter corresponds to the time needed to reach an OD600 of 0.4 at 1 cm from the top of the liquid surface.
Cell fractionation.
IPTG-induced bacterial cultures of 100 ml (OD600 of ∼1.5) were centrifuged, and the pellet was resuspended in 20 ml of sonication buffer (SB) (10 mM Tris-HCl [pH 8.0] with a cocktail of EDTA-free protease inhibitors [Roche]) Bacteria were lysed by sonication (Labsonic M; Sartorius Stedim), and the protein extracts were centrifuged (3,300 × g for 10 min at 4°C) to remove unlysed bacteria. Two milliliters of the soluble extract was subjected to ultracentrifugation (100,000 × g for 1 h at 4°C) to obtain the soluble and envelope fractions. An aliquot of the envelope fraction was resuspended in 2 ml of SB containing 1.5% (vol/vol) Triton X-100 (Sigma) and incubated for 30 min on ice to solubilize inner membrane proteins. Next, the sample was ultracentrifuged (100,000 × g for 1 h at 4°C) to obtain the soluble Triton X-100 fraction. The resulting pellet was resuspended in 2 ml of SB containing 4 M urea, incubated for 30 min on ice, and ultracentrifuged (100,000 × g for 1 h at 4°C). The solubilized proteins correspond to the urea fraction. The pellet corresponding to integral OMPs was resuspended in 2 ml of SB.
RESULTS
Selection of AT C-terminal domains from proteobacteria and structural predictions.
At least one AT from each class of proteobacteria that includes human pathogens was chosen (i.e., gamma-, beta-, alpha-, and epsilonproteobacteria), combining both poorly characterized and well-characterized ATs. The selected ATs are shown in Table 3 and are briefly described here. Since their expression was planned using E. coli K-12, the host strain most frequently employed for the heterologous expression of ATs, we included one AT from a pathogenic E. coli strain, the adhesin EhaA from enterohemorrhagic E. coli (EHEC) O157:H7 strain EDL933 (65), and another AT from a different gammaproteobacterial species, the adhesin ShdA from Salmonella enterica serovar Typhimurium strain LT2 (28, 29). From the betaproteobacteria, we chose IgAP from Neisseria gonorrhoeae strain MS11, our model AT in previous studies, and NalP from Neisseria meningitidis strain MC58, since the crystal structure of its C-terminal domain is known (41). From the alphaproteobacteria, we selected one uncharacterized AT predicted for the genome of Brucella abortus strain 2308 (9), which we named BruA. Finally, from the epsilonproteobacteria, the cytotoxin VacA from Helicobacter pylori strain 60190 was selected (15, 52). An in silico analysis of the amino acid sequences of these ATs with the protein secondary structure predictors PSI-pred (22), Pred-TMBB (1), and ProfTMB (4) allowed us to predict the putative transporter C-terminal domains of EhaA, ShdA, IgAP, BruA, and VacA as having one α-helix followed by 12 amphipathic β-strands (Fig. 1 A). In contrast to the amphipathicity of the β-strands, the predicted α-helices do not have amphipathic characteristics, except in the case of VacA (data not shown). Thus, despite low homology between their primary sequences, their secondary structures appear to be conserved, suggesting an overall three-dimensional (3D) structure similar to that of NalP. Indeed, tertiary-structure predictions with the Genesilico Metaserver-meta2 server (34) originated 3D models of EhaA, ShdA, IgAP, BruA, and VacA resembling the crystal structure of the C-terminal domain of NalP (Fig. 1B).
TABLE 3.
Selected AT C-terminal domainsa
| Bacterial strain | Class | AT | GenBank accession no. | AT size (aa) | Function | C-terminal domain size (aa) |
|---|---|---|---|---|---|---|
| Escherichia coli O157:H7 EDL933 | Gammaproteobacteria | EhaA | Q7AH89 | 1,327 | Adhesin | 339 |
| Salmonella enterica serovar Typhimurium LT2 | Gammaproteobacteria | ShdA | AAL21407 | 2,039 | Adhesin | 320 |
| Neisseria gonorrhoeae MS11 | Betaproteobacteria | IgAP | CAA28538 | 1,532 | Protease | 307 |
| Neisseria meningitidis MC58 | Betaproteobacteria | NalP | AAN71715 | 1,083 | Protease | 308 |
| Brucella abortus 2308 | Alphaproteobacteria | BruA | Q576H6 | 3,422 | Unknown | 338 |
| Helicobacter pylori 60190 | Epsilonproteobacteria | VacA | AAA17657 | 1,287 | Cytotoxin | 373 |
aa, amino acids.
FIG. 1.

Secondary- and tertiary-structure predictions of the selected AT C-terminal domains. (A) Alignment of the amino acid sequences of the C-terminal domains of the EhaA, ShdA, IgAP, NalP, BruA, and VacA ATs. The sequence predicted by the PSI-pred program (22) to fold as a hydrophilic α-helix is represented in red. The transmembrane amphipathic β-strands predicted with the Pred-TMBB (1) and ProfTMB (4) programs are shown in yellow. For clarity, the regions adopting α-helix or β-strand structures are also indicated with a cylinder or arrows on top, respectively. The roman numerals inside the arrows indicate the numbers of the β-strand (I to XII). Predicted inner loops (IL) and outer loops (OL) are labeled. Symbols indicating identity and similarity in the sequence alignment, according to the Tcoffe server (44), are indicated: *, 100% identity; :, >60% similarity; ·, >40% similarity. The conserved amino acid residues shown in boldface type correspond to those conserved in the alignment of the autotransporter family (Pfam accession number PF03797) from the Pfam server (12). (B) 3D structures of the C-terminal domains of selected ATs obtained with the Genesilico Metaserver-meta2 server (34). The secondary and tertiary structures of C-NalP correspond to the crystal structure data for NalP (41).
Expression of selected AT C-terminal domains in E. coli.
DNA fragments encoding the C-terminal domain of the selected ATs, as shown in Fig. 1A, were cloned into vector pHEβ, replacing the 45-kDa C-terminal region of IgAP found in this vector (63). The plasmids obtained, named pHEA (C-terminal domain of EhaA), pHES (C-terminal domain of ShdA), pHEI (C-terminal domain of IgAP), pHEN (C-terminal domain of NalP), pHEBA (C-terminal domain of BruA), and pHEV (C-terminal domain of VacA), encode polypeptides containing the N-terminal signal peptide (sp) of PelB (27), followed by the His tag (H) and E tag (E) epitopes and their respective C-terminal domains (Fig. 2 A). The expression of these HE-tagged C-terminal domains was induced with IPTG in E. coli K-12 strain UT5600 (Table 1), which is defective in OmpT protease (16). Western blot analysis of whole-cell protein extracts obtained upon induction showed that all HE-tagged C-terminal domains except HEV, the product of pHEV, had a high-mobility band in SDS-PAGE gels when protein samples were not boiled (Fig. 2B). These high-mobility bands are a characteristic of folded OMPs with a β-barrel structure, which are frequently resistant to SDS denaturation at room temperature (33). Therefore, these data indicate that all selected AT C-terminal domains, except that of VacA, are able to fold as SDS-resistant β-barrels in E. coli. However, a fraction of HES (ca. 2%), HEI (ca. 10%), HEN (ca. 50%), and HEBA (ca. 50%) was found in bands of low mobility in the unboiled samples (Fig. 2B), indicating a degree of sensitivity to SDS denaturation and/or the actual existence of a fraction of these polypeptides that do not fold correctly in vivo.
FIG. 2.

