

Abstract
The predisposition for scleroderma, defined as fibrosis and hardening of the skin, is poorly understood. We report that stiff skin syndrome (SSS), an autosomal dominant congenital form of scleroderma, is caused by mutations in the sole Arg-Gly-Asp (RGD) sequence-encoding domain of fibrillin-1 that mediates integrin binding. Ordered polymers of fibrillin-1 (termed microfibrils) initiate elastic fiber assembly and bind to and regulate the activation of the pro-fibrotic cytokine transforming growth factor β (TGFβ). Altered cell-matrix interactions in SSS accompany excessive microfibrillar deposition, impaired elastogenesis, and increased TGFβ concentration and signaling in the dermis. The observation of similar findings in systemic sclerosis (SSc), a more common acquired form of scleroderma, suggests broad pathogenic relevance.
INTRODUCTION
Scleroderma, defined as hardening of the skin due to pathologic fibrosis, has many clinical presentations. A common and severe form, termed systemic sclerosis (SSc), typically shows adult onset that associates with the presence of autoantibodies, progressive visceral fibrosis, pulmonary hypertension and peripheral vasoconstriction (Raynaud’s phenomenon) (1). Obstacles in understanding and treating SSc include the absence of a well-defined genetic contribution or validated animal models. We reasoned that the study of rare but tractable presentations of scleroderma, with clear evidence for a major single gene effect, might provide a foothold to elucidate disease pathogenesis of systemic sclerosis, a disease affecting one in five thousand individuals in the general population. First described by Esterly and McKusick in 1971, stiff skin syndrome (SSS) is characterized by hard, thick skin, usually over the entire body, that limits joint mobility and causes flexion contractures (2). Other occasional findings include focal lipodystrophy and muscle weakness. About forty cases have been reported in the literature (2–18). Although families with multiple instances of SSS have been described (2–18), the lack of large multiplex families has precluded definitive assignment of the inheritance pattern. Prior work interrogating the pathogenesis of SSS has been largely observational, with few derived mechanistic insights (2–18). Findings include increased collagen production and DNA synthesis in the dermis and increased circulating cytokines including TNFα, IL-6 and TGFβ2(13, 19, 20).
Multiple lines of evidence have suggested a contribution of the connective tissue protein fibrillin-1 to the pathogenesis of profibrotic phenotypes. First, cutaneous fibrosis in the Tight skin (Tsk) mouse is caused by a large in-frame duplication in the fibrillin-1 gene (Fbn1) (21, 22). Tsk mice also show manifestations of Marfan syndrome (MFS), a condition caused by FBN1 mutations that lead to a deficiency of extracellular fibrillin-1 (23). The mechanism of disease in Tsk mice remains elusive due to widely disparate findings including a dominant negative effect on the assembly of fibrillin-1 microfibrils, autoantibodies directed against fibrillin-1, and even apparent passive transfer of phenotype after bone marrow transplantation (24–31). Second, a genome-wide microsatellite screen at 10 cM resolution (400 markers) suggested a link between variation at the FBN1 locus and autoimmune systemic sclerosis (SSc) in Choctaw Indians (p ~ 0.0032, odds ratio 3.15) (32), however no pathogenic mutation has been identified (32, 33). Third, antibodies against fibrillin-1 have been described in patients with autoimmune scleroderma (34, 35), but whether these antibodies recognize native fibrillin-1 is controversial (36). Finally, fibrillin-1 contributes to the regulation of TGFβ, a family of profibrotic cytokines that has been linked to many fibrotic diseases including both SSS and SSc (37, 38).
TGFβ is secreted from the cell in a large latent complex (LLC) that includes the active cytokine, a dimer of its processed N-terminal propeptide (latency associated peptide or LAP) and one of three latent transforming growth factor β binding proteins (LTBP-1, -3, or -4). As implied by mouse models and confirmed biochemically, fibrillin-1 rich microfibrils contribute to targeting of the LLC to the extracellular matrix by direct interaction with LTBPs (39, 40). Failed matrix sequestration of the LLC in fibrillin-1 deficient patients (MFS) and mice promotes increased activation of the TGFβ family of cytokines. Indeed many features of MFS can be attenuated or prevented by systemic administration of TGFβ antagonists in mouse models (40–44). Despite the indirect evidence suggesting that mutations in FBN1 may underlie SSS, patients with SSS do not show any of the systemic manifestations of MFS (e.g. ocular lens dislocation, bone overgrowth, joint laxity, aortic aneurysm), and patients with MFS do not show skin fibrosis.
In this paper, we show that mutations affecting the integrin-binding TB4 domain of fibrillin-1 cause SSS in humans via mechanisms distinct from those seen in MFS. Our observation of similar events in skin from patients with SSc suggests that the study of SSS has the potential to derive treatment strategies with broad application.
RESULTS
Phenotypic characterization of SSS families
Assessment of four families with SSS, including one with 10 affected individuals in 5 generations, revealed autosomal dominant inheritance with high penetrance (Fig. 1A). All affected individuals showed diffusely thick and hard skin from the time of birth and joint contracture but lacked the typical skeletal, ocular and cardiovascular findings of MFS. Additional clinical features not previously described for SSS (2–18) include cutaneous nodules that predominantly affect the distal interphalangeal joints (Fig. 1A), relative short stature (mean adult male and female height < 15th and < 5th percentile, respectively), and diffuse entrapment neuropathy (nerve injury and dysfunction due to local compression) (Suppl. Table 1).
Figure 1. Characterization of SSS and hybrid patients.
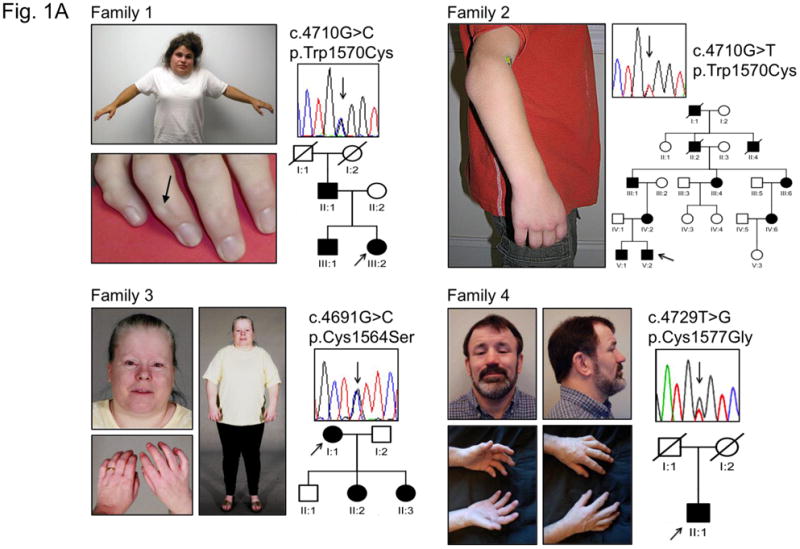
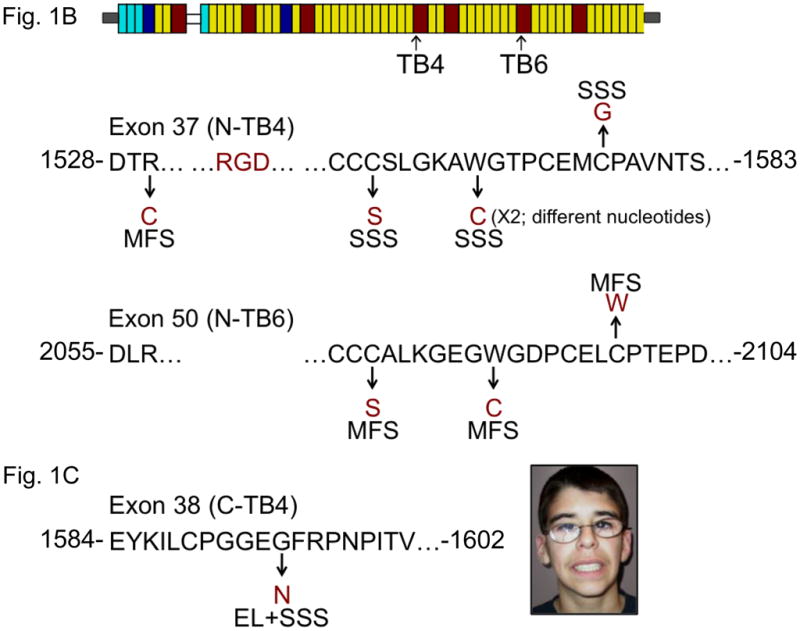
