

Abstract
In adipocytes, PDE3B (phosphodiesterase 3B) is an important regulatory effector in signalling pathways controlled by insulin and cAMP-increasing hormones. Stimulation of 3T3-L1 adipocytes with insulin or the β3-adrenergic receptor agonist CL316243 (termed CL) indicated that insulin preferentially phosphorylated/activated PDE3B associated with internal membranes (endoplasmic reticulum/Golgi), whereas CL preferentially phosphorylated/activated PDE3B associated with caveolae. siRNA (small interfering RNA)-mediated KD (knockdown) of CAV-1 (caveolin-1) in 3T3-L1 adipocytes resulted in down-regulation of expression of membrane-associated PDE3B. Insulin-induced activation of PDE3B was reduced, whereas CL-mediated activation was almost totally abolished. Similar results were obtained in adipocytes from Cav-1-deficient mice. siRNA-mediated KD of CAV-1 in 3T3-L1 adipocytes also resulted in inhibition of CL-stimulated phosphorylation of HSL (hormone-sensitive lipase) and perilipin A, and of lipolysis. Superose 6 gel-filtration chromatography of solubilized membrane proteins from adipocytes stimulated with insulin or CL demonstrated the reversible assembly of distinct macromolecular complexes that contained 32P-phosphorylated PDE3B and signalling molecules thought to be involved in its activation. Insulin- and CL-induced macromolecular complexes were enriched in cholesterol, and contained certain common signalling proteins [14-3-3, PP2A (protein phosphatase 2A) and cav-1]. The complexes present in insulin-stimulated cells contained tyrosine-phosphorylated IRS-1 (insulin receptor substrate 1) and its downstream signalling proteins, whereas CL-activated complexes contained β3-adrenergic receptor, PKA-RII [PKA (cAMP-dependent protein kinase)-regulatory subunit] and HSL. Insulin- and CL-mediated macromolecular complex formation was significantly inhibited by CAV-1 KD. These results suggest that cav-1 acts as a molecular chaperone or scaffolding molecule in cholesterol-rich lipid rafts that may be necessary for the proper stabilization and activation of PDE3B in response to CL and insulin.
Keywords: adipocyte, β3-adrenergic receptor, caveolin-1, insulin, protein kinase A (PKA), phosphodiesterase 3 (PDE3)
INTRODUCTION
Cyclic nucleotide PDEs (phosphodiesterases) are critical regulators of intracellular concentrations and physiological effects of cAMP and cGMP. The PDE superfamily contains 11 structurally related, but functionally distinct, gene families (PDE1–11) [1]. PDE3A and PDE3B isoforms, encoded by two similarly organized genes, hydrolyse cAMP and cGMP with high affinity in a mutually competitive manner [2]. PDE3B is relatively abundant in tissues that maintain energy homoeostasis [3]; previous results from PDE3B-transgenic mice indicate that PDE3B plays an important role in modulation of energy metabolism [4–6].
PKB (protein kinase B), a downstream effector of PI3K (phosphoinositide 3-kinase), mediates many metabolic, mitogenic and anti-apoptotic effects of insulin and IGF (insulin-like growth factor)-1 [3,7,8]. In adipocytes [9,10] and FDCP2 myeloid cells [11,12], insulin [9,10] and IGF-1 [11,12] phosphorylate/activate PDE3B via PI3K- and PKB-dependent signals. In adipocytes, insulin-mediated phosphorylation/activation of membrane-associated PDE3B leads to a reduction in PKA (cAMP-dependent protein kinase) activity and inhibition of lipolysis [3,13]. Inhibition of adipocyte PDE3B blocks the antilipolytic action of insulin, and also reduces insulin-stimulated lipogenesis and glucose uptake [13]. Stimulation of adipocytes with isoprenaline (isoproterenol) and other cAMP-increasing agents also induces phosphorylation/activation of PDE3B [2,3].
In adipocytes, PDE3B is found in both PM (plasma membrane)/caveolae and ER (endoplasmic reticulum)/Golgi membrane fractions [14,15]. As found in the present study, in 3T3-L1 adipocytes, insulin or CL (CL316243) preferentially phosphorylate/activate PDE3B associated with ER/Golgi or PM/caveolae fractions respectively, and induce formation of different macromolecular complexes [HMWCs (high-molecular-mass macromolecular complexes)] that contain 32P-phosphorylated PDE3B and signalling proteins thought to be involved in its activation. Targeted inactivation of Cav-1 (caveolin-1) in mice [16], and, as reported in the present paper, siRNA (small interfering RNA)-induced CAV-1 KD (knockdown) in 3T3-L1 adipocytes, resulted in reduction of β3-AR (β3-adrenergic receptor) agonist-stimulated phosphorylation of perilipin and lipolysis. In adipocytes from Cav-1−/− mice and in CAV-1-KD 3T3-L1 adipocytes, expression of membrane-associated PDE3B was reduced, and activation of PDE3B by CL was inhibited to a much greater extent than its activation by insulin. Formation of insulin- and CL-induced HMWCs was significantly attenuated in CAV-1-KD adipocytes, which might be attributed to a chaperone or scaffolding role of cav-1 in cholesterol-rich lipid domains that may be necessary for proper stabilization and activation of PDE3B within these macromolecular signalling complexes.
MATERIALS AND METHODS
Materials
3T3-L1 cells were purchased from American Type Culture Collection; DMEM (Dulbecco’s modified Eagle’s medium) and FBS (fetal bovine serum) were from Gemini Bio-Products; IBMX (isobutylmethylxanthine), insulin and dexamethasone were from Sigma; [3H]cAMP was from New England Nuclear; [γ -32P]ATP (3000 Ci/mmol) and [32P]Pi (1000 mCi/mmol) were from ICN Radiochemicals; SuperSignal® Westpico and Westfemto chemiluminescent reagents were from Pierce; polyclonal anti-p85 PI3K, -β3-AR and -PKB antibodies were from Upstate Biotechnology; anti-IRS (insulin receptor substrate)-1, -PKB1, -pPKB (phosphorylated PKB; Ser473), −pHSL [phosphorylated HSL (hormone-sensitive lipase); Ser563], −CREB (cAMP-response-element-binding protein), −pCREB (phosphorylated CREB) and −phospho (Ser/Thr) PKA substrate (p-perilipin) antibodies were from Cell Signaling Technology; an anti-perilipin antibody was from Abcam; anti-cav-1, −PKA-RII (PKA regulatory subunit), −BiP (immunoglobulin heavy-chain-binding protein), −GM130 (Golgi matrix protein 130), -nucleoporin p62,-β-catenin and -phosphotyrosine antibodies were from BD Biosciences; anti-A-cyclase V/VI, -14-3-3 and -HSP-90 (heat-shock protein 90) antibodies were from Santa Cruz Biotechnology; prestained molecular-mass markers were from Bio-Rad; and the anti-HSL antibody was a gift from Dr Cecilia Holm (Lund University, Lund, Sweden). Rabbit polyclonal antibodies against mouse PDE3B (GenBank® accession number AAN52086) were generated against peptides corresponding to the CT (C-terminal) domain (amino acids 1076–1095; NASLPQADEIQVIEEADEEE), the RD (regulatory) domain (amino acids 266–280; VIRPRRRSSCVSLGE) and the NT (N-terminal) domain (amino acids 2–16; RKDERERDAPAM-RSP). Affinity-purified anti-PDE3B-NT and anti-PDE3B-CT antibodies were used for Western blotting and immunoprecipitation experiments. Other materials were obtained as indicated, and were of the highest grade available.