Expression of the selected AT C-terminal domains in E. coli K-12. (A) Schematic representation of a native AT, bearing the passenger domain and the C-terminal domain, and of the HE-tagged C-terminal domain AT constructs (HE-AT) containing the pelB signal peptide (sp) followed by the His tag (H) and E tag (E) epitopes and the corresponding AT C-terminal domain. (B) Western blot probed with anti-E tag MAb of whole-cell protein extracts obtained from IPTG-induced E. coli UT5600 cells expressing the HE-ATs indicated at the top. Prior to lysis, intact bacteria were incubated with (+) or without (−) trypsin (10 μg/ml), as indicated. The samples were boiled (+) or not (−) before loading onto the 10% SDS-polyacrylamide gel. The protein bands with faster mobility correspond to the folded conformation of the polypeptides, and the bands with slower mobility correspond to the unfolded conformation. The mass of protein standards is shown on the left (in kDa). Images shown here were obtained with different exposure times to reveal bands of approximately similar intensities for all AT C-terminal domains, although the intensity of the protein bands of HEA and HES was found to be ∼10-fold higher than those of other HE-ATs, according to quantification with a Chemi-doc device (Bio-Rad). (C) Flow cytometry analysis of E. coli UT5600 cells expressing the indicated HE-ATs, or bearing a control plasmid (pAK-Not), with anti-E tag MAb and secondary anti-mouse IgG-Alexa 488-conjugated antibody. The fluorescence intensities of bacteria carrying the indicated plasmids are shown in the histograms.
The translocation of the HE epitopes to the bacterial surface by the selected AT C-terminal domains was first evaluated by monitoring their sensitivity to trypsin protease added exogenously to intact bacteria. Western blot analysis of trypsin-treated bacteria (Fig. 2B, third lane for each construct) revealed complete, or almost complete, digestion of the HE epitopes in all constructs except for HEV, indicating that the C-terminal domain of VacA is not functional in E. coli. Low-intensity trypsin-resistant bands were observed only for HES and HEBA, indicating that a small fraction of these polypeptides are not exposed on the bacterial surface. Translocation was also evaluated by flow cytometry analysis in which induced bacteria were incubated with an anti-E tag MAb and a fluorophore-labeled anti-mouse antibody. This experiment (Fig. 2C) showed the presence of the HE epitopes on the surface of bacteria expressing HEA, HES, HEI, HEN, and HEBA but not in bacteria expressing HEV, which have fluorescence signals similar those of the negative-control bacteria carrying an empty vector (pAK-Not). The mean intensities of the fluorescence signals obtained (Fig. 2C) indicated different levels of expression of each construct on the surface of E. coli, i.e., HEA > HES > HEI ∼ HEN > HEBA (>10-fold higher for HEA than for HEBA). Finally, induced bacteria were subjected to ELISA with an anti-peptidoglycan (PG) serum, demonstrating the integrity of the bacterial OM (data not shown). Taken together, these results indicate that the selected C-terminal sequences of EhaA, ShdA, IgAP, NalP, and BruA are sufficient to fold and function as actual AT C-terminal domains in E. coli, translocating peptides to the bacterial surface. On the contrary, the C-terminal domain of VacA does not fold correctly in E. coli and is unable to function in peptide transport. As a consequence, the C-terminal domain of VacA was not used further in this study.
Translocation of globular Ig domains with disulfide bonds.
We have previously reported the translocation of Ig domains with disulfide bonds from the variable domain of heavy-chain-only (VHH) antibodies fused to the 45-kDa C-terminal region of IgAP to the E. coli surface (62), but this transport capacity has not been evaluated for other AT C-terminal domains. VHHs have higher solubility and are less prone to aggregation than standard VH or VL domains (e.g., from mouse or human origin) while maintaining conserved structural features of Ig domains (e.g., globular β-sandwich of ca. 2 nm in diameter with an intradomain disulfide bond) (38, 39). Thus, a VHH domain was cloned, replacing the His tag epitope in all HE-tagged vectors, and the resulting constructs (named VHH-A, VHH-S, VHH-I, VHH-N, and VHH-BA) were expressed in E. coli UT5600 cells to analyze their folding and display on the bacterial surface. These experiments showed that the VHH fusions have the expected mobility shift in SDS-PAGE gels of proteins containing folded β-barrels (Fig. 3 A) and have their VHH domains fully accessible to trypsin digestion (Fig. 3A, third lane of each construct), except for VHH-BA, which is only partially digested by the protease. Flow cytometry analysis (Fig. 3B) demonstrated the display of these constructs on the cell surface, with VHH-A having the highest display level and VHH-BA having the lowest display level. A whole-cell ELISA developed with anti-PG serum gave similar signals for bacteria expressing VHH-AT constructs or carrying the empty vector (pAK-Not), indicating that the integrity of the OM is not altered (data not shown).
FIG. 3.

Surface display in E. coli of functional Ig VHH domains with disulfide bonds fused to AT C-terminal domains. (A) Western blot probed with anti-E tag MAb of whole-cell protein extracts obtained from IPTG-induced E. coli UT5600 cells expressing the VHH-ATs indicated at the top. Intact bacteria were incubated with (+) or without (−) trypsin (10 μg/ml), as indicated. The samples were boiled (+) or not (−) before loading onto a 10% SDS-polyacrylamide gel. The masses of protein standards are shown on the left (in kDa). (B) Flow cytometry analysis of E. coli UT5600 cells expressing the indicated VHH-ATs or bearing a control plasmid (pAK-Not) incubated with anti-E tag MAb and secondary anti-mouse IgG-Alexa 488-conjugated antibody. (C) ELISA to determine the antigen binding activity of the exposed VHH on E. coli UT5600 cells. Induced bacteria expressing the indicated VHH-AT fusion or bearing the empty vector (pAK-Not) were bound to ELISA plates coated with human fibrinogen (black bars) or BSA as a control (white bars). Binding signals in the ELISA developed with the anti-E tag MAb are shown. (D) ELISA (as in C) in which the VHH-A fusion was induced in wild-type (wt) E. coli UT5600 or its isogenic dsbA strain. (E) Western blot developed with anti-E tag MAb of wild-type and dsbA bacteria expressing the VHH-A fusion that were incubated (+) or not (−) with mPEG-MAL-5000 and DTT (as indicated) to analyze disulfide bond formation in the translocated VHH. The band corresponding to the PEGylated VHH-A fusion is labeled with an arrow.
The folding of the displayed VHH domains was evaluated by ELISA to test their antigen binding capacities. Bacteria expressing VHH-AT fusions or carrying the empty vector were incubated with immunoplates coated with the cognate antigen of the VHH clone used (human fibrinogen) or with a negative-control antigen (BSA), and the VHH-antigen interaction was developed with an anti-E tag MAb (Fig. 3C). This experiment revealed a specific binding of the displayed VHH to its cognate antigen in all VHH-AT constructs, thus indicating the correct folding of the translocated VHH domain. The formation of an intradomain disulfide bond in Ig domains in E. coli occurs in the periplasmic space, catalyzed by the action of DsbA (26). In order to test whether DsbA plays a role in the oxidation of the translocated VHH, we chose the VHH-A fusion as a model, given its high display level and antigen binding activity. As reported previously for fusions to C-IgAP (62), the expression of VHH-A in a dsbA mutant strain strongly diminished its antigen binding activity (Fig. 3D). To obtain direct evidence of the oxidation state of the VHH, E. coli UT5600 and isogenic dsbA mutant bacteria expressing VHH-A were incubated in vivo with mPEG-MAL, which covalently PEGylates free sulfhydryl groups in proteins. PEGylated proteins have a slower mobility in nonreducing SDS-PAGE gels than the corresponding unreactive oxidized form containing disulfide bonds (35). Western blot analysis showed that in the wild-type strain, VHH-A was not PEGylated by mPEG-MAL, indicating the presence of disulfide bonds in its VHH domain (Fig. 3E, lane 3). As a control, wild-type bacteria were incubated with 100 mM dithiothreitol (DTT) at 60°C to reduce disulfide bonds prior to mPEG-MAL treatment. In this situation, a complete PEGylation of VHH-A was observed (Fig. 3E, lane 4). In contrast, in the dsbA mutant, a significant fraction of VHH-A was reactive to mPEG-MAL in vivo, forming a high-molecular-weight PEGylated band (Fig. 3E, lane 7). However, the PEGylation of VHH-A was not complete in the dsbA mutant in vivo, which could indicate a partial oxidation of the VHH by a DsbA-independent mechanism or a partial accessibility of the sulfhydryl groups of the VHH in vivo. A full PEGylation of VHH-A was obtained with this mutant under denaturing and reducing conditions (100 mM DTT at 60°C) (Fig. 3E, lane 8). These data demonstrate that AT C-terminal domains have the conserved capacity to translocate to the surface of E. coli cells' functional Ig domains with disulfide bonds formed by the action of periplasmic DsbA.