(A) Phenotypic characteristics, FBN1 mutation and pedigree for families 1–4. Pedigrees of families 1–3 document an autosomal dominant pattern of inheritance. Individual 1-III:2 shows decreased facial expression due to tightness of the skin and limited shoulder elevation. Note nodules at the distal interphalangeal joints (arrow). Individual 2-V:2 demonstrates limited extension of the elbows. Individual 3-I:1 shows a tight facial expression and limited extension of fingers and elbows. Individual 4-II:1 shows tightness of facial skin and limited flexion/extension of the fingers along with the presence of multiple interphalangeal nodules. The position of the nucleotide substitutions is indicated by arrows. Impact of mutations at the protein level are indicated using the three letter amino acid code. Circle, female; square, male; open symbol, unaffected; shaded symbol, affected; diagonal line, deceased; arrows below symbols indicate probands. (B) Diagram representing the domain structure of the fibrillin-1 protein. Yellow rectangles, calcium binding epidermal growth factor domain; red rectangles, transforming growth factor beta binding protein-like domain (TB domain); light blue rectangles, non-calcium binding epidermal growth factor-like module, dark blue rectangles, hybrid domains. Arrows indicate the fourth and sixth TB domains (TB4 and TB6, respectively), encoded by exons 37-38 and exons 50–51 of the FBN1 gene. The location of the RGD motif (arginine-glycine-aspartic acid) is indicated within a partial peptide sequence (positions of the first and last amino acid residues indicated) encoded by exon 37. In exon 37, encoding the N-terminal portion of TB4 (N-TB4), the substituted amino acid residues in the four SSS families are indicated. Mutations affecting the corresponding amino acid residues in exon 50 (encoding TB6) result in typical MFS syndrome (55). A mutation N-terminal to the RGD sequence in TB4 (p.Arg1530Cys) also causes MFS. (C) Position of FBN1 mutation (p.Gly1594Asn) in exon 38 in the patient with the hybrid (stiff skin and ectopia lentis) phenotype (inset). Exon 38 encodes the C-terminal portion of the fourth TB domain (C-TB4). The single letter amino acid code is utilized (R-arginine, C-cysteine, G-glycine, D-aspartic acid, S-serine, W-tryptophan, N-asparagine).
The phenotypic similarities between SSS and Tsk mice suggested that SSS might be caused by alterations of fibrillin-1. This hypothesis was strengthened upon our recognition of a patient with a hybrid phenotype including ocular lens dislocation (a cardinal manifestation of MFS), glaucoma, retinal detachment, and tight skin with diffuse joint contracture. This 14 year old male, one of fraternal triplets, was considerably shorter than his unaffected brothers and showed no skeletal or cardiovascular manifestations of MFS (Fig. 1C).
Altered fibrillin-1, elastin and collagen deposition
Pulse-chase analysis of dermal fibroblasts from SSS patients and controls showed equivalent secretion of fibrillin-1 (Suppl. Fig. 1). Recombinant fibrillin-1 peptides harboring SSS mutations also showed appropriate secretion into the media of stably-transfected cells (Suppl. Fig. 1). Confocal immunofluorescence analysis of skin biopsies from patients with SSS revealed increased deposition of both fibrillin-1 and elastin in the dermis, when compared to age- and gender-matched control samples (Fig. 2A). Although both patients and controls showed accumulation of microfibrils at the dermal-epidermal junction, these microfibrillar bundles had a stubby appearance in SSS, without the deep projections into the underlying dermis seen in controls (Fig. 2A). Dermal deposition of elastin was seen immediately adjacent to the epidermis in SSS, a zone that shows relative exclusion of elastin in controls (Fig. 2A). These data suggest that pathogenic events in SSS alter the amount and architecture of microfibrillar deposits and are abnormally permissive for the association of fibrillin-1 and elastin at the dermal-epidermal junction. Trichrome staining of skin biopsies revealed a wide zone of increased collagen deposition in the papillary dermis of patients with SSS when compared to control samples (Fig. 2B).
Figure 2. Elastin, fibrillin-1 and collagen composition of skin biopsies.

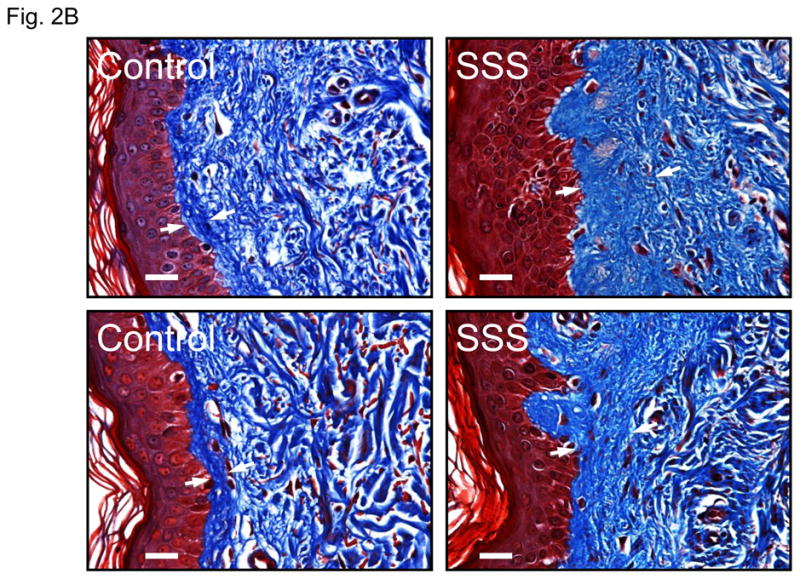
(A) Confocal microscopy of skin biopsies (with keratin in the epithelium delineated in red) from a patient with SSS and a control, seen at low magnification (upper panel) and high magnification (lower panel). The control sample shows long microfibrillar projections from the dermal-epidermal junction (DEJ, arrow) into the superficial (papillary) dermis with relatively sparse fibrillin-1 deposition in the deeper dermis. There is relative exclusion of elastin in the superficial dermis immediately adjacent to the DEJ. The SSS sample shows stubby microfibrillar deposits immediately adjacent to the DEJ that co-localize with elastin. There is also increased deposition of elastin in the deeper dermis. Scale bars = upper row of low magnification images, 50 microns; lower row of high magnification images, 20 microns. (B) Trichrome staining of skin biopsies from two controls and two SSS patients. Note the widened zone of increased deposition of collagen (blue) in the papillary dermis of SSS patients, as delineated by white arrows. Scale bars, 20 microns
Sequencing of FBN1 in SSS families
Sequencing of all exons of the FBN1 gene from each of the four probands with SSS revealed a heterozygous mutation in each of the four patients (Fig. 1A). Remarkably, all mutations occurred in exon 37, encoding the N-terminal portion of the 4th (of 7) transforming growth factor β-binding protein like-domain (N-TB4) in fibrillin-1 (Fig. 1B). Two patients were heterozygous for the identical amino acid substitution (p.Trp1570Cys) created by different nucleotide substitutions (c.4710G>C and c.4710G>T). A third patient was heterozygous for amino acid substitution p.Cys1564Ser (c.4691G>C), and the fourth was heterozygous for p.Cys1577Gly (c.4729T>G). Interestingly, the patient with the hybrid SSS-ectopia lentis (SSS with dislocated eye lenses) phenotype had a mutation (c.4781G>A) in exon 38, encoding the C-terminal portion of TB4 (C-TB4), leading to amino acid substitution p.Gly1594Asn (Fig. 1C).