Differentiation of 3T3-L1 adipocytes
3T3-L1 preadipocytes were differentiated as previously described [14,17]. After incubation with MDI (0.3 mM IBMX, 1 μM dexamethasone and 5 μg/ml insulin) for 72 h, medium was changed to complete DMEM/10 %(v/v) FBS containing 5 μg/ml insulin, and replaced every 48 h. Experiments with differentiated 3T3-L1 adipocytes were usually performed 13–14 days after initiation of differentiation.
Preparation of adipocytes from Cav-1+/+ and Cav-1−/− mice
All animals were handled in strict accordance with good animal practice as defined by the national and local animal welfare bodies, and all animal work was approved by the Ethics Committee at Lund University and Linkoping University, Sweden. Adipocytes were prepared from epididymal fat pads from 10–12-week-old Cav-1−/− and Cav-1+/+ mice by collagenase digestion [18], with some modifications [19]. Cells (3–8 % cell suspension) were resuspended at 37 °C in KRH (Krebs–Ringer-Hepes medium), pH 7.4, containing 25 mM Hepes, 200 nM adenosine, 2 mM glucose and 1 % BSA. Adipocytes were stimulated with insulin (2 nM for 10 min) or CL (10 μM for 15 min), homogenized in 1 ml of buffer [40 mM Hepes (pH 7.4), 10 mM NaF, 1 mM PMSF, 0.25 mM sodium orthovanadate, 20 nM calyculin A, 1 mg/ml pepstatin A, 10 mg/ml leupeptin and 10 mg/ml antipain], and centrifuged (50 000 g for 60 min). The fat cake was removed and the pellet was resuspended in buffer [50 mM Tes (pH 7.4), 50 mM sucrose, 1 mM EDTA, 0.1 mM EGTA, 1 mg/ml pepstatin A, 10 mg/ml leupeptin and 10 mg/ml antipain] for BCA (bicinchoninic acid) protein measurement and PDE assays.
cAMP PDE assay
PDE3 activity {that portion of total PDE activity inhibited by 1.0 μM cilostamide, a selective PDE3 inhibitor (IC50 ~17–80 nM); [20]}, was measured by modification of our previously published method [21], using 0.1 μM [3H]cAMP (35000 c.p.m.) as a substrate. PDE activity is expressed as pmol of cAMP hydrolysed/min per mg of protein.
Fractionation of membranes on continuous 10–45 % sucrose gradients
3T3-L1 adipocytes (100-mm culture dishes) were incubated in serum-free DMEM (16 h at 37 °C), to reduce the possible effects of growth factors in serum. After incubation without or with insulin (100 nM for 10 min) or CL (10 μM for 15 min), adipocytes were washed and homogenized (in a Dounce homogenizer) in ice-cold buffer A [50 mM Hepes, 50 mM sucrose, 1 mM EDTA, 10 mM pyrophosphate, 5 mM NaF, 100 mM NaCl, 0.1 μM okadaic acid, 1 mM sodium orthovanadate and Roche protease inhibitor cocktail (pH 7.5)]. After centrifugation at 500 g, total membrane and cytosol fractions were prepared by centrifugation of supernatants [37 600 rev./min (in a SW41 Ti rotor; Beckman) for 30 min at 4 °C] [17]. Membrane fractions were centrifuged on continuous 10–45 % sucrose gradients, as described previously [15]. After centrifugation, gradients were manually divided into 17 equal fractions (each of 0.7 ml), collected sequentially from the top.
Superose 6 chromatography of solubilized 3T3-L1 adipocyte membrane fractions
Adipocytes were incubated in serum-free DMEM (for 16 h), and then without or with insulin (100 nM for 10 min) or CL (10 μM for 15 min), washed twice, and homogenized in ice-cold buffer B (buffer A containing 250 mM sucrose). Total membrane and cytosolic fractions were prepared as described above. Membrane proteins were solubilized by homogenization (using a Dounce homogenizer) and sonication of membrane pellets in buffer B containing 1 % (v/v) Nonidet P40, and after incubation/rotation by centrifugation [28 000 rev./min for 30 min at 4 °C, using a SW41 Ti rotor (Beckman)]. Portions of solubilized membranes (1.0 ml, 3 mg of protein) were applied to a Superose 6 HR 10/30 column (Amersham), which was equilibrated and eluted with buffer B (without sucrose) containing 150 mM NaCl and 1 %Nonidet P40. Portions of fractions (0.5 ml) were used for immunoblotting and immunoprecipitations, and for assay of PDE3 activity. The cholesterol content of fractions was assayed using Amplex® Red cholesterol assay kits (Molecular Probes).
32P-phosphorylation of PDE3B
Adipocytes were incubated overnight in serum-free DMEM, then with and without [32P]Pi (200 μCi/ml) for 90 min, and then without or with insulin (100 nM for 10 min) or CL (10 μM for 15 min). Samples of solubilized membrane proteins from subcellular fractions, sucrose-gradient fractions or Superose 6 fractions were immunoprecipitated with 20–25 μl of anti-PDE3B-CT antibodies as described below. Immunoprecipitated proteins were separated by SDS/PAGE, gels were washed, and [32P]-labelled PDE3B was detected using phosphoimager (Amersham) analysis of wet gels, after which proteins were electrophoretically transferred on to nitrocellulose membranes for immunoblotting.
Immunoprecipitation and immunoblotting
In some experiments, total lysates were prepared by homogenization (using a Dounce homogenizer, 15–20 strokes) of adipocytes in buffer B containing 1 % Nonidet P40 and centrifugation (10 000 g for 10 min at 4 °C). After the fat cake was removed, samples were resuspended, extracted (30 min on ice) by rotation, and centrifuged (10 000 g for 10 min at 4 °C). Portions of supernatants containing whole-cell extracts were subjected to SDS/PAGE and Western blotting, or analysed for protein concentration using BCA protein assay kits (Pierce), with BSA as a standard.