Characterization of the quaternary structures of the selected C-terminal domains.
We tested whether the expression of HEA, HES, HEI, HEN, or HEBA increased the sensitivity of E. coli to hydrophilic antibiotics, which may indicate the formation of hydrophilic channels in the OM by these C-terminal domains. An E. coli strain lacking major OM porins [BL21(DE3) omp8] (Table 1) was used as a host for the induction of HE-tagged constructs. Induced bacteria were spread onto plates containing 50 μM IPTG and discs with the hydrophilic antibiotics erythromycin (Mr, 733), bacitracin (Mr, 1,421), and vancomycin (Mr, 1,485). The expression and surface display of AT constructs in BL21(DE3) omp8 cells were confirmed by Western blotting and trypsin digestion (data not shown). The controls included bacteria of the same strain harboring the empty vector (pAK-Not) or a plasmid encoding the large monomeric porin OmpG, whose expression increased the permeability of the OM to hydrophilic antibiotics (11), grown and plated under identical conditions. The diameter of the growth inhibition halo for each construct and antibiotic is shown in Table 4. These data show that the expression of the different AT constructs does not increase significantly the permeability of the E. coli OM. On the contrary, the expression of the OmpG porin increases antibiotic sensitivity with respect to the negative control. Therefore, these data suggest either that AT C-terminal domains do not form stable hydrophilic channels in the OM or that any channels that are formed are in a “closed” conformation.
TABLE 4.
Antibiotic sensitivities of E. coli BL21(DE3) omp8 cells expressing AT C-terminal domains
| Antibiotic | Mr | Amt of antibiotic (μg/disc) | Mean inhibition halo (mm) ± SDa |
||||||
|---|---|---|---|---|---|---|---|---|---|
| pAK-Not | pOmpG | pHEA | pHES | pHEI | pHEN | pHEBA | |||
| Erythromycin | 733 | 200 | 8 ± 3 | 12 ± 3 | 7 ± 3 | 8 ± 1 | 7 ± 1 | 10 ± 2 | 9 ± 1 |
| Bacitracin | 1,421 | 500 | 6 ± 3 | 10 ± 3 | 7 ± 3 | 3 ± 2 | 5 ± 1 | 8 ± 4 | 6 ± 3 |
| Vancomycin | 1,485 | 200 | 8 ± 2 | 13 ± 0 | 7 ± 0 | 5 ± 6 | 7 ± 4 | 9 ± 1 | 7 ± 2 |
The growth inhibition zone, caused by sensitivity to the antibiotic, was measured in mm from the rim of the disc. Values are the means of data from three independent experiments.
To gain some evidence of the oligomeric state of the AT C-terminal domains in vivo, we performed chemical cross-linking experiments by the incubation of intact E. coli UT5600 cells expressing the HE-tagged constructs with dithiobis-succinimidyl propionate (DSP), a bifunctional amine-reactive N-hydroxysuccinimide (NHS) ester with a spacer arm of 12 Å containing a disulfide bond that can be cleaved by incubation with 2-mercaptoethanol (2-ME). After incubation with DSP, bacterial suspensions were mixed with either reducing or nonreducing SDS-PAGE sample buffer, boiled, and subjected to Western blotting with anti-E tag MAbs. This experiment revealed that only HES was cross-linked by DSP, forming a protein band of the expected size for a dimer (Fig. 4 A). On the contrary, all other HE constructs were very ineffectively cross-linked (HEA and HEBA) or not cross-linked at all (HEI and HEN). In the case of HEA and HEBA, although most of the produced protein remained as monomers, faint protein bands of high molecular weight were observed after film overexposure (Fig. 4A). According to their mobility, these minor bands could correspond to dimers (ca. 80 to 90 kDa) and other larger oligomeric forms of HEA and HEBA (e.g., trimers and tetramers). As expected for DSP, cross-linked protein bands were reverted by boiling in SDS sample buffer containing 2-ME (Fig. 4B). Thus, the predominant band of the selected AT C-terminal domains detected by in vivo DSP cross-linking corresponds to their monomeric form.
FIG. 4.
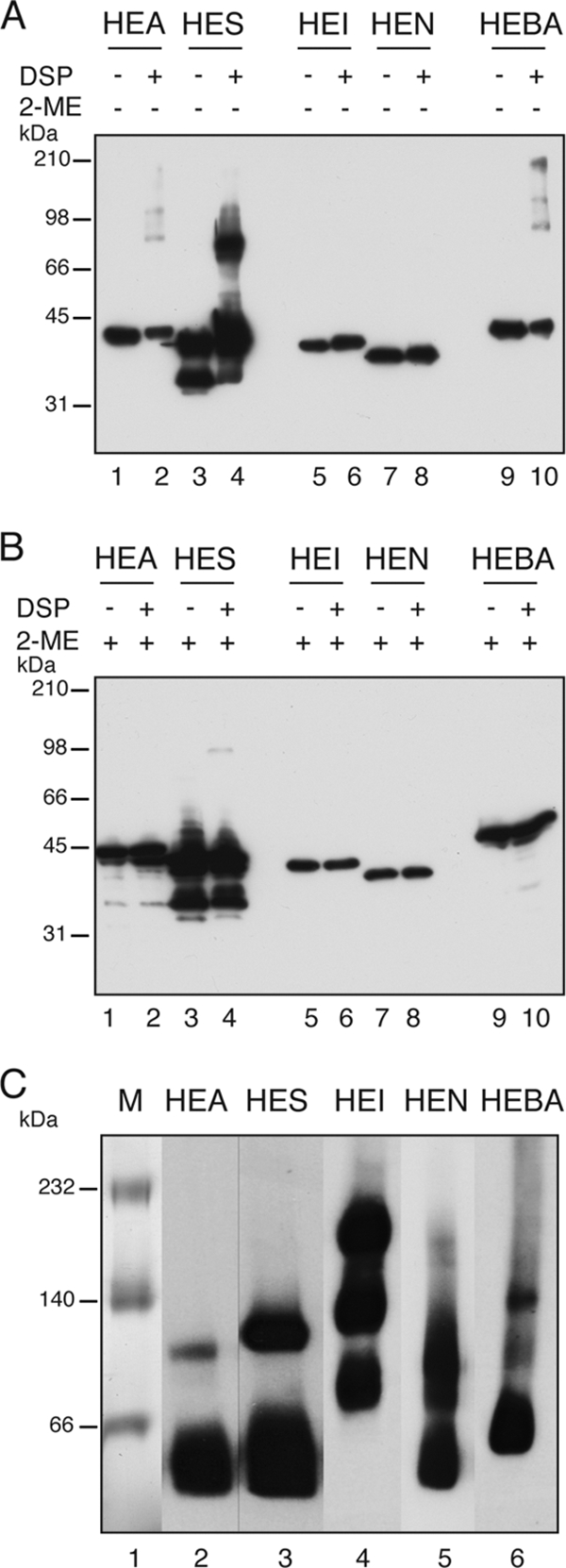
Analysis of the quaternary structure of the AT C-terminal domains in vivo and in vitro. (A and B) Cross-linking with DSP of HE-ATs expressed in E. coli to determine the formation of oligomeric complexes of AT C-terminal domains in vivo. DSP-treated (+) and untreated (−) samples were subjected to nonreducing SDS-PAGE, and the Western blot was probed with anti-E tag MAb. Samples incubated with the reducing agent 2-ME are indicated (+). (C) Blue native PAGE of purified HE-ATs from the outer membrane of E. coli bacteria. The polypeptides were detected by Western blotting with an anti-E tag MAb. The masses of protein standards are shown on the left (in kDa).