Disturbed cell-matrix interactions
In order to test the hypothesis that SSS mutations alter integrin binding, expression constructs were prepared that encode TB4 and adjacent calcium-binding epidermal growth factor-like domains (cbEGF22-TB4-cbEGF23). SSS mutations that cause the W1570C and C1564S substitutions were introduced by site-directed mutagenesis. We found that the RGD loop is exposed in both mutant recombinant peptides and is therefore available for integrin interactions (Suppl. Fig. 2). Previous in vitro studies have shown that in human endometrial stromal fibroblasts (hESF cells), RGD-dependent adhesion to recombinant fibrillin-1 RGD fragments is mediated mainly through αvβ3 integrins, whereas in VB6 keratinocytes, a keratinocyte cell line expressing high levels of integrin αvβ6, it is largely dependent on αvβ6 (45) (Suppl. Fig. 3). Human foreskin dermal fibroblasts (FS2 cells) express both α5β1 and αvβ3 integrins, with only the latter localizing to focal adhesions (Suppl. Fig. 3). Therefore, we assessed integrin-mediated events when different cell types were plated on dishes coated with either wild-type or mutant recombinant fibrillin-1 fragments. VB6 keratinocytes showed normal attachment and spreading when plated on mutant as well as wild-type substrates, consistent with preserved αvβ6 integrin-fibrillin-1 interaction in this system (Fig. 3A). In contrast, both SSS mutations induced a dramatic loss of both attachment and spreading of FS2 cells, suggesting impairment of interaction with αvβ3 and possibly α5β1 integrins. Informatively, hESF cells also failed to attach or spread when plated on mutant substrates (Suppl. Fig. 3). Furthermore, cultured dermal fibroblasts from patients with SSS showed reduced amounts of the activated (phosphorylated) form of focal adhesion kinase (pFAK), an event mediated by the interaction of RGD ligands with integrins concentrated at focal adhesions (Fig. 3B). Taken together, these data suggest that FBN1 mutations that cause SSS can impair integrin binding and signaling.
Figure 3. Cell spreading and attachment.


(A) Cell attachment (upper panel) and spreading (lower panel) of FS2 dermal fibroblasts (left panel) and VB6 keratinocytes (right panel) adherent on recombinant wild-type and mutant cbEGF22-TB4-cbEGF23 W1570C and C1564S protein constructs. Wild-type cbEGF22-TB4-cbEGF23 was used as a positive control and BSA as negative control. In contrast to FS2 cells, similar attachment and spreading profiles were observed for VB6 cells plated onto wild-type and mutant fragments. Data are expressed as mean ±S.D. (attachment, n=9; spreading, n=5), from three independent experiments. (B) Assessment of phosphorylated focal adhesion kinase protein (phosphorylation at Y397) at steady state comparing six controls and four SSS patients. Quantification shows decreased pFAK in SSS.
Microfibrillar accumulation
It is unlikely SSS is caused solely by the loss of integrin signaling due to absence of interactions with the extracellular matrix, since a deficiency of fibrillin-1 does not cause skin fibrosis in MFS. Although the difference in phenotype might relate to the integrin subtype-specific deficit seen in SSS, the contribution of some other mutation-imposed event could not be excluded. Therefore, we used ultrastructural analysis to further interrogate the consequence of SSS mutations on microfibrillar deposition and homeostasis. Compared to control skin, immuno-electron microscopy (EM) of all SSS biopsies showed haphazard labeling of giant and dense chords of stubby microfibrils that lacked deep dermal projections or apparent basement membrane associations seen in normal skin (Fig. 4A). Lower magnification revealed that the dermis in SSS patients is packed tightly with abnormally dense microfibrillar aggregates and collagen bundles (Fig. 4B). Whereas normal microfibrillar projections from the dermal-epidermal junction are devoid of elastin, we observed the presence of elastin in microfibrillar aggregates near this junction in SSS (Fig. 4C). This observation confirms the abnormal colocalization of elastin and fibrillin-1 just below the dermal-epidermal junction in SSS (Fig. 2A). In the deeper dermis in control skin, elastin assemblies appear as homogeneous dense cores of elastin surrounded by thin mantles of microfibrils (Fig. 4C). In SSS, microfibrils are more abundant, and the elastin core appears poorly assembled (Fig. 4C). We also observed normal-appearing elastic fibers in SSS that were surrounded by microfibrillar assemblies with normal periodic labelling (Fig. 4D), highlighting the potential for context-specific consequences of SSS mutations.
Figure 4. Electron microscopy (EM) and immuno-EM of skin biopsies.




(A) Immuno-gold labeling of fibrillin-1 at the dermal-epidermal junction (DEJ) in control skin reveals periodic labeling of lacey microfibrillar bundles which make contact with the basement membrane at zones adjacent to hemidesmosomes (open arrowheads) and extend deeply into the papillary dermis (black arrow). In the SSS, microfibrils with periodic labeling (black arrows) are seen at the periphery of giant microfibrillar aggregates but do not make contact with the basement membrane, instead we did see an increased number and wider distribution of anchoring fibrils (white arrows), that are largely composed of type VII collagen. Scale bars, 500 nm. (B) The skin in SSS is densely packed with microfibrillar chords (white arrows). Dense microfibrillar accumulation is also shown at higher magnification in the inset. Chords are embedded in dense collagen bundles (black arrows) in SSS patients compared to the control. Scale bars, 2 μm. (C) Immuno-gold labeling reveals the atypical presence of elastin (arrowheads) within fibrillin-1 deposits immediately adjacent to the DEJ (black arrow) within SSS skin (left panel). Homogeneously-dense deposits of elastin (black material) are surrounded by a thin mantle of microfibrils (arrows) in the papillary and reticular dermis of control skin (middle panel). Within SSS skin, patchy accumulations of elastin are often sparsely distributed within dense microfibrillar aggregates (right panel). Scale bars, 500 nm. (D) Immuno-EM reveals that SSS patients retain the capacity to form some normal elastic fibers, particularly in the reticular dermis, as demonstrated by homogeneous elastin cores with a sheath of microfibrils (arrows) that label with normal periodicity using an antibody specific for fibrillin-1 (mAb 69). Scale bar, 500 nm.
Altered TGFβ signaling
TGFβ is a critical profibrotic cytokine that has been linked to the pathogenesis of SSc (46). We hypothesized that the predominant influence of excessive microfibrillar deposition in SSS might be excessive concentration of TGFβ in the LLC (Suppl. Fig. 4). Examination of dermal biopsies revealed accumulation of the LLC component LTBP4 throughout the dermis of patients with SSS (Fig. 5A). We also observed increased nuclear accumulation of phosphorylated Smad2 (pSmad2) and expression of connective tissue growth factor (CTGF), a direct effector and a target of TGFβ signaling, respectively, in the dermis of SSS patients compared to controls (Fig. 5B, C). Enhanced nuclear accumulation of pSmad2 was seen in both isolated cells scattered throughout the dermis and in cells clustered around the microvasculature (Fig. 5B).
Figure 5. Immunohistochemical assessment of TGFβ concentration and signaling.



(A, C) Increased immunostaining for latent transforming growth factor binding protein 4 (LTBP4) and connective tissue growth factor (CTGF) throughout the dermis of two SSS patients compared to control. Scale bars in panel A, C 30 micron and 80 micron, respectively (B) Immunostaining for phosphorylated Smad2 (pSmad2) in the dermis of two control individuals and two SSS patients. Note the increased number nuclei intensely stained for pSmad2 (black arrowheads) in samples from the SSS patients compared to controls, indicative of increased TGFβ signaling. Quantification was performed by counting the number of nuclei (not associated with blood vessels, arrows) with intense pSmad2 signal in at least ten high power fields in two SSS patients by three independent observers blinded to phenotype. Data with SEM expressed after normalization to a randomly selected control sample. Scale bar, 100 micron
Pathological mesenchymal transition
During normal wound healing, TGFβ induces cells of epithelial or endothelial origin to lose cell contacts and polarity. These cells then migrate, invade surrounding tissues and alter their synthetic repertoire to adopt a more mesenchymal character through epithelial-or endothelial-to-mesenchymal transition (collectively EMT) (47). The resultant myofibroblasts express alpha-smooth muscle actin (αSMA) and contribute to fibrosis through collagen secretion. These cells typically disappear upon resolution of the healing response, but can be pathologically produced and maintained in many fibrotic diseases (48). We observed increased expression of αSMA in basal keratinocytes of the epidermis in patients with SSS (Fig. 6A, B). These basal keratinocytes also lacked the columnar organization seen in control skin.