For immunoprecipitations, solubilized membrane, cytosol or column fractions were adjusted, when necessary, to 1 %Nonidet P40 (final concentration). After solubilization of membrane fractions and centrifugation [28 000 rev./min (using a SW41 Ti rotor; Beckman) for 30 min at 4 °C], supernatants were usually adjusted to 3 mg of protein/ml. For most experiments, samples were cleared by incubation [1 h at room temperature (20 °C)] with 5 μg of non-immune IgG, and then with 50 μl of Protein G–Sepharose (Amersham) for 30 min, before centrifugation (2800 g at 4 °C for 5 min). Cleared fractions were incubated (overnight at 4 °C) with the specified antibodies, followed by incubation (for 1 h) with fresh Protein G–Sepharose before centrifugation (2800 g at 4 °C for 5 min). Washed immunoprecipitates were subjected to SDS/PAGE, electrotransferred on to membranes, and immunoblotted with the appropriate primary antibody, and then with HRP (horseradish peroxidase)-labelled secondary antibody (Pierce). Immunoreactive proteins were reacted with Supersignal® Westpico or Westfemto chemiluminescent reagents; signals were detected with a Fuji Imagereader LAS3000.
siRNA KD of caveolin
siRNA duplex oligonucleotides (Dharmacon ‘smartpool’, catalogue number L-058415-00) and a control, scrambled, non-targetting siRNA oligonucleotide (catalogue number D-001810-10), used as a negative control, were purchased from Dharmacon. The CAV-1 siRNA oligonucleotides (a pool of four siRNAs for CAV-1) were targeted to sequences in the CAV-1 mRNA (GenBank® accession number NM_007616) that started at positions 91, 454, 534 and 564. Information concerning the siRNA ‘smartpool’ is as follows: (i) GenBank® accession number NM_007616, pool catalogue number L-058415-00, duplex catalogue number J-058415-05, sequence (564) 5′-GCUAUUGGCAAGAUAUUCA-3′; (ii) GenBank® accession number NM_007616, pool catalogue number L-058415-00, duplex catalogue number J-058415-06, sequence (454) 5′-GCACAUCUGGGCGGUUGUA-3′; (iii) GenBank® accession number NM_007616, pool catalogue number L-058415-00, duplex catalogue number J-058415-07, sequence (91) 5′-GCAAAUACGUGGACUCCGA-3′; and (iv) GenBank® accession number NM_007616, pool catalogue number L-058415-00, duplex catalogue number J-058415-08, sequence (534) 5′-GUCCAUACCUU-3′.
Optimal conditions for siRNA KD involved transfecting adipocytes with siRNA using MBS (modified bovine serum) mammalian transfection reagent (Stratagene) in DMEM, following the manufacturer’s protocol. After 10 h, adipocytes were supplemented with 10 % (v/v) FBS, and further incubated for 46 h. After 56 h, adipocytes were incubated (16 h) in serum-free DMEM, and then without or with insulin and/or CL as indicated. Additional experiments with a second set of siRNA duplex oligonucleotides or Ad (adenoviral) siRNA vectors are described in the Supplementary Online Data (at http://www.BiochemJ.org/bj/424/bj4240399add.htm).
Lipolysis assay
All in vitro lipolysis experiments were performed with 14-day post-confluent 3T3-L1 adipocytes, grown and differentiated in 12-well tissue culture plates. Adipocytes were equilibrated (0.5–1 h at 37 °C) in fresh DMEM/10 % (v/v) FBS, and then washed twice with PBS (prewarmed to 37 °C). To initiate lipolysis, PBS was removed, and replaced with 0.5 ml of KRH buffer [25 mM Hepes (pH 7.4), 125 mM NaCl, 5 mM KCl, 1 mM KH2PO4, 2.5 mM CaCl2 and 2.5 mM MgCl2], containing 3 % fatty-acid-free BSA (Intergen) and 5 mM glucose. Treatment with CL and/or insulin was carried out as indicated at 37 °C in a 5 % CO2 atmosphere for periods ranging between 10 min and 2 h, after which medium was collected for glycerol analyses, and whole-cell lysates were prepared for protein analysis. Glycerol content, determined using a lipolysis assay kit (Zen-Bio), was normalized for total cellular protein, which was quantified using BCA kits.
RESULTS
Insulin or CL preferentially phosphorylate/activate PDE3B in Golgi/ER or PM/caveolae respectively
As described previously [14,15] and as seen in Figure 1, based on the distribution of molecular ‘markers’, PM (β3-AR and cav-1), Golgi (GM130), ER (BiP) and nuclear (P62) proteins were separated during centrifugation of 3T3-L1 adipocyte membranes on 10–45 %continuous sucrose gradients. Membrane-associated PDE3B was found in the PM and ER/Golgi fractions (Figure 1) [17]. Cav-1, present in different subcellular compartments [22], was distributed in the PM and internal membranes (Figure 1). We have reported previously that PDE3B is associated with PM/caveolae in rodent adipocytes [14,15]; in extending these studies, we demonstrated localization of human PDE3B to caveolae via immunogold labelling and electron microscopy of PM sheets from primary human adipocytes (Supplementary Figure S1 at http://www.BiochemJ.org/bj/424/bj4240399add.htm). Consistent with Western blot analysis (Figure 1), approx. 35 % of total membrane PDE3 activity was found in the PM/caveolae, and 65 %in ER/Golgi fractions (Figure 2A) [17].
Figure 1. Sucrose-gradient fractionation of 3T3-L1 adipocyte membranes.

Adipocytes were incubated without (control) or with insulin (100 nM for 10 min) or CL (10 μM for 15 min). Total membranes were prepared and centrifuged on 10–45 % continuous sucrose gradients, and 17 fractions (each of 0.7 ml) were collected manually from the top of the tube. Portions (25 μl) of fractions were subjected to SDS/PAGE, and immunoblotted with specific antibodies as indicated, including BiP (ER marker), GM130 (Golgi marker), β3-AR (PM marker) and P62 (nuclear marker). Results shown are representative of four experiments for control and insulin-stimulated adipocytes and two experiments for CL-stimulated adipocytes.
Figure 2. Insulin or CL differentially phosphorylate and activate PDE3B in ER/Golgi or PM/caveolae fractions respectively.