Since the above-described results do not rule out the existence of oligomers that do not react to the cross-linker in vivo, we analyzed the capacity of the selected C-terminal domains to oligomerize in vitro. The HE-tagged polypeptides were expressed in E. coli UT5600 cells, solubilized under native conditions from the OM fraction, and purified by metal affinity chromatography by means of their His tag epitope. The folding of the purified HE-tagged polypeptides was evaluated by their mobility shift in SDS-PAGE gels (data not shown). Next, the oligomeric state of the purified proteins was assessed by blue native PAGE (BN-PAGE) (51). To increase the sensitivity of this assay, proteins were transferred onto a PVDF membrane for immunodetection with an anti-E tag MAb after BN-PAGE. As shown in Fig. 4C, all the AT C-terminal domains except that of IgAP (HEI) exist predominantly as monomers in vitro, although bands with the mobility expected for dimers were detectable in all of them and were especially abundant in the case of HES, which supports the dimerization of HES detected by in vivo cross-linking (Fig. 4B). In agreement with the data reported previously for the crystal of the transporter domain of NalP (41), HEN was found mostly in the monomeric form, although a putative dimer was also detected by BN-PAGE. Finally, in the case of the C-terminal domain of IgAP (HEI), three protein bands with estimated molecular masses of ∼200 kDa, ∼120 kDa, and ∼80 kDa were detected with similar intensities, but no band with the expected size of the monomer was found (Fig. 4C). The protein band with the highest molecular weight of HEI could correspond to a hexamer, whereas the other bands may be intermediate oligomeric forms. The formation of large oligomers of HEI in vitro contrasts with the absence of DSP-cross-linked bands of HEI in vivo but agrees with the previously reported oligomerization of the 45-kDa fragment of C-IgAP (63). Thus, out of the five AT C-terminal domains analyzed, only the C-terminal domain of IgAP was found in large oligomeric forms, whereas the rest were found mostly as monomers with some capacity to dimerize in vitro.
Role of the α-helix of AT C-terminal domains in folding and transport.
The presence of an α-helix at the N terminus of the β-barrel is a conserved feature in the available X-ray crystallographic structures (2, 41, 59) and is also predicted for our selected C-terminal domains (Fig. 1). To assess the role of this α-helix, we first analyzed the folding and translocation of mutant AT C-terminal domains having α-helix deletions. To this end, the His tag epitope in the HE constructs was replaced by the leucine zipper peptide from the transcription factor c-Jun, originating the constructs JunA, JunS, JunI, JunN, and JunBA, and derivatives of these constructs with internal in-frame deletions of the predicted α-helix were obtained and named JundA, JundS, JundI, JundN, and JundBA, respectively. Previous work done by our group showed that E. coli cells displaying the Jun peptide with the 45-kDa C-terminal fragment of IgAP adhere to bacteria displaying the Fos peptide, from the leucine zipper of c-Fos, on their surface, forming large bacterial aggregates that sediment in liquid culture (60). This aggregation phenotype was used to monitor macroscopically the display of α-helix deletion constructs induced in E. coli UT5600 bacteria. As shown in Fig. 5 A, none of the α-helix deletion mutants induced bacterial aggregation, whereas all Jun wild-type constructs induced a clear bacterial aggregation of the cultures, although bacteria with the highly expressed C-terminal domains JunA and JunS exhibited shorter aggregation times (τ) than those induced by JunI, JunN, and JunBA (Fig. 5A).
FIG. 5.

Expression and cell surface translocation of α-helix deletion mutants. (A) Aggregation assay of E. coli bacteria expressing Jun fusions to the C-terminal domain of the ATs, either wild-type Jun-AT polypeptides, depicted as a small cylinder (α-helix) on top of a large cylinder (β-barrel), or the α-helix-deleted Jun-dAT mutants (large cylinder). The picture was taken at 24 h after the mixing of two independent cultures of E. coli cells expressing either Fosβ or each Jun fusion. The τ parameter is indicated in hours. NA, not applicable. (B) Western blot probed with anti-E tag MAb of whole-cell protein extracts obtained from IPTG-induced E. coli cells expressing the Jun fusion constructs indicated at the top. The samples were boiled (+) or not (−) before loading onto a 10% SDS-polyacrylamide gel. The protein bands with faster mobility correspond to the folded conformation of the polypeptides, and the band with slower mobility corresponds to the unfolded conformation. Intact bacteria were incubated with (+) or without (−) trypsin (10 μg/ml). The masses of protein standards are shown on the left (in kDa). (C) Flow cytometry analysis to determine the presence of the indicated Jun-AT fusions on the surface of E. coli cells with anti-E tag MAb. The bacterial controls and secondary antibodies used are described in the legend of Fig. 2.
Western blot analysis of whole-cell extracts demonstrated that wild-type Jun constructs had the expected heat-dependent mobility shift and sensitivity to trypsin digestion of intact bacteria, except for JunBA, which was only partially digested by trypsin, and its unfolded band was the predominant form in the unboiled sample (Fig. 5B). On the other hand, the α-helix-deleted polypeptides were expressed at lower levels in vivo than their wild-type counterparts, being JundA and JundS, expressed at ∼3-fold-lower levels, and JundI and JundN were barely detectable (Fig. 5B). The deletion of the α-helix of JunBA showed little effect on protein levels compared to the wild-type protein (Fig. 5B). Importantly, the α-helix deletions that were accumulated in vivo behave mostly as unfolded polypeptides in the heat-dependent mobility assay and showed resistance to trypsin digestion (Fig. 5B). Only JundA exhibited some residual capacity to fold as a β-barrel resistant to SDS denaturation under nonboiling conditions (Fig. 5B). Flow cytometry analysis with an anti-E tag MAb (Fig. 5C) demonstrated that the AT C-terminal domains without an α-helix were not competent for passenger (E tag) translocation to the bacterial surface, since their expression elicited fluorescence histograms similar to that of the negative control (pAK-Not). These data indicate that the observed requirement of the α-helix region for passenger translocation could be a secondary effect of its active role in the folding and stability of the AT C-terminal domain.
Analysis of chimeric AT C-terminal domains with heterologous α-helices.
In order to assess whether the expression and transport defects of an α-helix-deleted AT C-terminal domain could be rescued by a heterologous α-helix from a different AT, we produced chimeric derivatives of JunI in which the α-helix of IgAP was replaced either by the α-helix of EhaA to produce JunAI (α-helix from EhaA and β-barrel from IgAP) or by the α-helix of NalP to produce JunNI (α-helix from NalP and β-barrel from IgAP). The expression of the JunAI chimera, but not of JunNI, induced a positive-aggregation phenotype for the bacterial culture (Fig. 6 A), indicating that the Jun peptide of JunAI is accessible on the bacterial surface. Nonetheless, cultures expressing JunAI have longer aggregation times than those expressing wild-type JunI (Fig. 6A), suggesting low expression levels and/or a partial functionality of this chimera. Flow cytometry analysis demonstrated the display of the JunAI chimera on the bacterial surface (Fig. 6B) albeit at lower levels than those of wild-type JunI. Bacterial display of the JunNI chimera was not detected by flow cytometry (Fig. 6B). Western blot analysis confirmed that JunAI is expressed at lower levels than wild-type JunI, with JunNI not being detectable (Fig. 6C). JunAI is completely degraded by trypsin in intact bacteria, which also suggests the translocation of the Jun peptide to the bacterial surface (Fig. 6C). However, JunAI is sensitive to SDS denaturation at low temperatures and does not show any heat-modifiable mobility in SDS-PAGE gels (Fig. 6C), probably indicating a lower stability of its β-barrel. To assess whether the expression of JunAI affects the permeability of the OM, we tested the peptidoglycan (PG) accessibility of the induced bacteria by ELISA with anti-PG serum. This assay showed that the expression of JunAI slightly increased the detection of PG in intact compared to wild-type JunI or the α-helix deletion JundI (Fig. 6D). Nevertheless, the level of PG accessibility detected by the expression of JunAI was similar to that elicited by the expression of the α-helix deletion JundA (Fig. 6D), which produced an undetectable display of the Jun peptide in all assays (i.e., bacterial aggregation, trypsin digestion, and flow cytometry). Thus, the JunAI chimera does not induce a significant increase in the permeability of the OM that could explain its detection on the bacterial surface. Therefore, these data support the actual display of the JunAI chimera on the bacterial surface.