Figure 6. Immunohistochemistry for α smooth muscle actin.

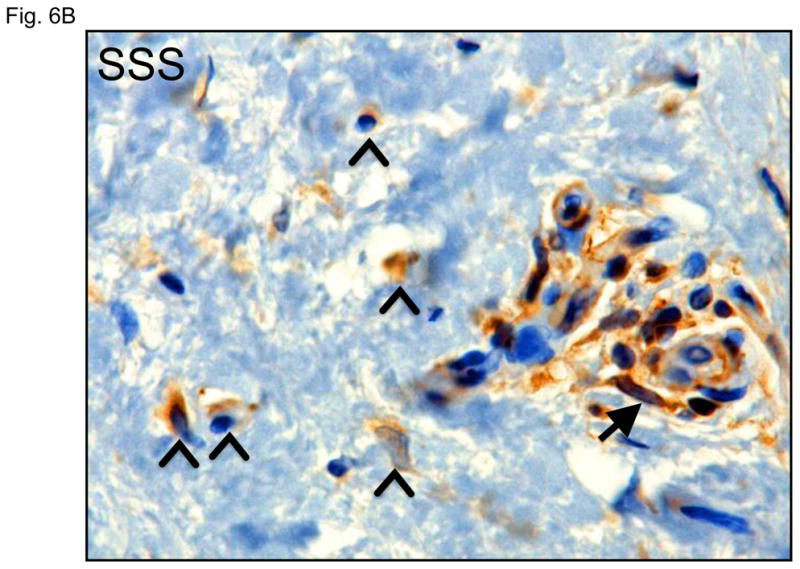
(A) Increased alpha-smooth muscle actin (αSMA) in basal keratinocytes within the epidermis (arrows) and in cells in the superficial dermis (open arrowheads) of two SSS patients compared to two controls. Note that the intensity of the signal around small blood vessels (closed arrowheads) is comparable in both patients and controls, confirming specificity of the abnormally intense staining of basal keratinocytes and isolated dermal cells in SSS. Scale bars top panel, 50 micron, bottom panel, 20 micron (B) In addition, SSS samples also occasionally show αSMA-positve cells occluding small vessels (arrow) and migrating around their periphery (open arrowheads).
Potential relevance to SSc
In order to investigate whether our findings in SSS have broader relevance for scleroderma, we examined skin biopsies from five patients with typical SSc (Suppl. Table 2), including adult-onset and progressive fibrosis of the skin and viscera, circulating autoantibodies, and peripheral vasoconstriction (Raynaud’s phenomenon). SSc patient biopsies showed giant and disorganized microfibrillar aggregates throughout the dermis that retained the ability to bind and concentrate the LLC of TGFβ (Fig. 7A-E). As in SSS, elastin desposition upon abnormal microfibrillar aggregates is often sparse and patchy (Fig. 7E). Other alterations in elastin morphology were also present including electron-lucent areas within elastic cores, giving them a mottled appearance (Fig. 7D).
Figure 7. Electron microscopy of skin biopsies from patients with systemic sclerosis.

(A,B) Electron microscopy reveals abnormally organized and dense macroaggregates of microfibrils (arrows) in the papillary and reticular dermis of patients with systemic sclerosis (SSc) that are reminiscent of those seen in SSS. (B) These abnormal aggregates of microfibrils in SSc are often embedded within dense deposits of collagen. (C) Immuno-EM demonstrates labeling of microfibrillar aggregates with an antibody directed against LTBP4 in SSc. (D, E) Abnormal appearance of elastic fibers in SSc, including a mottled appearance (D) or an overt paucity of elastin (E) compared to the homogeneously-dense elastin core in an age- and gender-matched control. Images are representative of those seen in five SSc patients. Scale bar, 500 nm except in (C), 200 nm.
DISCUSSION
We describe domain-specific mutations in the FBN1 gene as the genetic cause for a congenital form of scleroderma, SSS. Whereas prior work has demonstrated genetic alterations and environmental exposures that can contribute to fibrotic phenotypes (49), we report a genetic alteration that is sufficient to initiate and maintain cutaneous profibrotic programs in people, culminating in a scleroderma phenotype with early onset and complete penetrance. We propose that FBN1 mutations and the consequent perturbation of both microfibrillar assembly and microfibril-integrin interactions contribute to the pathogenesis of SSS, at least in part, through dysregulation of TGFβ signaling.
Several lines of evidence indicate that the defined FBN1 mutations cause SSS. First, the mutations segregated with the phenotype in families and were absent in unaffected family members. The mutation associated with the hybrid phenotype occurred de novo in the context of sporadic disease. Second, none of the mutations were observed in over 400 ethnically-matched control chromosomes. Third, all of the mutations causing typical SSS involve the creation or removal of a cysteine residue within a domain-type known to be highly dependent upon a defined spacing of eight cysteines for proper folding via intra-domain disulfide linkages (50–52). Indeed, the presence of eight cysteines is a defining feature of all TB domains (in fibrillins and LTBPs) throughout evolution (53, 54). Finally, the mutated tryptophan residue in families 1 and 2 is a key structural residue in TB domains and helps to stabilize the hydrophobic core (52). FBN1 mutations at the extreme N-terminus of TB4 cause classic MFS, as do substitutions identical to those seen in SSS at orthologous residues in other TB domains (like TB6; Fig. 1B) (55). TB4 substitutions associated with SSS may cause different effects on domain folding, stability and protein secretion than those seen with MFS-causing substitutions. Furthermore, the presence of an RGD motif in TB4 and the high affinity calcium binding site (Kd~16nM) formed between TB4 and cbEGF23 (48) likely confer unique functional properties to this TB domain within fibrillin-1 and hence specific phenotypic consequences upon disruption of domain structure and function.
We also provide evidence that these SSS-specific FBN1 mutations alter cytokine regulation. The contribution of microfibrils to TGFβ regulation is complicated. Studies of MFS have demonstrated that microfibrillar sequestration of the TGFβ LLC limits cytokine activation and signaling (40). Fibrillin-2 deficient mice show fusion of digits (syndactyly) and were found to have a decreased amount of selected TGFβ superfamily members (e.g. bone morphogenetic protein 7) in developing autopods. This suggests that microfibrils can also serve to concentrate TGFβ ligands in order to achieve the signaling threshold needed to mediate critical morphogenic events (56). While apparently at odds with events in MFS and perhaps context-specific, the level of TGFβ signaling might simply integrate the divergent roles of microfibrils with other determinants of cytokine bioavailability and activation. Several possibilities may explain why increased TGFβ signaling causes skin fibrosis in SSS but not in MFS. In MFS it is thought that gradual loss of microfibrils (largely during postnatal life) achieves a critical threshold that allows loss of negative regulation of TGFβ activation (40). Increased activation supports excessive signaling, which leads to changes in gene expression that result in many of the phenotypes in MFS such as pulmonary emphysema, myxomatous changes of the atrioventricular valves, dural ectasia, aortic aneurysm, and muscle hypoplasia and weakness (40–42). Consistent with this model, postnatal TGFβ antagonism with systemic delivery of a neutralizing antibody attenuates or prevents many clinical phenotypes in fibrillin-1 deficient mice. In our model of this microfibril-deficient state, decreased TGFβ concentration is offset by increased activation (Fig. 5A) (40–42). An increase in signaling is dependent on ongoing production of TGFβ.
In contrast, given the accumulation of aberrant microfibrillar assemblies in SSS, increased concentration of latent TGFβ may sustain a chronic increased level of TGFβ signaling whether or not the abnormal character of microfibrils promotes increased TGFβ activation. This model is also compatible with the restricted location and small repertoire of mutations in SSS, which is more typical of gain (rather than loss) of function. This same line of reasoning could explain the hybrid phenotype (SSS-MFS) of Tsk mice where the large internal fibrillin-1 duplication (encompassing exon 37) both impairs the context and function of the integrin-interacting RGD sequence and the overall structure and stability of fibrillin-1.