Adipocytes were incubated without or with [32P]Pi for 90 min, and then without (c, control) or with insulin (I/Ins) (100 nM for 10 min) or CL (10 μM for 15 min) as indicated. Total membranes were prepared and fractionated on 10–45 % continuous sucrose gradients, and 17 fractions (each of 0.7 ml) were collected manually. (A) Portions (10 μl) of fractions from adipocytes not incubated with [32P]Pi were assayed for PDE3 activity (pmol of cAMP hydrolysed/min per mg of protein), as described in the Materials and methods section, and are presented as means ± S.E.M. (n = 3 experiments). (B) Portions (400 μl) of fractions from adipocytes labelled with [32P]Pi were analysed, after immunoprecipitation of solubilized PDE3B with an anti-PDE3B-CT antibody, for [32P]PDE3B by phosphoimager analysis on wet gels (right-hand panels) and for PDE3B protein by immunoblotting (left-hand panels). The location of the PM and ER is indicated at the bottom. A representative experiment is shown (n = 2). IB, immunoblotting.
Incubation of 3T3-L1 adipocytes with insulin (100 nM for 10 min) increased PDE3 activity more than 2-fold in ER/Golgi, compared with a much smaller increase in PM fractions (Figure 2A), similar to our previous findings [17]. On the other hand, CL-stimulation of β3-ARs activated PDE3 more than 2-fold in PM fractions compared with a much smaller increase in ER/Golgi (Figure 2A). There did not appear to be significant translocation of PDE3B between membrane fractions after stimulation of adipocytes with insulin or CL (Figure 1). Consistent with spatial segregation of the effects of insulin and CL on activation of PDE3B, during stimulation of [32P]Pi-labelled adipocytes with insulin or CL, PDE3B was phosphorylated to a greater extent in ER/Golgi or caveolin-enriched PM fractions respectively (Figure 2B). As seen in Figure 1, insulin stimulation also resulted in enrichment of PKB and pPKB in both PM and ER/Golgi membranes compared with unstimulated adipocytes, whereas CL stimulation was associated with enrichment of PKA-RII in PM fractions. Taken together, these results strongly suggest different signalling roles for PDE3Bs, located in different microdomains of the same cell.
Insulin or CL induce the formation of different macromolecular complexes that contain PDE3B
Previously we have demonstrated that insulin induced formation of macromoleclar complexes that contained PDE3B [17]. To characterize and compare HMWC-ins (HMWCs induced by insulin) or HMWC-CL (HMWCs induced by CL), solubilized total membranes were fractionated by gel filtration on Superose 6 [(HR 10/30) columns (exclusion molecular mass > 4 × 107)]. As seen in Supplementary Figure S2 (at http://www.BiochemJ.org/bj/424/bj4240399add.htm), 1 % Nonidet P40 was more efficient in solubilizing membrane-associated PDE3B than octyl methyl glucoside or zwitterion (CHAPS) detergents. During chromatography on Superose 6 (Figure 3), solubilized PDE3B from unstimulated adipocytes exhibited a molecular mass of ~ 600–900 kDa, much larger than its free monomeric molecular mass of ~ 135 kDa. After incubation of adipocytes with either insulin or CL, PDE3 activity increased and approx. 50 %of PDE3B eluted at a higher apparent molecular mass of ≥ 3000 kDa, consistent with its presence in HMWCs (Figure 3A, top-left panel) induced by insulin or CL. Cholesterol was detected in both HMWCs and low-molecular-mass protein peaks. Formation of HMWC-ins or HMWC-CL coincides with elution of activated PDE3B and increased amounts of cholesterol with the multiprotein complex(es) (Figure 3A, top-right panel). Insulin treatment altered the elution patterns of several signalling proteins, including IRS-1, PI3K p85, PKB, HSP-90, 14-3-3, PP2A (protein phosphatase 2A), perilipin and cav-1, to the extent that portions of these molecules co-eluted with PDE3B at ≥3000 kDa (Figure 3A, bottom panel). The migration of some of these proteins, including 14-3-3, PP2A, perilipin and cav-1, were altered in the same way in response to CL. CL treatment specifically altered the migration pattern of PKA-RII, β3-AR and HSL. CL treatment selectively altered migration of adenylate cyclase in three out of four experiments (Figure 3A); in one experiment elution was altered by insulin as well as CL. Little or no insulin receptor (results not shown), β3-AR or PKA-RII co-eluted with HMWC-ins, whereas little or no IRS, PI3K or PKB co-eluted with HMWC-CL.
Figure 3. Formation of different PDE3B macromolecular signalling complexes in response to insulin and CL.

(A) CL or insulin stimulates assembly of different macromolecular complexes. Solubilized total membranes from adipocytes (3 mg of protein, 1 ml), prepared as described in the Materials and methods section, incubated without (Control, C) or with CL (10 μM for 15 min) or insulin (Ins) (100 nM for 10 min), were subjected to chromatography on Superose 6. Fractions from control (●,○) and CL- (▲,△) or insulin- (■,□) stimulated adipocytes were analysed. Top-left-hand panel: portions (10 μl) of fractions were assayed for PDE3 activity (pmol of cAMP hydrolysed/0.5 ml per min); top-middle panel: protein content [AU (absorption units) 280 nm]; and top-right-hand panel: cholesterol content [RFU (relative fluorescence units)/5 μl]. Molecular mass standards are seen as peaks in the top-left-hand and middle panels: 1, thyroglobulin (670 kDa); 2, γ -globulin (158 kDa); 3, ovalbumin (44 kDa); 4, myoglobin (17 kDa); 5, vitamin B12 (1.35 kDa). Bottom panels: samples (20 μl) of indicated fractions (0.5 ml) were subjected to SDS/PAGE and immunoblotted with specific antibodies as indicated. Void volume (Vo)>3000 kDa and molecular mass standards, thyroglobulin (670 kDa) and γ -globulin (158 kDa), are indicated. One representative experiment is shown (n = 4). (B) CL or insulin induces formation of macromolecular complexes which contain phosphorylated/activated PDE3B. Solubilized total membranes (3 mg of protein, 1 ml), prepared from adipocytes incubated without (C, control) or with CL (10 μM for 15 min) or insulin (Ins) (100 nM for 10 min), were subjected to chromatography on Superose 6. Fractions from control (●,○), CL- (▲,△) and insulin- (■,□) stimulated adipocytes were analysed. Top-left-hand panel: portions (10 μl) of fractions were assayed for PDE3 activity (pmol of cAMP hydrolysed/0.5 ml per min), means ± S.E.M. (n = 3); top-right-hand panel: protein content [AU (absorption units) 280 nm]. Bottom panels: PDE3B was immunoprecipiated from samples (400 μl) of fractions from adipocytes labelled with [32P]Pi as described in the Materials and methods section. Immunoprecipitates were subjected to SDS/PAGE, and 32P-labelled PDE3B was detected in wet gels by phosphoimager analysis (left-hand panels) before the same gels were used for immunoblotting with anti-PDE3B-CT antibody (right-hand panels). One representative experiment is shown (n = 3). IB, immunoblotting.