FIG. 6.

Expression and cell surface translocation of AT C-terminal domains with heterologous α-helices. (A) Aggregation assay of E. coli bacteria expressing the indicated Jun fusions (JunI, JunAI, and JunNI). The picture was taken at 24 h after the mixing of the indicated E. coli cultures with a culture of E. coli cells expressing Fosβ. The τ parameter is indicated in hours. NA, not applicable. (B) Flow cytometry analysis to determine the presence of the indicated Jun-AT constructs on the surface of E. coli cells with anti-E tag MAb. Bacterial controls and secondary antibodies used are described in the legends of Fig. 2 and 5. (C) Western blot probed with anti-E tag MAb of whole-cell protein extracts obtained from IPTG-induced E. coli cells expressing the indicated Jun-AT constructs. Intact bacteria were incubated with (+) or without (−) trypsin (10 μg/ml), and the samples were either boiled (+) or not (−) before loading onto a 10% SDS-polyacrylamide gel. The masses of protein standards are shown on the left (in kDa). (D) ELISA developed with anti-PG serum to test the peptidoglycan accessibility of bacteria expressing Jun-ATs or bearing the empty vector pAK-Not (black bars). EDTA-permeabilized bacteria harboring pAK-Not were used to monitor the reactivity of the anti-PG serum (white bar).
Next, the insertion of the JunAI chimera in the OM was analyzed and compared to those of JunI, JunA, and the α-helix deletion JundA (JundI was not detected in vivo) (Fig. 5B). We fractionated “soluble” and “envelope” proteins from whole-cell bacterial lysates. The inner membrane proteins and nonintegral OM proteins were then extracted from the “envelope” fraction by consecutive incubations with 1.5% TX-100 and 4 M urea. The insoluble fraction obtained after TX-100 and urea extraction contained integral OM proteins (“OMP” fraction). The presence of the different Jun polypeptides in these fractions was assessed by Western blotting with the anti-E tag MAb. As shown in Fig. 7, the JunAI chimera was present mostly in the “envelope” and “OMP” fractions and was weakly extracted by TX-100 or urea, similarly to the wild-type proteins JunI and JunA. In contrast, the α-helix deletion JundA was extracted from the “envelope” fraction by TX-100 and urea, indicating that a major portion of this deletion mutant was not properly inserted into the OM. The reliability of the fractionation method was confirmed by detecting the periplasmic maltose binding protein (MBP), which was mostly in the “soluble” fraction, and OmpA in the “envelope” and “OMP” fraction. Thus, the above-described results indicate that the JunAI chimera is properly localized in the OM. These data demonstrate that some defects caused by the deletion of the α-helix in AT C-terminal domains (e.g., transport, protein levels, and OM insertion) can be partially restored with a heterologous α-helix.
FIG. 7.
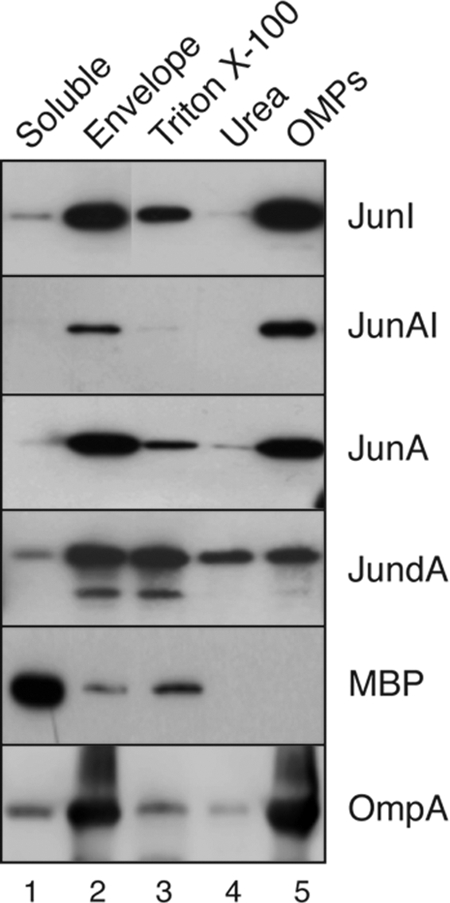
Subcellular localization of AT C-terminal domains with a deleted or heterologous α-helix. Western blots were probed with anti-E tag MAb to detect Jun-AT constructs in the indicated subcellular fractions obtained from induced E. coli cells: soluble, envelope, TX-100-solubilized, urea-solubilized, and integral OMPs (see Materials and Methods). The detection of the periplasmic MBP and OmpA with specific antibodies (indicated in the panels) was done to control the E. coli fractionation method. The masses of protein standards are shown on the left (in kDa).
DISCUSSION
In this work we have studied the structural and functional properties of six AT C-terminal domains selected from gamma-, beta-, alpha-, and epsilonproteobacteria: EhaA from EHEC O157:H7, ShdA from Salmonella enterica, IgAP from Neisseria gonorrhoeae, NalP from Neisseria meningitidis, BruA from Brucella abortus, and VacA from Helicobacter pylori. Out of them, the 3D structure of the C-terminal domain of NalP was known from crystallographic data obtained after its purification from inclusion bodies and in vitro refolding. The NalP C-terminal domain crystal structure revealed a distinct β-barrel composed of 12 amphipathic β-strands and one α-helix running through the internal hydrophilic channel (41). By in silico analysis, similar secondary and tertiary structures were predicted for the C-terminal sequences of EhaA, ShdA, IgAP, BruA, and VacA (Fig. 1). The crystal structures of the C-terminal domain of EspP from E. coli (2) and, recently, of EstA from Pseudomonas aeruginosa (59) have also shown β-barrels with 12 amphipathic β-strands and one α-helix inside the β-barrel. Therefore, AT C-terminal domains are highly conserved in their overall secondary and tertiary structures despite low similarity in their primary sequences. Our results demonstrate that the selected C-terminal domain regions (except that of VacA) are functional units for the translocation of peptides (HE and Jun) and small globular domains (Ig VHH domains) with disulfide bonds to the surface of E. coli. Domains with a deleted α-helix accumulate at low levels in vivo and are sensitive to SDS, indicating that the α-helix plays an important role in the folding and stability of the AT C-terminal domains.