What is the nature of the proposed gain of function in SSS? The unique feature of TB4 is that it encodes the only RGD sequence in fibrillin-1, a motif that mediates cell-matrix interactions via integrin binding. Indeed, the RGD in TB4 of fibrillin-1 is known to interact with α5β1, αvβ3 and αvβ6 integrins (57, 58). Our in vitro data suggest that SSS mutations impair integrin-mediated cellular events such as the promotion of cellular adhesion and spreading. This does not seem sufficient to initiate fibrosis as such events would also be lost in a microfibrillar deficiency state (i.e. MFS). It is notable that both SSS and SSc associate with excessive accumulation of dermal microfibrillar deposits that concentrate TGFβ in the skin (59, 60). Increased matrix deposition of abnormal microfibrils may be a consequence of perturbed integrin interactions in SSS. There is precedence for this hypothesis. Takahashi and colleagues showed that upon replacement of the RGD sequence in fibronectin with RGE, cells deposit abnormally short and thickened fibronectin fibrils, reminiscent of the abnormal microfibrillar deposits in SSS (61). The presumed gain of microfibrillar function in SSS (i.e. concentration of TGFβ LLC) would be imposed early in development and could explain the congenital onset of fibrosis.
It remains unknown whether physiologic or pathologic levels of TGFβ activation accompany the increased concentration of latent TGFβ in the dermis of patients with SSS. The fact that all three integrins that interact with fibrillin-1 (αvβ3, αvβ6 and α5β1) are also known to activate TGFβ (45, 62–64) suggests a potential mechanism for enhanced TGFβ activation. Integrins activate TGFβ by two processes, which may act independently or in concert, depending on the tissue context as well as the cell-type involved (37). First, integrins can simultaneously interact with the latent TGFβ complex (via an RGD sequence in LAP) and other proteins such as matrix metalloproteases and TGFβ receptors to promote TGFβ activation (63–67). In a second more mechanical event, integrin binding to the RGD domain of LAPβ1 within the LLC transmits traction forces that conformationally change the LLC and liberate active TGFβ (68). A mouse with a knock-in mutation of the RGD sequence in LAPβ1 (preventing integrin-mediated activation of TGFβ1) bears striking phenotypic similarity to the TGFβ1 knockout mouse, demonstrating the importance of integrin binding to the LLC in overall TGFβ activation (69).
Several studies have implicated aberrant integrin expression or function in SSc and other fibrotic phenotypes. In culture, integrin αvβ3 is upregulated in SSc dermal fibroblasts. Furthermore, its inhibition prevents collagen expression and reverses the myofibroblastic phenotype of SSc fibroblasts in a TGFβ1 – dependent manner (70). In this light, it is possible (but unproven) that impairment of integrin interaction with fibrillin-1 in SSS induces increased integrin expression and/or bioavailabllity to participate in TGFβ activation, and that this contributes to downstream events including tissue fibrosis.
A number of our observations suggest involvement of basal keratinocytes in the pathogenesis of SSS. These basal keratinocytes showed increased expression of α-SMA and lacked the columnar organization seen in control skin, suggesting the loss of attachments and polarity characteristic of cells undergoing EMT. There was also increased representation of αSMA-positive cells, distinct from the microvasculature, within the peripheral dermis of patients with SSS (Fig. 6A). SSS samples also showed αSMA-positive cells occluding small vessels and migrating around their periphery (Fig. 6B). These data suggest a contribution of pathologic EMT in SSS, an event plausibly initiated and/or sustained by excessive TGFβ signaling. A specific perturbation of the assembly of microfibrils elaborated by keratinocytes in SSS would explain the normal microfibrillar assemblies found in the deeper dermis of these patients and their lack of manifestations of MFS or fibrosis in other tissues. Human keratinocytes physiologically express integrin α5β1, while they express αvβ6 during wound healing (71). Although our in vitro experiments using recombinant SSS fibrillin-1 peptides did not reveal altered interaction with VB6 keratinocytes, this does not exclude such perturbations in the context of full-length mutant fibrillin-1 and normal keratinocytes during tissue development or homeostasis in vivo. Informatively, basal keratinocytes have been shown to contribute to pathologic EMT in hypertrophic scar development (72).
Our finding of altered microfibrillar assembly in SSc is in keeping with a prior report (73), but requires further validation in scleroderma-spectrum disorders. The mechanistic basis for this observation remains to be elucidated. Multiple prior reports have described the presence of auto-antibodies to fibrillin-1 in patients with typical presentations of SSc (34, 35, 74, 75). Although the presence of antibodies that recognize native fibrillin-1 remains controversial (36), it will be interesting to determine if autoantibodies or another circulating factor in SSc alters the interaction between integrins and fibrillin-1, mimicking the effect of fibrillin-1 mutations in SSS. Modeling of SSS in mice will provide an ideal platform to further interrogate pathogenesis and to test potential therapeutic strategies including TGFβ or EMT antagonists.
MATERIALS and METHODS
Subjects and clinical evaluation
This study was approved by the Institutional Review Board of the Johns Hopkins University School of Medicine and the Belfast City Hospital. We obtained informed consent from all subjects involved in the study and specific consent for the publication of photographs that show identity.
Mutation analysis
We amplified genomic DNA by PCR using primers complementary to the flanking introns of all FBN1 exons, as previously described (76). We carried out sequence analysis using an Applied Biosystems automated DNA sequencer and protocols provided by the manufacturer. If family members were available, segregation of the mutation was shown. None of the SSS mutations were found in a panel of at least 400 control chromosomes.
Immunohistochemistry
Punch skin biopsies were performed on the forearm of SSS patients and age/sex matched controls. The tissue was paraffin-embedded and immunohistochemical staining was performed using antibodies directed against phosphorylated Smad2 (Cell Signaling Technology), CTGF (Abcam) and LTBP-4 (Santa Cruz) using previously described methods (40).
Electron Microscopy
Fresh biopsies were either fixed immediately in 1.5% glutaraldehyde/1.5% paraformaldehyde with 0.05% tannic acid in 0.1M cacodylate, pH 7.4 for 2 hours, then rinsed in cacodylate buffer overnight, then fixed in 1% OsO4, dehydrated, and embedded in Spurrs epoxy. For en-bloc immuno-electron microscopy, samples were rinsed overnight in DMEM, then soaked overnight in mAb 69 (specific to fibrillin-1) or pAb 2101 (specific to LTBP4) (77), rinsed, then soaked overnight in the appropriate secondary antibody conjugate (5nM). Samples were then fixed and embedded as above. Tissue sections labeled using mAb 10B8 (specific for elastin) were fixed in 0.1% glutaraldehyde, dehydrated at low temperature, and embedded in LRWhite media, then immunolabeled as described previously(78).
Confocal Microscopy
Fresh biopsies stored in DMEM overnight were snap frozen in liquid nitrogen cooled hexanes and embedded in OCT. 30μm cryostat sections were fixed in 100% acetone at −20° C, rinsed in PBS and then simultaneously labeled with pAb 9543 (specific for fibrillin-1) and a mAb specific for cytokeratin to label the dermis, followed by GAM Alexa 546 combined with GAR Alexa 633, and viewed with laser light at 543 nm and 633 nm. Within the images, fibrillin-1 labeling was assigned a “green” color and cytokeratin “red”. Elastin labeling was similarly accomplished with mAb 10B8 and GAM Alexa 546 and viewed with laser light at 543 nm.
Cell culture
We derived fibroblast cultures from skin biopsies taken from the forearm. Cells were cultured in minimal essential media with 10% fetal bovine serum in the presence of antibiotics and passaged at 80% confluence. The cells were grown in 2% fetal bovine serum for two weeks at confluency to allow for extracellular matrix elaboration.
Western blot analysis
Protein was extracted from whole cell lysates of dermal fibroblasts using M-Per (Pierce). Protein concentration was determined using a BCA assay kit (Pierce) and 10 μg protein were separated under reducing conditions by standard gel electrophoresis on 10% Bis-Tris gels (Biorad). The separated proteins were then transferred onto nitrocellulose membranes (Biorad). The membranes were blocked for 1 h in LiCor blocking buffer and washes were performed with phosphate-buffered saline. Antibodies used were directed against phospho-FAK (Millipore) and alpha-tubulin (Sigma) with appropriate secondary antibodies (Licor). Membranes were scanned and signals were quantified with the Licor Odyssey System using instructions provided by the manufacturer.