After insulin or CL stimulations (Figure 3B), fractionation of solubilized membranes from 32P-labelled adipocytes on Superose 6, and phosphoimager analysis of immunoprecipitated PDE3B, confirmed that the HMWCs contained most of the 32P-phosphorylated and activated PDE3B. This effect of insulin or CL on phosphorylation/activation of PDE3B and formation of macromolecular complexes was prevented by prior treatment with either the PI3K inhibitor wortmannin [17] or the PKA inhibitor H89 respectively (results not shown).
Interactions between PDE3B and other proteins in HMWCs were studied in pull-down assays. Samples of Superose 6 fractions from (Figure 3A) control, and insulin- or CL-treated adipocytes were pooled into three groups (group A, fractions 17–21; group B, fractions 22–30; group C, fractions 32–40). Pooled fractions were immunoprecipitated with anti-PDE3B-CT antibody and immunoblotted. Several signalling proteins co-immunoprecipitated with PDE3B in the high-molecular-mass (>3000 kDa) fraction (group A) of the insulin- or CL-stimulated complexes. Signalling proteins in pooled fractions from groups B and C from control and insulin- or CL-stimulated cells did not co-immunoprecipitate with PDE3B (Figure 4). These studies suggest that PDE3B is a component of multi-molecular signalling complexes in adipocytes, containing IRS-1, PI3K p85, PKB and HSP-90 in insulin-induced cells, and PKA-RII and β3-AR in CL-treated cells, in addition to other unidentified proteins. Molecules common to both include 14-3-3, PP2A and cav-1.
Figure 4. Insulin- or CL-induced assembly of macromolecular complex(es) containing PDE3B and signalling proteins: co-immunoprecipitation with PDE3B.

Superose 6 fractions, as in Figure 3, were divided into three groups (A, B and C), and pooled for co-immunoprecipitation (Group A, fractions 17–21; Group B, fractions 22, 24, 26, 28 and 30; Group C, fractions 32, 34, 36, 38 and 40). Pooled fractions were cleared, and complexes containing PDE3B were immunoprecipitated with 20 μl of anti-PDE3B-CT antibodies. Group A fractions were also immunoprecipitated with control rabbit IgG. Protein G–Sepharose-bound proteins were eluted in 100 μl of Laemmli’s sample buffer. Samples (15 μl) were subjected to SDS/PAGE and immunoblotting with specific antibodies as indicated. One representative experiment is shown (n = 3), with the exception that immunoprecipitation/Western blot analysis for β3-AR were conducted in two experiments. IP, immunoprecipitation.
Effects of down-regulation of CAV-1 on PDE3B expression and activation
Differentiated 3T3-L1 adipocytes (~ day 10) were transfected with increasing concentrations of control (non-targeting siRNA) or CAV-1 siRNA (50–150 nM) (Figure 5A). Transfection with control siRNA did not affect the expression of CAV-1, compared with untransfected control cells (Figure 5A and Supplementary Figure S3 at http://www.BiochemJ.org/bj/424/bj4240399add.htm); CAV-1 expression was efficiently knocked-down in a dose-dependent manner by CAV-1 siRNA (Figure 5A). Specific CAV-1 KD (100 nM siRNA) was confirmed by qRT-PCR (quantitative real-time PCR) analysis (Figure 5B). As seen in Supplementary Figure S3, another synthetic CAV-1 siRNA and an Ad CAV-1 siRNA similarly inhibited expression of CAV-1 in a concentration-dependent manner, confirming the specificity of CAV-1 KD.
Figure 5. siRNA-mediated KD of CAV-1 expression in 3T3-L1 adipocytes.
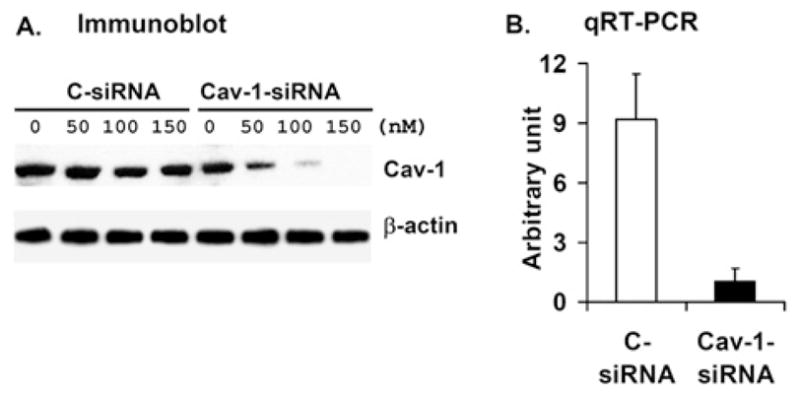
Differentiated 3T3-L1 adipocytes were transfected as described in the Materials and methods section with the indicated concentrations of duplex siRNA targeted against CAV-1 (cav-1 siRNA) or a control, scrambled sequence (C-siRNA). (A) At 72 h after transfection, cell lysates were prepared as described in the Materials and methods section, and subjected to SDS/PAGE and Western blot analysis with anti-cav-1 and anti-β-actin antibodies. One representative Western blot is shown (n = 2). (B) After transfection with 100 nM siRNA, CAV-1 KD was confirmed by qRT-PCR analysis (see Supplementary Online Data at http://www.BiochemJ.org/bj/424/bj4240399add.htm). Data are presented as means±half of the range (n = 2). For each experiment, RNA was isolated from three different plates in each experimental group, and was assayed using qRT-PCR in duplicate.