The predicted AT C-terminal domains were expressed in E. coli K-12 fused to small peptides in order to minimize the influence of the passenger on the biochemical and functional properties of the polypeptides. The C-terminal domains of EhaA, ShdA, IgAP, NalP, and BruA were functional and showed expression and display levels with an inverse relationship to their phylogenetic distance from E. coli K-12. ATs from gammaproteobacteria (EhaA and ShdA) were expressed at higher levels than those of betaproteobacteria (IgAP and NalP) or alphaproteobacteria (BruA) (Fig. 2). A fraction of the BruA C-terminal domain was not displayed on E. coli K-12 cells, which may indicate a limitation for BruA recognition by the protein-folding and OM assembly factors of E. coli (e.g., SurA, DegP, and BamA) (32). The C-terminal domain of VacA, from the distant epsilonproteobacteria, was neither folded nor displayed in E. coli (Fig. 2). It was reported previously that a fusion of CtxB to a C-terminal fragment of VacA was not functional in E. coli but was correctly expressed in Helicobacter pylori (13). Thus, the phylogenetic distance between E. coli and H. pylori seems to prevent the correct folding of the VacA C-terminal domain in E. coli. Putative orthologues of E. coli SurA and BamA are found in the genome of H. pylori strain 26695 (open reading frames [ORFs] HP0175 and HP0655) albeit with low sequence identity (15% and 21%, respectively). E. coli BamA has a reported preference for the recognition of polar (but not positively charged) residues in the penultimate position of β-barrel OMPs, before the conserved aromatic amino acid (F or W) found at the C terminus (49). This preference could partially explain the lower expression levels of IgAP, NalP, and BruA in E. coli than those of EhaA and ShdA. The last two residues of IgAP (RF), NalP (RF), and BruA (RW) contain a positive charge in the penultimate position, whereas this is not the case for EhaA (QF) and ShdA (NW). However, the VacA C-terminal sequence (SF) does not contain a positively charged residue and is identical to the C terminus of E. coli OmpG. It was also reported that E. coli SurA recognizes a short motif (aromatic-X-aromatic) frequently found in the primary sequence of integral OMPs (5), but the VacA C-terminal domain contains this short motif repeated 10 times. Therefore, it is unclear to us why the VacA C-terminal domain is not folded in E. coli despite its overall structural similarities with other AT C-terminal domains and E. coli OMPs.
We found that the expression of the C-terminal domains of EhaA, ShdA, IgAP, NalP, and BruA in E. coli does not induce an increased sensitivity to large hydrophilic antibiotics, such as bacitracin and vancomycin (Table 4). These data suggest that channels formed by AT C-terminal domains in the OM are mostly in the “closed” conformation. Interestingly, in vivo cross-linking experiments revealed a dimerization of the C-terminal domain of ShdA but did not show any evidence of multimeric forms of the AT C-terminal domains analyzed (Fig. 4). Also, the purified AT C-terminal domains behave mostly as monomers and dimers in vitro, with the dimer being especially abundant in the case of ShdA. An important exception is the C-terminal domain of IgAP (HEI), which was found in dimers and multimers but mostly in tetramers and hexamers (Fig. 4C). The multimerization of HEI is in agreement with the previously reported multimerization of the 45-kDa C-terminal fragment of IgAP (63). Thus, contrary to other AT C-terminal domains, IgAP has a clear tendency to multimerize. The absence of multimeric cross-linked bands of HEI in vivo contrasts with previous cross-linking data for the 45-kDa C-IgAP fragment, which was found as multimers with the same cross-linker reagent (DSP) (63). This discrepancy could be due to the instability of multimeric HEI in vivo or by a lower reactivity of HEI due to its shorter exposed N region. It seems clear that multimerization is not a conserved feature of the AT C-terminal domains. Monomeric structures have been reported for the crystal structures of NalP, EspP, and EstA (2, 41, 59), and monomers and dimers have been detected using biochemical methods for EspP (53), AIDA-I (37), and Tsh (18).
In addition, our data indicate an important role for the conserved α-helix of ATs in the stability of the β-barrel and its OM insertion. The α-helix deletion mutants analyzed were expressed at lower levels in vivo than their wild-type counterparts and were not functional in transport. A mutant AT C-terminal domain lacking the α-helix (JundA), which was expressed in vivo at significant levels, was not inserted properly in the OM and was extracted from the envelope fractions with neutral detergents and urea (Fig. 7). Interestingly, the α-helix of EhaA can partially restore the transport function of the α-helix-deleted IgAP C-terminal domain, improving its expression level and OM insertion. The chimera JunAI is not extracted with neutral detergents and urea from the OM (Fig. 7). However, this chimeric polypeptide is sensitive to SDS, pointing out the importance of the natural α-helix for the full stability of the β-barrel. This is also emphasized by the JunNI chimera, in which the α-helix of NalP is not able to restore any of the defects of the α-helix-deleted IgAP C-terminal domain (Fig. 6). Since AT C-terminal domains without an α-helix do not fold or insert properly in the OM, it is unlikely that these unstable β-barrels could act as hydrophilic channels for the OM translocation of polypeptides. In addition, it was reported previously that the α-helix of EspP is placed inside its β-barrel before the C-terminal domain is inserted into the OM (20). Together, these data do not appear to agree with self-translocation models proposing passenger transport across the hydrophilic pore of a β-barrel. However, the possibility that the α-helical segment is first inserted into the β-barrel in an extended conformation and promotes passenger domain transport by a hairpin mechanism cannot be excluded.
Finally, we have shown that AT C-terminal domains tolerate the secretion of globular Ig domains of ∼2 nm in diameter containing disulfide bonds. Structural tolerance for folded protein domains was also reported previously for natural passengers of ATs (7, 53). Therefore, models for AT secretion should take into account these observations. In this sense, the translocation of a folded protein domain across a monomeric β-barrel by a hairpin-like mechanism appears to be a less plausible scenario than an assisted-translocation mechanism in which the Bam complex would drive OM insertion and provide the channel for passenger translocation (19, 54). A similar mechanism could also apply for intimins, which contain a surface-exposed passenger with globular Ig- and lectin-like domains containing a DsbA-dependent disulfide bond and an N-terminal region that require BamA for OM insertion (6).
Acknowledgments
We thank Gad Frankel, Francisco García del Portillo, Ignacio López-Goñi, David Turner, and Dlawer Ala'Aldeen for materials used in this study. We thank Esteban Veiga, Hilde De Reuse, and Kim Hardie for helpful comments and discussions. We thank Valencio Salema for the English editing.
This work was supported by grants to L.A.F. from the Spanish Ministry of Science (grant BIO2008-05201), the Autonomous Community of Madrid (grant S-BIO-236-2006), and the VI Framework Program from the European Union (grant FP6-LSHB-CT-2005-512061 NoE EuroPathogenomics). E.M. is a holder of a research assistant contract of the Autonomous Community of Madrid. G.B. was a holder of postdoctoral contract Juan de la Cierva from the Spanish Ministry of Science.
We declare that there is no conflict of interest. Data mentioned in the manuscript and not shown due to space limitations can be obtained upon request to L.A.F.
Footnotes
Published ahead of print on 27 August 2010.