Cell adhesion and spreading assays
The cell attachment data are derived from the intensity of the crystal violet stain taken up specifically by attached cells. The signal intensity at OD 620 is directly proportional to the number of adherent cells, as described previously (45). The spreading assay is performed by counting 200 cells from each well of a 96-well plate, and grading them as spreading or non-spreading cells based upon morphology, as described previously (45). We represent the data as the % of cells showing a spreading morphology indexed to the total number of attached cells. VB6 is a keratinocyte cell line and FS2 cells are primary dermal fibroblasts established from foreskin.
Supplementary Material
Acknowledgments
We thank John Marshall for VB6 cells, Bryan Sykes for FS2 cells, and Kay Kielty for the anti-fibrillin-1 RGD antibody. Fibroblast cell lines were established and provided by George Thomas (Kennedy Krieger Institute MRDD Research Center grant P30 HD024061). This work was supported in part by the Scleroderma Research Foundation, the Howard Hughes Medical Institute, the Smilow Center for Marfan Syndrome Research, the National Marfan Foundation, the National Institutes of Health (AR41135), and Shriners Hospital for Children. H. Dietz is an Investigator in the Howard Hughes Medical Institute. B. Loeys is a Senior Clinical Investigator of the Fund for Scientific Research Flanders. E. Davis is a Canada Research Chair and is supported by Canadian Institute of Health grant M0P86713. P. Handford, H. Mardon and C. Chillakuri acknowledge support from the Medical Research Council (G9828023). P. Whiteman is supported by the EPA Cephalosporin Trust.
References
- 1.Wigley FM. Vascular disease in scleroderma. Clin Rev Allergy Immunol. 2009;36:150–175. doi: 10.1007/s12016-008-8106-x. [DOI] [PubMed] [Google Scholar]
- 2.Esterly NB, McKusick VA. Stiff skin syndrome. Pediatrics. 1971;47:360–369. [PubMed] [Google Scholar]
- 3.Amoric JC, Stalder JF, David A, Bureau B, Pierard GE, Litoux P. Dysmorphism in Stiff Skin syndrome. Ann Dermatol Venereol. 1991;118:802–804. [PubMed] [Google Scholar]
- 4.Bodemer C, Habib K, Teillac D, Munich A, de Prost Y. A new case of Stiff Skin syndrome. Ann Dermatol Venereol. 1991;118:805–806. [PubMed] [Google Scholar]
- 5.Bundy SE, Lie K. Stiff skin syndrome. Birth Defects Orig Artic Ser. 1975;11:360–361. [PubMed] [Google Scholar]
- 6.DiRocco M. Clinical images: Stiff skin syndrome. Arthritis Rheum. 2000;43:1542. [PubMed] [Google Scholar]
- 7.Esterly NB. The stiff skin syndrome. Birth Defects Orig Artic Ser. 1971;7:306–308. [PubMed] [Google Scholar]
- 8.Ferrari D, Rossi R, Donzelli O. Stiff-skin syndrome. Chir Organi Mov. 2005;90:69–73. [PubMed] [Google Scholar]
- 9.Geng S, Lei X, Toyohara JP, Zhan P, Wang J, Tan S. Stiff skin syndrome. J Eur Acad Dermatol Venereol. 2006;20:729–732. doi: 10.1111/j.1468-3083.2006.01619.x. [DOI] [PubMed] [Google Scholar]
- 10.Gilaberte Y, Saenz-de-Santamaria MC, Garcia-Latasa FJ, Gonzalez-Mediero I, Zambrano A. Stiff skin syndrome: a case report and review of the literature. Dermatology. 1995;190:148–151. doi: 10.1159/000246666. [DOI] [PubMed] [Google Scholar]
- 11.Helm TN, Wirth PB, Helm KF. Congenital fascial dystrophy: the stiff skin syndrome. Cutis. 1997;60:153–154. [PubMed] [Google Scholar]
- 12.Jablonska S, Blaszczyk M. Stiff skin syndrome is highly heterogeneous, and congenital fascial dystrophy is its distinct subset. Pediatr Dermatol. 2004;21:508–510. doi: 10.1111/j.0736-8046.2004.21422.x. [DOI] [PubMed] [Google Scholar]
- 13.Jablonska S, Groniowski J, Krieg T, Nerlich A, Peltonen L, Oikarinen A, Dabrowski J, Pietrow D. Congenital fascial dystrophy--a noninflammatory disease of fascia: the stiff skin syndrome. Pediatr Dermatol. 1984;2:87–97. doi: 10.1111/j.1525-1470.1984.tb00453.x. [DOI] [PubMed] [Google Scholar]
- 14.Jablonska S, Schubert H, Kikuchi I. Congenital fascial dystrophy: stiff skin syndrome--a human counterpart of the tight-skin mouse. J Am Acad Dermatol. 1989;21:943–950. doi: 10.1016/s0190-9622(89)70280-2. [DOI] [PubMed] [Google Scholar]
- 15.Kikuchi I, Inoue S, Hamada K, Ando H. Stiff skin syndrome. Pediatr Dermatol. 1985;3:48–53. doi: 10.1111/j.1525-1470.1985.tb00486.x. [DOI] [PubMed] [Google Scholar]
- 16.Le T, Pierard GE. Stiff skin syndrome. Ann Dermatol Venereol. 1989;116:807–809. [PubMed] [Google Scholar]
- 17.Liu T, McCalmont TH, Frieden IJ, Williams ML, Connolly MK, Gilliam AE. The stiff skin syndrome: case series, differential diagnosis of the stiff skin phenotype, and review of the literature. Arch Dermatol. 2008;144:1351–1359. doi: 10.1001/archderm.144.10.1351. [DOI] [PubMed] [Google Scholar]
- 18.Pages ON, Maliszewicz P, Lefebvrel F, Poli-Merol ML, Morville P. Visceral involvement in stiff skin syndrome. Pediatr Dermatol. 2007;24:327. doi: 10.1111/j.1525-1470.2007.00417.x. [DOI] [PubMed] [Google Scholar]
- 19.Fidzianska A, Jablonska S. Congenital fascial dystrophy: abnormal composition of the fascia. J Am Acad Dermatol. 2000;43:797–802. doi: 10.1067/mjd.2000.107961. [DOI] [PubMed] [Google Scholar]
- 20.Richard MA, Grob JJ, Philip N, Rey J, Chamson A, Mege JL, Andrac L, Faure F, Basseres N, Bonerandi JJ. Physiopathogenic investigations in a case of familial stiff-skin syndrome. Dermatology. 1998;197:127–131. doi: 10.1159/000017983. [DOI] [PubMed] [Google Scholar]
- 21.Green MC, Sweet HO, Bunker LE. Tight-skin, a new mutation of the mouse causing excessive growth of connective tissue and skeleton. Am J Pathol. 1976;82:493–512. [PMC free article] [PubMed] [Google Scholar]
- 22.Siracusa LD, McGrath R, Ma Q, Moskow JJ, Manne J, Christner PJ, Buchberg AM, Jimenez SA. A tandem duplication within the fibrillin 1 gene is associated with the mouse tight skin mutation. Genome Res. 1996;6:300–313. doi: 10.1101/gr.6.4.300. [DOI] [PubMed] [Google Scholar]
- 23.Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, Puffenberger EG, Hamosh A, Nanthakumar EJ, Curristin SM, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–339. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- 24.Adachi Y, Oyaizu H, Taketani S, Minamino K, Yamaguchi K, Shultz LD, Iwasaki M, Tomita M, Suzuki Y, Nakano K, Koike Y, Yasumizu R, Sata M, Hirama N, Kubota I, Fukuhara S, Ikehara S. Treatment and transfer of emphysema by a new bone marrow transplantation method from normal mice to Tsk mice and vice versa. Stem Cells. 2006;24:2071–2077. doi: 10.1634/stemcells.2005-0575. [DOI] [PubMed] [Google Scholar]