As shown in Figure 6, CAV-1 KD reduced expression of PDE3B by ~ 40 % (top panel); in a total of seven similar experiments (including those discussed in Supplementary Figure S4 at http://www.BiochemJ.org/bj/424/bj4240399add.htm), PDE3B expression was reduced by 34.6±2.8 % (mean±S.E.M., n = 7, P < 0.01) in CAV-1-KD cells. This may reflect a decreased half-life of PDE3B protein, since a recent report has demonstrated increased degradation of insulin receptor and GLUT4 (glucose transporter 4) proteins in 3T3-L1 KD adipocytes, stably transfected with lenteviral CAV-1 siRNA [23]. As seen in Figure 6 (middle panel), insulin (100 nM for 10 min) or CL (10 μM for 15 min) increased PDE3B activity ~ 2-fold in cells treated with scrambled control siRNA (S-siRNA). In KD adipocytes, insulin-induced activation was partially inhibited, whereas CL-induced activation was almost completely inhibited. These results were confirmed by transfection/infection with a second oligonucleotide CAV-1 siRNA and Ad CAV-1 siRNA (Supplementary Figure S4). As shown in Figure 6 (lower panel), when PDE3B activity was normalized to PDE3B expression, basal PDE3B activity was similar in control and KD cells. CL-induced activation was almost completely inhibited; insulin-induced activation was reduced by 36±1.9 % (mean±S.E.M., n = 7, P < 0.01). In support of these results, we found that in adipocytes isolated from Cav-1−/− mice, PDE3B was down-regulated and CL-, but not insulin-, induced activation of PDE3B was inhibited (Figure 7).
Figure 6. KD of CAV-1 is associated with reduced PDE3B expression and impaired activation of PDE3B.

At 72 h after transfection with control (C-) or CAV-1 siRNA, control and CAV-1-KD adipocytes were incubated without or with CL (10 μM for 15 min), insulin (100 nM for 10 min) or both. For the combination treatment, the cells were treated with insulin for 10 min followed by CL for 10 min. Total membranes were prepared as described in the Materials and methods section. Membrane proteins were solubilized by homogenization (using a Dounce homogenizer) and sonication of membrane pellets in buffer B containing 1 % (v/v) Nonidet P40, and, after incubation/rotation (30 min for 4 °C), by centrifugation (10 000 g for 30 min at 4 °C). Solubilized membrane proteins were assayed for PDE3 activity, or subjected to SDS/PAGE (30 μg) and Western blot analysis. Upper panel: one representative Western blot is shown (n = 7, including those shown in the Supplementary Online Data at http://www.BiochemJ.org/bj/424/bj4240399add.htm). The scanning densitometric ratio of PDE3B/β-actin (means±S.E.M.) for this experiment is 0.99±0.07 (n = 4) and 0.59±0.05 (n = 4) for S-siRNA control and CAV-1-KD adipocytes respectively; for the seven experiments, taken together, values were 0.99±0.02 and 0.65±0.03 (n = 7) for S-siRNA control and CAV-1-KD adipocytes respectively. Middle panel: PDE3 activity data (pmol of cAMP hydrolysed/min per mg of protein) is presented as mean±S.E.M. (n = 7, including those experiments shown in the Supplementary Online Data at http://www.BiochemJ.org/bj/424/bj4240399add.htm). *P < 0.001 and +P = NS (not significant), compared with their respective controls. For each experiment, cells were harvested from two plates and PDE3 activity assays were carried out in duplicate. Bottom panels: PDE3 activity was normalized to PDE3 expression (0.99 for S-siRNA control; 0.65 for CAV-1-KD adipocytes, n = 7), which was quantified as described above.
Figure 7. Effects of insulin or CL on PDE3 activity in adipocytes from WT (wild-type) and Cav-1−/− mice.
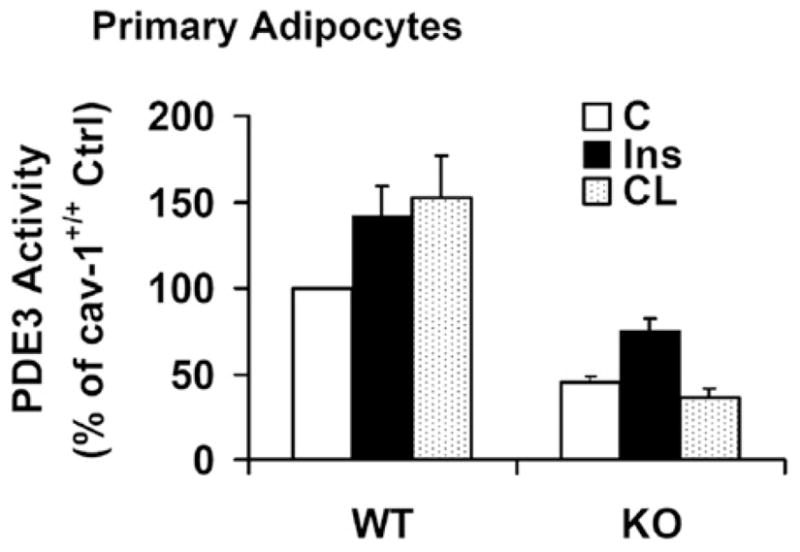
Adipocytes from Cav-1+/+ (WT) and Cav-1−/− (KO) mice were prepared, incubated without (Ctrl) or with insulin (Ins) (2 nM for 10 min) or CL (10 μM for 15 min), and membrane-associated PDE3 activity was measured as described in the Materials and methods section. Results are presented as the percentage of wild-type control PDE3 activity (means + S.E.M., n = 4–7); absolute values were 20–90 pmol/min per mg of protein.
Down-regulation of CAV-1 impairs CL-stimulated lipolysis and phosphorylation of PKA substrate proteins
Although several kinases can phosphorylate CREB, its phosphorylation generally correlates with PKA activity in cells [24]. As seen in Figure 8(A), CAV-1 KD inhibited phosphorylation of CREB in adipocytes incubated with and without CL or insulin, compared with control cells. Using a perilipin-specific antibody and a phospho-PKA-motif-specific antibody [P-RXX(pS/T)], we found that perilipin phosphorylation was dramatically decreased in CAV-1-KD cells in response to CL stimulation, without any significant changes in perilipin content (Figure 8A), consistent with studies in Cav-1−/− mouse adipocytes [25]. Phosphorylation of HSL, another PKA substrate, was also dramatically decreased, with a small decrease in HSL protein. Insulin inhibited CL-stimulated phosphorylation of HSL, but not of perilipin. There was no change in the expression of PKA-RII, β3-AR or β-actin (Figure 8A). In control 3T3-L1 adipocytes, CL increased lipolysis in a concentration-dependent manner (0.0001–1 μM) (results not shown). As shown in Figure 8(B), CL (100 nM) markedly stimulated lipolysis in control adipocytes (~ 6.5-fold), but was much less effective (~ 1.8-fold) in CAV-1-KD adipocytes, consistent with inhibition of CL-stimulated lipolysis in adipocytes from Cav-1−/− mice [25]. These studies were confirmed by transfection/infection with a second CAV-1 siRNA or Ad-CAV-1 siRNA, (Supplementary Figures S5 and S6 at http://www.BiochemJ.org/bj/424/bj4240399add.htm). Thus, in adipocytes, caveolin may be a scaffolding factor that is normally required for PKA-mediated signalling, including phosphorylation/activation of PDE3B as well as phosphorylation of perilipin and HSL, and activation of lipolysis.