REFERENCES
- 1.Bagos, P. G., T. D. Liakopoulos, I. C. Spyropoulos, and S. J. Hamodrakas. 2004. PRED-TMBB: a Web server for predicting the topology of beta-barrel outer membrane proteins. Nucleic Acids Res. 32:W400-W404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barnard, T. J., N. Dautin, P. Lukacik, H. D. Bernstein, and S. K. Buchanan. 2007. Autotransporter structure reveals intra-barrel cleavage followed by conformational changes. Nat. Struct. Mol. Biol. 14:1214-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bernstein, H. D. 2007. Are bacterial ‘autotransporters’ really transporters? Trends Microbiol. 15:441-447. [DOI] [PubMed] [Google Scholar]
- 4.Bigelow, H. R., D. S. Petrey, J. Liu, D. Przybylski, and B. Rost. 2004. Predicting transmembrane beta-barrels in proteomes. Nucleic Acids Res. 32:2566-2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bitto, E., and D. B. McKay. 2003. The periplasmic molecular chaperone protein SurA binds a peptide motif that is characteristic of integral outer membrane proteins. J. Biol. Chem. 278:49316-49322. [DOI] [PubMed] [Google Scholar]
- 6.Bodelón, G., E. Marín, and L. A. Fernández. 2009. Role of periplasmic chaperones and BamA (YaeT/Omp85) in folding and secretion of intimin from enteropathogenic Escherichia coli strains. J. Bacteriol. 191:5169-5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brandon, L. D., and M. B. Goldberg. 2001. Periplasmic transit and disulfide bond formation of the autotransported Shigella protein IcsA. J. Bacteriol. 183:951-958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bullock, W. O., J. M. Fernández, and J. M. Short. 1987. XL1-Blue: a high efficiency plasmid transforming recA Escherichia coli strain with beta-galactosidase selection. Biotechniques 4:376-378. [Google Scholar]
- 9.Chain, P. S., D. J. Comerci, M. E. Tolmasky, F. W. Larimer, S. A. Malfatti, L. M. Vergez, F. Aguero, M. L. Land, R. A. Ugalde, and E. Garcia. 2005. Whole-genome analyses of speciation events in pathogenic brucellae. Infect. Immun. 73:8353-8361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dautin, N., and H. D. Bernstein. 2007. Protein secretion in Gram-negative bacteria via the autotransporter pathway. Annu. Rev. Microbiol. 61:89-112. [DOI] [PubMed] [Google Scholar]
- 11.Fajardo, D. A., J. Cheung, C. Ito, E. Sugawara, H. Nikaido, and R. Misra. 1998. Biochemistry and regulation of a novel Escherichia coli K-12 porin protein, OmpG, which produces unusually large channels. J. Bacteriol. 180:4452-4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finn, R. D., J. Mistry, J. Tate, P. Coggill, A. Heger, J. E. Pollington, O. L. Gavin, P. Gunasekaran, G. Ceric, K. Forslund, L. Holm, E. L. Sonnhammer, S. R. Eddy, and A. Bateman. 2009. The Pfam protein families database. Nucleic Acids Res. 38:D211-D222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fischer, W., R. Buhrdorf, E. Gerland, and R. Haas. 2001. Outer membrane targeting of passenger proteins by the vacuolating cytotoxin autotransporter of Helicobacter pylori. Infect. Immun. 69:6769-6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garmendia, J., A. D. Phillips, M. F. Carlier, Y. Chong, S. Schuller, O. Marches, S. Dahan, E. Oswald, R. K. Shaw, S. Knutton, and G. Frankel. 2004. TccP is an enterohaemorrhagic Escherichia coli O157:H7 type III effector protein that couples Tir to the actin-cytoskeleton. Cell. Microbiol. 6:1167-1183. [DOI] [PubMed] [Google Scholar]
- 15.Gebert, B., W. Fischer, E. Weiss, R. Hoffmann, and R. Haas. 2003. Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science 301:1099-1102. [DOI] [PubMed] [Google Scholar]
- 16.Grodberg, J., and J. J. Dunn. 1988. OmpT encodes the Escherichia coli outer membrane protease that cleaves T7 RNA polymerase during purification. J. Bacteriol. 170:1245-1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henderson, I. R., F. Navarro-Garcia, M. Desvaux, R. C. Fernandez, and D. Ala'Aldeen. 2004. Type V protein secretion pathway: the autotransporter story. Microbiol. Mol. Biol. Rev. 68:692-744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hritonenko, V., M. Kostakioti, and C. Stathopoulos. 2006. Quaternary structure of a SPATE autotransporter protein. Mol. Membr. Biol. 23:466-474. [DOI] [PubMed] [Google Scholar]
- 19.Ieva, R., and H. D. Bernstein. 2009. Interaction of an autotransporter passenger domain with BamA during its translocation across the bacterial outer membrane. Proc. Natl. Acad. Sci. U. S. A. 106:19120-19125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ieva, R., K. M. Skillman, and H. D. Bernstein. 2008. Incorporation of a polypeptide segment into the beta-domain pore during the assembly of a bacterial autotransporter. Mol. Microbiol. 67:188-201. [DOI] [PubMed] [Google Scholar]
- 21.Jain, S., and M. B. Goldberg. 2007. Requirement for YaeT in the outer membrane assembly of autotransporter proteins. J. Bacteriol. 189:5393-5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones, D. T. 1999. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292:195-202. [DOI] [PubMed] [Google Scholar]
- 23.Jong, W. S. P., C. M. ten Hagen-Jongman, T. den Blaauwen, D. Jan Slotboom, J. R. H. Tame, D. Wickstrom, J.-W. de Gier, B. R. Otto, and J. Luirink. 2007. Limited tolerance towards folded elements during secretion of the autotransporter Hbp. Mol. Microbiol. 63:1524-1536. [DOI] [PubMed] [Google Scholar]
- 24.Junker, M., R. N. Besingi, and P. L. Clark. 2009. Vectorial transport and folding of an autotransporter virulence protein during outer membrane secretion. Mol. Microbiol. 71:1323-1332. [DOI] [PubMed] [Google Scholar]
- 25.Jurado, P., V. de Lorenzo, and L. A. Fernández. 2006. Thioredoxin fusions increase folding of single chain Fv antibodies in the cytoplasm of Escherichia coli: evidence that chaperone activity is the prime effect of thioredoxin. J. Mol. Biol. 357:49-61. [DOI] [PubMed] [Google Scholar]
- 26.Jurado, P., D. Ritz, J. Beckwith, V. de Lorenzo, and L. A. Fernández. 2002. Production of functional single-chain Fv antibodies in the cytoplasm of Escherichia coli. J. Mol. Biol. 320:1-10. [DOI] [PubMed] [Google Scholar]
- 27.Keen, N. T., and S. Tamaki. 1986. Structure of two pectate lyase genes from Erwinia chrysanthemi EC16 and their high-level expression in Escherichia coli. J. Bacteriol. 168:595-606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kingsley, R. A., R. L. Santos, A. M. Keestra, L. G. Adams, and A. J. Baumler. 2002. Salmonella enterica serotype Typhimurium ShdA is an outer membrane fibronectin-binding protein that is expressed in the intestine. Mol. Microbiol. 43:895-905. [DOI] [PubMed] [Google Scholar]
- 29.Kingsley, R. A., K. van Amsterdam, N. Kramer, and A. J. Baumler. 2000. The shdA gene is restricted to serotypes of Salmonella enterica subspecies I and contributes to efficient and prolonged fecal shedding. Infect. Immun. 68:2720-2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klauser, T., J. Pohlner, and T. F. Meyer. 1990. Extracellular transport of cholera toxin B subunit using Neisseria IgA protease b-domain: conformation-dependent outer membrane translocation. EMBO J. 9:1991-1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klauser, T., J. Pohlner, and T. F. Meyer. 1992. Selective extracellular release of cholera toxin B subunit by Escherichia coli: dissection of Neisseria Igab-mediated outer membrane transport. EMBO J. 11:2327-2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Knowles, T. J., A. Scott-Tucker, M. Overduin, and I. R. Henderson. 2009. Membrane protein architects: the role of the BAM complex in outer membrane protein assembly. Nat. Rev. Microbiol. 7:206-214. [DOI] [PubMed] [Google Scholar]
- 33.Koebnik, R., K. P. Locher, and P. Van Gelder. 2000. Structure and function of bacterial outer membrane proteins: barrels in a nutshell. Mol. Microbiol. 37:239-253. [DOI] [PubMed] [Google Scholar]