- 25.Gayraud B, Keene DR, Sakai LY, Ramirez F. New insights into the assembly of extracellular microfibrils from the analysis of the fibrillin 1 mutation in the tight skin mouse. J Cell Biol. 2000;150:667–680. doi: 10.1083/jcb.150.3.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kielty CM, Raghunath M, Siracusa LD, Sherratt MJ, Peters R, Shuttleworth CA, Jimenez SA. The Tight skin mouse: demonstration of mutant fibrillin-1 production and assembly into abnormal microfibrils. J Cell Biol. 1998;140:1159–1166. doi: 10.1083/jcb.140.5.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leszczynski D, Renkonen R, Hayry P. Bone marrow transplantation in the rat. III. Structure of the liver inflammatory lesion in acute graft-versus-host disease. Am J Pathol. 1985;120:316–322. [PMC free article] [PubMed] [Google Scholar]
- 28.Murai C, Saito S, Kasturi KN, Bona CA. Spontaneous occurrence of anti-fibrillin-1 autoantibodies in tight-skin mice. Autoimmunity. 1998;28:151–155. doi: 10.3109/08916939808996283. [DOI] [PubMed] [Google Scholar]
- 29.Muryoi T, Kasturi KN, Kafina MJ, Saitoh Y, Usuba O, Perlish JS, Fleischmajer R, Bona CA. Self reactive repertoire of tight skin mouse: immunochemical and molecular characterization of anti-topoisomerase I autoantibodies. Autoimmunity. 1991;9:109–117. doi: 10.3109/08916939109006746. [DOI] [PubMed] [Google Scholar]
- 30.Phelps RG, Daian C, Shibata S, Fleischmajer R, Bona CA. Induction of skin fibrosis and autoantibodies by infusion of immunocompetent cells from tight skin mice into C57BL/6 Pa/Pa mice. J Autoimmun. 1993;6:701–718. doi: 10.1006/jaut.1993.1059. [DOI] [PubMed] [Google Scholar]
- 31.Walker MA, Harley RA, DeLustro FA, LeRoy EC. Adoptive transfer of tsk skin fibrosis to +/+ recipients by tsk bone marrow and spleen cells. Proc Soc Exp Biol Med. 1989;192:196–200. doi: 10.3181/00379727-192-42979. [DOI] [PubMed] [Google Scholar]
- 32.Tan FK, Wang N, Kuwana M, Chakraborty R, Bona CA, Milewicz DM, Arnett FC. Association of fibrillin 1 single-nucleotide polymorphism haplotypes with systemic sclerosis in Choctaw and Japanese populations. Arthritis Rheum. 2001;44:893–901. doi: 10.1002/1529-0131(200104)44:4<893::AID-ANR146>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 33.Zhou X, Tan FK, Wang N, Xiong M, Maghidman S, Reveille JD, Milewicz DM, Chakraborty R, Arnett FC. Genome-wide association study for regions of systemic sclerosis susceptibility in a Choctaw Indian population with high disease prevalence. Arthritis Rheum. 2003;48:2585–2592. doi: 10.1002/art.11220. [DOI] [PubMed] [Google Scholar]
- 34.Tan FK, Arnett FC, Antohi S, Saito S, Mirarchi A, Spiera H, Sasaki T, Shoichi O, Takeuchi K, Pandey JP, Silver RM, LeRoy C, Postlethwaite AE, Bona CA. Autoantibodies to the extracellular matrix microfibrillar protein, fibrillin-1, in patients with scleroderma and other connective tissue diseases. J Immunol. 1999;163:1066–1072. [PubMed] [Google Scholar]
- 35.Tan FK, Arnett FC, Reveille JD, Ahn C, Antohi S, Sasaki T, Nishioka K, Bona CA. Autoantibodies to fibrillin 1 in systemic sclerosis: ethnic differences in antigen recognition and lack of correlation with specific clinical features or HLA alleles. Arthritis Rheum. 2000;43:2464–2471. doi: 10.1002/1529-0131(200011)43:11<2464::AID-ANR13>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 36.Brinckmann J, Hunzelmann N, El-Hallous E, Krieg T, Sakai LY, Krengel S, Reinhardt DP. Absence of autoantibodies against correctly folded recombinant fibrillin-1 protein in systemic sclerosis patients. Arthritis Res Ther. 2005;7:R1221–1226. doi: 10.1186/ar1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hinz B. Tissue stiffness, latent TGF-beta1 activation, and mechanical signal transduction: implications for the pathogenesis and treatment of fibrosis. Curr Rheumatol Rep. 2009;11:120–126. doi: 10.1007/s11926-009-0017-1. [DOI] [PubMed] [Google Scholar]
- 38.Zhou X, Tan FK, Milewicz DM, Guo X, Bona CA, Arnett FC. Autoantibodies to fibrillin-1 activate normal human fibroblasts in culture through the TGF-beta pathway to recapitulate the "scleroderma phenotype". J Immunol. 2005;175:4555–4560. doi: 10.4049/jimmunol.175.7.4555. [DOI] [PubMed] [Google Scholar]
- 39.Isogai Z, Ono RN, Ushiro S, Keene DR, Chen Y, Mazzieri R, Charbonneau NL, Reinhardt DP, Rifkin DB, Sakai LY. Latent transforming growth factor beta-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. J Biol Chem. 2003;278:2750–2757. doi: 10.1074/jbc.M209256200. [DOI] [PubMed] [Google Scholar]
- 40.Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, Ramirez F, Sakai LY, Dietz HC. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33:407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- 41.Cohn RD, van Erp C, Habashi JP, Soleimani AA, Klein EC, Lisi MT, Gamradt M, ap Rhys CM, Holm TM, Loeys BL, Ramirez F, Judge DP, Ward CW, Dietz HC. Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat Med. 2007;13:204–210. doi: 10.1038/nm1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, Myers L, Klein EC, Liu G, Calvi C, Podowski M, Neptune ER, Halushka MK, Bedja D, Gabrielson K, Rifkin DB, Carta L, Ramirez F, Huso DL, Dietz HC. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jones KB, Myers L, Judge DP, Kirby PA, Dietz HC, Sponseller PD. Toward an understanding of dural ectasia: a light microscopy study in a murine model of Marfan syndrome. Spine (Phila Pa 1976) 2005;30:291–293. doi: 10.1097/01.brs.0000152166.88174.1c. [DOI] [PubMed] [Google Scholar]
- 44.Ng CM, Cheng A, Myers LA, Martinez-Murillo F, Jie C, Bedja D, Gabrielson KL, Hausladen JM, Mecham RP, Judge DP, Dietz HC. TGF-beta-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J Clin Invest. 2004;114:1586–1592. doi: 10.1172/JCI22715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jovanovic J, Takagi J, Choulier L, Abrescia NG, Stuart DI, van der Merwe PA, Mardon HJ, Handford PA. alphaVbeta6 is a novel receptor for human fibrillin-1. Comparative studies of molecular determinants underlying integrin-rgd affinity and specificity. J Biol Chem. 2007;282:6743–6751. doi: 10.1074/jbc.M607008200. [DOI] [PubMed] [Google Scholar]
- 46.Varga J, Pasche B. Transforming growth factor beta as a therapeutic target in systemic sclerosis. Nat Rev Rheumatol. 2009;5:200–206. doi: 10.1038/nrrheum.2009.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic-Canic M. Growth factors and cytokines in wound healing. Wound Repair Regen. 2008;16:585–601. doi: 10.1111/j.1524-475X.2008.00410.x. [DOI] [PubMed] [Google Scholar]
- 48.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med. 2009;360:1989–2003. doi: 10.1056/NEJMra0806188. [DOI] [PubMed] [Google Scholar]
- 50.Dyer CE, Shuttleworth CA, Kielty CM. Conformation and function of fibrillin 8-cysteine motifs. Biochem Soc Trans. 1995;23:506S. doi: 10.1042/bst023506s. [DOI] [PubMed] [Google Scholar]