Figure 8. Effects of CAV-1 KD on lipolysis, and insulin- or CL-stimulated phosphorylation of PKB and PKA substrates.

3T3-L1 adipocytes (the same group of transfected adipocytes was used for experiments presented in Figures 8 and 9A–9C) were transfected with 100 nM CAV-1 siRNA or S-siRNA. After 72 h, adipocytes were incubated without or with 100 nM CL or 100 nM insulin (Ins) for 15 min, or a combination of the two as indicated. For the combination treatment the cells were treated with insulin for 10 min followed by CL for 15 min. (A) Cell lysates (30 μg) were prepared as described in the Materials and methods section, and subjected to SDS/PAGE and immunoblotted with antibodies against the indicated proteins. One representative experiment is shown (n = 3). (B) Adipocytes were incubated (in triplicate) without (control) or with the indicated concentrations of insulin (Ins) or CL for 1 h. Lipolysis (glycerol release) was measured as described in the Materials and methods section. Values (fold increase in glycerol release) represent the means+S.E.M. (n = 3). *P < 0.001 and +P = NS (not significant), compared with their respective controls.
Residual CL (100 nM)-stimulated lipolysis was inhibited by insulin (100 nM) in CAV-1-KD cells (Figure 8B). There was no change in the expression of total PKB, or of phosphorylation of PKB in response to insulin in CAV-1-KD adipocytes compared with control adipocytes (Figure 8A). These studies suggested that CAV-1 KD did not affect insulin-mediated activation of PKB, which is important in activation of PDE3B and the antilipolytic action of insulin. Although expression of PDE3B is reduced in CAV-1-KD adipocytes (Figure 6 and Supplementary Figure S4), activation of PDE3B by insulin/PI3K/PKB, while partially inhibited (Figure 6 and Supplementary Figure S4), is apparently sufficient to reduce cAMP, leading to inhibition of residual CL-stimulated lipolysis (Figure 8B and Supplementary Figure S5B).
CAV-1 KD inhibits formation of macromolecular complexes
Transfection with control S-siRNA did not inhibit insulin- or CL-induced formation of HMWC-ins or HMWC-CL (Figure 9B). On the other hand, CAV-1 KD (Figure 9A) not only inhibited insulin-and CL-induced activation of PDE3B (compare Figure 9C with Figure 6), but also inhibited recruitment of PDE3B into HMWCs (Figure 9C). In CAV-1-KD adipocytes (Figure 9C), CL-induced activation/recruitment of PDE3B was virtually completely blocked, whereas during incubation with insulin (Figure 9C; compare with Figure 9B), PDE3 was partially activated and recruited into modified HMWCs [smaller than HMWC-ins (Figure 9B), but larger than in control cells]. As seen in Figure 9(D), although insulin-induced phosphorylation/activation was reduced in CAV-1-KD adipocytes compared with control S-siRNA cells (top and middle panels), in 32P-labelled KD adipocytes, virtually all 32P-labelled PDE3B (middle panel) was recruited into modified HMWCs (Figure 9D), further suggesting that phosphorylation/activation of PDE3B is associated with its recruitment into HMWC-ins, and that cav-1 is important in the structural integrity (assembly or maintainence) of these complexes.
Figure 9. Effect of CAV-1 KD on CL- or insulin-induced formation of macromolecular complexes.

(A) Expression of CAV-1 after transfection of adipocytes with control scrambled (S)-siRNA or CAV-1-siRNA (cav). (B) PDE3 activities in Superose 6 fractions from adipocytes transfected with control scrambed (S)-siRNA, and incubated without (●; C-S), or with CL (▲; CL-S) or insulin (■; I-S). (C) Solubilized total membranes (3 mg of protein) were prepared from adipocytes transfected with CAV-1 siRNA and subjected to Superose 6 chromatography as described in the Materials and methods section. Fractions from control (●; C-cav), and CL- (▲; CL-cav) or insulin-stimulated (■; I-cav) adipocytes were analysed. Upper panel: PDE3 activities. Lower panel: PDE3B Western blot analysis. One representative experiment is shown (n = 2). (D) Upper panel: expression of CAV-1 after transfection of adipocytes with control scrambled (S)-siRNA or CAV-1 siRNA (cav). Second panel: PDE3 activities in Superose 6 fractions of solubilized membrane proteins from adipocytes transfected with CAV-1-siRNA (cav) and incubated without (●; Con-cav) or with insulin (▲; Ins-cav), or transfected with scrambled (S)-siRNA and stimulated with insulin (■; Ins-S). PDE3 activity (pmol of cAMP hydrolysed/min per 0.5 ml). Third panel: PDE3B was immunoprecipiated from samples (400 μl) of fractions from adipocytes labelled with [32P]Pi; immunoprecipitates were subjected to SDS/PAGE, and 32P-labelled PDE3B was detected in wet gels by phosphoimager analysis. Lower panel: the wet gels were transferred on to nitrocellulose membranes for immunoblotting with an anti-PDE3B-CT antibody. One representative experiment is shown (n = 2).
In summary, these experiments point to a chaperone or scaffolding role of cav-1 in cholesterol-rich lipid domains that may be necessary for membrane integrity and the recruitment, proper stabilization and phosphorylation/activation of PDE3B in hormone-sensitive, macromolecular signalling complexes.
DISCUSSION
Previously, we have reported that ~ 35 % of rodent adipocyte membrane-associated PDE3B was localized in caveolae-enriched PM, and ~ 65 % in internal membrane (Golgi/ER) fractions [17]. Our electron microscopy studies indicate that, in human primary adipocytes, PDE3B is also associated with PM/caveolae regions. A very important observation in the present study indicates that activation of distinct membrane pools of PDE3B by insulin or CL in 3T3-L1 adipocytes reflects formation of different macromolecular complexes, i.e. HMWC-ins or HMWC-CL. Before exposure of adipocytes to insulin or CL, PDE3B exhibited an apparent molecular mass of ~ 600–900 kDa during size-exclusion chromatography; after phosphorylation/activation by insulin or CL, the apparent molecular mass of approx. 50 % of the total PDE3B activity increased and eluted at ≥3000 kDa, indicating that phosphorylated PDE3B was recruited into HMWC-ins or HMWC-CL. PDE3B not recruited into HMWC-ins or HMWC-CL, or PDE3B from adipocytes not treated with insulin or CL, exhibited little or no phosphorylation. In addition to phosphorylated PDE3B, HMWC-ins included IRS-1, PI3K p85, PKB/pPKB, HSP-90, 14-3-3, PP2A and cav-1, all of which co-eluted and co-immunoprecipitated with PDE3B after stimulation of adipocytes with insulin. HMWC-CL contained, in addition to phosphorylated/activated PDE3B, several signalling molecules that are probably involved in activation of PDE3B via PKA-mediated signalling pathways. HMWC-CL included PKA-RII, 14-3-3, β3-AR and cav-1, all of which co-eluted and co-immunoprecipitated with PDE3B after stimulation of adipocytes with CL. HMWC-CL apparently lack IRS-1, PI3K p85, PKB and HSP-90, which were present in HMWC-ins. Molecules common to both HMWC-ins and HMWC-CL included 14-3-3, PP2A, cav-1 and cholesterol. Thus differential phosphorylation/activation of different pools of membrane-associated PDE3B most probably reflects formation of different macromolecular complexes. The consequences of differential regulation of PDE3B at different intracellular locations on the spatial and temporal regulation of cAMP pools and biological responses remain to be elucidated.