- 34.Kurowski, M. A., and J. M. Bujnicki. 2003. GeneSilico protein structure prediction meta-server. Nucleic Acids Res. 31:3305-3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu, J., and C. Deutsch. 2001. Pegylation: a method for assessing topological accessibilities in Kv1.3. Biochemistry 40:13288-13301. [DOI] [PubMed] [Google Scholar]
- 36.Maurer, J., J. Jose, and T. F. Meyer. 1999. Characterization of the essential transport function of the AIDA-I autotransporter and evidence supporting structural predictions. J. Bacteriol. 181:7014-7020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Muller, D., I. Benz, D. Tapadar, C. Buddenborg, L. Greune, and M. A. Schmidt. 2005. Arrangement of the translocator of the autotransporter adhesin involved in diffuse adherence on the bacterial surface. Infect. Immun. 73:3851-3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muyldermans, S., T. N. Baral, V. C. Retamozzo, P. De Baetselier, E. De Genst, J. Kinne, H. Leonhardt, S. Magez, V. K. Nguyen, H. Revets, U. Rothbauer, B. Stijlemans, S. Tillib, U. Wernery, L. Wyns, G. Hassanzadeh-Ghassabeh, and D. Saerens. 2009. Camelid immunoglobulins and nanobody technology. Vet. Immunol. Immunopathol. 128:178-183. [DOI] [PubMed] [Google Scholar]
- 39.Muyldermans, S., C. Cambillau, and L. Wyns. 2001. Recognition of antigens by single-domain antibody fragments: the superfluous luxury of paired domains. Trends Biochem. Sci. 26:230-235. [DOI] [PubMed] [Google Scholar]
- 40.Nguyen, V. Q., R. M. Caprioli, and T. L. Cover. 2001. Carboxy-terminal proteolytic processing of Helicobacter pylori vacuolating toxin. Infect. Immun. 69:543-546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oomen, C. J., P. Van Ulsen, P. Van Gelder, M. Feijen, J. Tommassen, and P. Gros. 2004. Structure of the translocator domain of a bacterial autotransporter. EMBO J. 23:1257-1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perna, N. T., G. Plunkett III, V. Burland, B. Mau, J. D. Glasner, D. J. Rose, G. F. Mayhew, P. S. Evans, J. Gregor, H. A. Kirkpatrick, G. Posfai, J. Hackett, S. Klink, A. Boutin, Y. Shao, L. Miller, E. J. Grotbeck, N. W. Davis, A. Lim, E. T. Dimalanta, K. D. Potamousis, J. Apodaca, T. S. Anantharaman, J. Lin, G. Yen, D. C. Schwartz, R. A. Welch, and F. R. Blattner. 2001. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature 409:529-533. [DOI] [PubMed] [Google Scholar]
- 43.Pohlner, J., R. Halter, K. Beyreuther, and T. F. Meyer. 1987. Gene structure and extracellular secretion of Neisseria gonorrhoeae IgA protease. Nature 325:458-462. [DOI] [PubMed] [Google Scholar]
- 44.Poirot, O., E. O'Toole, and C. Notredame. 2003. Tcoffee@igs: a Web server for computing, evaluating and combining multiple sequence alignments. Nucleic Acids Res. 31:3503-3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prilipov, A., P. S. Phale, P. Van Gelder, J. P. Rosenbusch, and R. Koebnik. 1998. Coupling site-directed mutagenesis with high-level expression: large scale production of mutant porins from E. coli. FEMS Microbiol. Lett. 163:65-72. [DOI] [PubMed] [Google Scholar]
- 46.Purdy, G. E., C. R. Fisher, and S. M. Payne. 2007. IcsA surface presentation in Shigella flexneri requires the periplasmic chaperones DegP, Skp, and SurA. J. Bacteriol. 189:5566-5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Purdy, G. E., M. Hong, and S. M. Payne. 2002. Shigella flexneri DegP facilitates IcsA surface expression and is required for efficient intercellular spread. Infect. Immun. 70:6355-6364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Quintela, J. C., M. A. de Pedro, P. Zöllner, G. Allmaier, and F. Garcia-del Portillo. 1997. Peptidoglycan structure of Salmonella typhimurium growing within cultured mammalian cells. Mol. Microbiol. 23:693-704. [DOI] [PubMed] [Google Scholar]
- 49.Robert, V., E. B. Volokhina, F. Senf, M. P. Bos, P. Van Gelder, and J. Tommassen. 2006. Assembly factor Omp85 recognizes its outer membrane protein substrates by a species-specific C-terminal motif. PLoS Biol. 4:e377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sauri, A., Z. Soprova, D. Wickstrom, J. W. de Gier, R. C. Van der Schors, A. B. Smit, W. S. Jong, and J. Luirink. 2009. The Bam (Omp85) complex is involved in secretion of the autotransporter haemoglobin protease. Microbiology 155:3982-3991. [DOI] [PubMed] [Google Scholar]
- 51.Schägger, H., W. A. Cramer, and G. von Jagow. 1994. Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal. Biochem. 217:220-230. [DOI] [PubMed] [Google Scholar]
- 52.Schmitt, W., and R. Haas. 1994. Genetic analysis of the Helicobacter pylori vacuolating cytotoxin: structural similarities with the IgA protease type of exported protein. Mol. Microbiol. 12:307-319. [DOI] [PubMed] [Google Scholar]
- 53.Skillman, K. M., T. J. Barnard, J. H. Peterson, R. Ghirlando, and H. D. Bernstein. 2005. Efficient secretion of a folded protein domain by a monomeric bacterial autotransporter. Mol. Microbiol. 58:945-958. [DOI] [PubMed] [Google Scholar]
- 54.Stegmeier, J. F., and C. Andersen. 2006. Characterization of pores formed by YaeT (Omp85) from Escherichia coli. J. Biochem. 140:275-283. [DOI] [PubMed] [Google Scholar]
- 55.Subbarao, G. V., and B. van den Berg. 2006. Crystal structure of the monomeric porin OmpG. J. Mol. Biol. 360:750-759. [DOI] [PubMed] [Google Scholar]
- 56.Suzuki, T., M. C. Lett, and C. Sasakawa. 1995. Extracellular transport of VirG protein in Shigella. J. Biol. Chem. 270:30874-30880. [DOI] [PubMed] [Google Scholar]
- 57.Turner, D. P., K. G. Wooldridge, and D. A. Ala'Aldeen. 2002. Autotransported serine protease A of Neisseria meningitidis: an immunogenic, surface-exposed outer membrane, and secreted protein. Infect. Immun. 70:4447-4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Valls, M., S. Atrian, V. de Lorenzo, and L. A. Fernández. 2000. Engineering a mouse metallothionein on the cell surface of Ralstonia eutropha CH34 for immobilization of heavy metals in soil. Nat. Biotechnol. 18:661-665. [DOI] [PubMed] [Google Scholar]
- 59.van den Berg, B. 2010. Crystal structure of a full-length autotransporter. J. Mol. Biol. 396:627-633. [DOI] [PubMed] [Google Scholar]
- 60.Veiga, E., V. De Lorenzo, and L. A. Fernández. 2003. Autotransporters as scaffolds for novel bacterial adhesins: surface properties of Escherichia coli cells displaying Jun/Fos dimerization domains. J. Bacteriol. 185:5585-5590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Veiga, E., V. de Lorenzo, and L. A. Fernández. 1999. Probing secretion and translocation of a beta-autotransporter using a reporter single-chain Fv as a cognate passenger domain. Mol. Microbiol. 33:1232-1243. [DOI] [PubMed] [Google Scholar]
- 62.Veiga, E., V. De Lorenzo, and L. A. Fernández. 2004. Structural tolerance of bacterial autotransporters for folded passenger protein domains. Mol. Microbiol. 52:1069-1080. [DOI] [PubMed] [Google Scholar]
- 63.Veiga, E., E. Sugawara, H. Nikaido, V. de Lorenzo, and L. A. Fernández. 2002. Export of autotransported proteins proceeds through an oligomeric ring shaped by C-terminal domains. EMBO J. 21:2122-2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wagner, J. K., J. E. Heindl, A. N. Gray, S. Jain, and M. B. Goldberg. 2009. Contribution of the periplasmic chaperone Skp to efficient presentation of the autotransporter IcsA on the surface of Shigella flexneri. J. Bacteriol. 191:815-821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wells, T. J., O. Sherlock, L. Rivas, A. Mahajan, S. A. Beatson, M. Torpdahl, R. I. Webb, L. P. Allsopp, K. S. Gobius, D. L. Gally, and M. A. Schembri. 2008. EhaA is a novel autotransporter protein of enterohemorrhagic Escherichia coli O157:H7 that contributes to adhesion and biofilm formation. Environ. Microbiol. 10:589-604. [DOI] [PubMed] [Google Scholar]