- 51.Lee SS, Knott V, Jovanovic J, Harlos K, Grimes JM, Choulier L, Mardon HJ, Stuart DI, Handford PA. Structure of the integrin binding fragment from fibrillin-1 gives new insights into microfibril organization. Structure. 2004;12:717–729. doi: 10.1016/j.str.2004.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yuan X, Downing AK, Knott V, Handford PA. Solution structure of the transforming growth factor beta-binding protein-like module, a domain associated with matrix fibrils. EMBO J. 1997;16:6659–6666. doi: 10.1093/emboj/16.22.6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Corson GM, Chalberg SC, Dietz HC, Charbonneau NL, Sakai LY. Fibrillin binds calcium and is coded by cDNAs that reveal a multidomain structure and alternatively spliced exons at the 5' end. Genomics. 1993;17:476–484. doi: 10.1006/geno.1993.1350. [DOI] [PubMed] [Google Scholar]
- 54.Maslen CL, Corson GM, Maddox BK, Glanville RW, Sakai LY. Partial sequence of a candidate gene for the Marfan syndrome. Nature. 1991;352:334–337. doi: 10.1038/352334a0. [DOI] [PubMed] [Google Scholar]
- 55.Qin Y, Yan J, Simpson JL, Gu HF, Wang LC, Chen ZJ. Novel non-synonymous mutation in the transforming growth factor beta binding protein-like (TB) domain of the fibrillin-1 (FBN1) gene in a Han Chinese family with Marfan syndrome (MFS) Neuro Endocrinol Lett. 2007;28:629–632. [PubMed] [Google Scholar]
- 56.Arteaga-Solis E, Gayraud B, Lee SY, Shum L, Sakai L, Ramirez F. Regulation of limb patterning by extracellular microfibrils. J Cell Biol. 2001;154:275–281. doi: 10.1083/jcb.200105046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pfaff M, Reinhardt DP, Sakai LY, Timpl R. Cell adhesion and integrin binding to recombinant human fibrillin-1. FEBS Lett. 1996;384:247–250. doi: 10.1016/0014-5793(96)00325-0. [DOI] [PubMed] [Google Scholar]
- 58.Sakamoto H, Broekelmann T, Cheresh DA, Ramirez F, Rosenbloom J, Mecham RP. Cell-type specific recognition of RGD- and non-RGD-containing cell binding domains in fibrillin-1. J Biol Chem. 1996;271:4916–4922. [PubMed] [Google Scholar]
- 59.Bouzeghrane F, Reinhardt DP, Reudelhuber TL, Thibault G. Enhanced expression of fibrillin-1, a constituent of the myocardial extracellular matrix in fibrosis. Am J Physiol Heart Circ Physiol. 2005;289:H982–991. doi: 10.1152/ajpheart.00151.2005. [DOI] [PubMed] [Google Scholar]
- 60.Fleischmajer R, Jacobs L, Schwartz E, Sakai LY. Extracellular microfibrils are increased in localized and systemic scleroderma skin. Lab Invest. 1991;64:791–798. [PubMed] [Google Scholar]
- 61.Takahashi S, Leiss M, Moser M, Ohashi T, Kitao T, Heckmann D, Pfeifer A, Kessler H, Takagi J, Erickson HP, Fassler R. The RGD motif in fibronectin is essential for development but dispensable for fibril assembly. J Cell Biol. 2007;178:167–178. doi: 10.1083/jcb.200703021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jovanovic J, Iqbal S, Jensen S, Mardon H, Handford P. Fibrillin-integrin interactions in health and disease. Biochem Soc Trans. 2008;36:257–262. doi: 10.1042/BST0360257. [DOI] [PubMed] [Google Scholar]
- 63.Bax DV, Bernard SE, Lomas A, Morgan A, Humphries J, Shuttleworth CA, Humphries MJ, Kielty CM. Cell adhesion to fibrillin-1 molecules and microfibrils is mediated by alpha 5 beta 1 and alpha v beta 3 integrins. J Biol Chem. 2003;278:34605–34616. doi: 10.1074/jbc.M303159200. [DOI] [PubMed] [Google Scholar]
- 64.D'Arrigo C, Burl S, Withers AP, Dobson H, Black C, Boxer M. TGF-beta1 binding protein-like modules of fibrillin-1 and -2 mediate integrin-dependent cell adhesion. Connect Tissue Res. 1998;37:29–51. doi: 10.3109/03008209809028898. [DOI] [PubMed] [Google Scholar]
- 65.Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- 66.Galliher AJ, Schiemann WP. Beta3 integrin and Src facilitate transforming growth factor-beta mediated induction of epithelial-mesenchymal transition in mammary epithelial cells. Breast Cancer Res. 2006;8:R42. doi: 10.1186/bcr1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Scaffidi AK, Petrovic N, Moodley YP, Fogel-Petrovic M, Kroeger KM, Seeber RM, Eidne KA, Thompson PJ, Knight DA. alpha(v)beta(3) Integrin interacts with the transforming growth factor beta (TGFbeta) type II receptor to potentiate the proliferative effects of TGFbeta1 in living human lung fibroblasts. J Biol Chem. 2004;279:37726–37733. doi: 10.1074/jbc.M403010200. [DOI] [PubMed] [Google Scholar]
- 68.Wipff PJ, Hinz B. Integrins and the activation of latent transforming growth factor beta1 - an intimate relationship. Eur J Cell Biol. 2008;87:601–615. doi: 10.1016/j.ejcb.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 69.Yang Z, Mu Z, Dabovic B, Jurukovski V, Yu D, Sung J, Xiong X, Munger JS. Absence of integrin-mediated TGFbeta1 activation in vivo recapitulates the phenotype of TGFbeta1-null mice. J Cell Biol. 2007;176:787–793. doi: 10.1083/jcb.200611044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Asano Y, Ihn H, Yamane K, Jinnin M, Mimura Y, Tamaki K. Increased expression of integrin alpha(v)beta3 contributes to the establishment of autocrine TGF-beta signaling in scleroderma fibroblasts. J Immunol. 2005;175:7708–7718. doi: 10.4049/jimmunol.175.11.7708. [DOI] [PubMed] [Google Scholar]
- 71.Raja, Sivamani K, Garcia MS, Isseroff RR. Wound re-epithelialization: modulating keratinocyte migration in wound healing. Front Biosci. 2007;12:2849–2868. doi: 10.2741/2277. [DOI] [PubMed] [Google Scholar]
- 72.Werner S, Krieg T, Smola H. Keratinocyte-fibroblast interactions in wound healing. J Invest Dermatol. 2007;127:998–1008. doi: 10.1038/sj.jid.5700786. [DOI] [PubMed] [Google Scholar]
- 73.Davis EC, Blattel SA, Mecham RP. Remodeling of elastic fiber components in scleroderma skin. Connect Tissue Res. 1999;40:113–121. doi: 10.3109/03008209909029107. [DOI] [PubMed] [Google Scholar]
- 74.Arnett FC, Tan FK, Uziel Y, Laxer RM, Krafchik BR, Antohi S, Bona C. Autoantibodies to the extracellular matrix microfibrillar protein, fibrillin 1, in patients with localized scleroderma. Arthritis Rheum. 1999;42:2656–2659. doi: 10.1002/1529-0131(199912)42:12<2656::AID-ANR22>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 75.Pandey JP, Page GP, Silver RM, LeRoy EC, Bona CA. Anti-fibrillin-1 autoantibodies in systemic sclerosis are GM and KM allotype-restricted. Exp Clin Immunogenet. 2001;18:123–129. doi: 10.1159/000049191. [DOI] [PubMed] [Google Scholar]
- 76.Loeys B, De Backer J, Van Acker P, Wettinck K, Pals G, Nuytinck L, Coucke P, De Paepe A. Comprehensive molecular screening of the FBN1 gene favors locus homogeneity of classical Marfan syndrome. Hum Mutat. 2004;24:140–146. doi: 10.1002/humu.20070. [DOI] [PubMed] [Google Scholar]
- 77.Ono RN, Sengle G, Charbonneau NL, Carlberg V, Bachinger HP, Sasaki T, Lee-Arteaga S, Zilberberg L, Rifkin DB, Ramirez F, Chu ML, Sakai LY. Latent transforming growth factor beta-binding proteins and fibulins compete for fibrillin-1 and exhibit exquisite specificities in binding sites. J Biol Chem. 2009;284:16872–16881. doi: 10.1074/jbc.M809348200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sakai LY, Keene DR. Fibrillin: monomers and microfibrils. Methods Enzymol. 1994;245:29–52. doi: 10.1016/0076-6879(94)45004-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.