PM caveolae are a specialized subset of membrane lipid rafts which are enriched in specific lipids, especially cholesterol and glycosphingolipids, and scaffolding proteins, e.g. caveolins, that interact with numerous proteins and serve as organizers of, or platforms for, diverse signalling pathways [22,26]. Recent findings in rodent cardiac myocytes, for example, suggest that bradykinin protects the heart from ischaemia/reperfusion injury by inducing formation of, and internalization of its receptor into, caveolar vesicular signalling complexes, called signalosomes. These signalosomes recruit additional signalling molecules, migrate to mitochondria, and induce opening of the mitoKATP channel, resulting in cardioprotection [27]. In adipocytes, caveolae have been implicated in compartmentalization and regulation of cAMP- and insulin-regulated signalling pathways, e.g. lipolysis, lipogenesis and glucose uptake [13,25,26,28–31]. Although caveolae represent highly ‘ordered’ lipid rafts with decreased fluidity and greater buoyancy than other PM domains, caveolin also associates, in adipocytes, with other membrane lipid rafts with higher densities, different molar ratios of cholesterol to phospholipid, and different protein compositions, suggesting differential functional segregation in these caveolar or lipid raft subclasses [31]. Although caveolae are generally resistant to solubilization by non-ionic detergents, changes in glycosphingolipid content, especially ganglioside GM3, can increase solubilization of membrane-associated cav-1 by non-ionic detergents [32–34]. We cannot be certain whether this accounts for solubilization of some membrane-associated cav-1 with 1 % Nonidet P40 in our experiments. It is clear, however, that solubilized cav-1 co-elutes with low-molecular-mass material from control cells; after exposure of 3T3-L1 adipocytes to insulin or CL, some (not all) solubilized cav-1 co-elutes with the HMWCs.
The presence of cav-1 and cholesterol in HMWC-ins or HMWC-CL led us to evaluate the role of cav-1 in regulation of PDE3B. Cav-1−/− mice [16,25] have allowed the study of cav-1 functions in vivo. siRNA-mediated down-regulation of CAV-1 in 3T3-L1 adipocytes provided an alternative approach, avoiding possible compensatory mechanisms developed in Cav-1−/− mice. These two models provide complementary information about the effects of short- and long-term down-regulation of CAV-1 in adipocytes. The results of the present study indicate that siRNA-mediated KD of CAV-1 in 3T3-L1 adipocytes led to reduced expression of PDE3B, perhaps due to increased degradation of PDE3B protein [23]. Furthermore, in these cells, insulin-induced activation of PDE3B was partially inhibited, whereas CL-induced activation was completely inhibited. Similar results were obtained in primary adipocytes from Cav-1−/− mice. In CAV-1-KD adipocytes, CL-induced activation/recruitment of PDE3B was virtually completely blocked, whereas, in the presence of insulin, PDE3 was partially activated and recruited into modified HMWCs (smaller than HMWC-ins, but larger than in control cells).
Activation of lipolysis involves production of cAMP, activation of PKA and translocation of HSL to lipid droplets, elicited by phosphorylated perilipin [35]. Previous reports suggested that ligand-induced lipolysis in adipocytes may be mediated by association (assessed by co-immunoprecipitation) between cav-1, PKA and perilipin [25], which facilitates PKA-induced phosphorylation of perilipin. In adipocytes from Cav-1−/− mice, ligand-induced formation of the complex of PKA, perilipin and cav-1 was prevented, and CL-stimulated lipolysis was inhibited [25]. Thus reduced phosphorylation of perilipin and, perhaps, of HSL, and reduced lipolysis in CAV-1-KD adipocytes could be due to defects in the cav-1-mediated association between perilipin and PKA. We suggest that PDE3B may associate/interact with this complex, and disruption of the complex due to down-regulation of cav-1 might prevent CL-induced phosphorylation/activation of PDE3B, as well as of perilipin and HSL. In CAV-1-KD cells, even if down-regulation of PDE3B expression resulted in increased cAMP, disruption of the PKA–perilipin complex would limit access of PKA to its substrates and inhibit phosphorylation of PKA effectors and cAMP/PKA signalling. With regard to the anti-lipolytic action of insulin, our studies suggest fewer changes in insulin-induced phosphorylation/activation of PKB in CAV-1-KD adipocytes, where insulin, presumably via PKB, induces partial phosphorylation/activation of PDE3B and its recruitment into modified HMWCs, and inhibits residual CL-stimulated lipolysis. Thus cav-1 may be more important for regulation of cAMP/PKA-mediated activation of PDE3B than for insulin/PKB-mediated activation, in addition to its importance in stabilization of PDE3B expression, and its function as a scaffold for insulin- and CL-induced activation of PDE3B and recruitment of signalling molecules into HMWC-ins and HMWC-CL.
Our previous studies in 3T3-L1 adipocytes suggested that siRNA-induced PDE3B KD did not prevent insulin-stimulated recruitment of signalling molecules into HMWC-ins, indicating that PDE3B is not necessary for HMWC-ins [17]. In adipocytes, however, recruitment of recombinant PDE3B into HMWC-ins did require the presence of its NT regulatory region [17], which, in another study, was shown to contain sites phosphorylated in recombinant PDE3B in adipocytes and hepatoma cells stimulated by insulin and cAMP-elevating agents [36]. In CAV-1-KD adipocytes, CL-induced phosphorylation/activation/recruitment of PDE3B was virtually completely blocked. In the presence of insulin, however, PDE3B was partially phosphorylated/activated and recruited into modified HMWCs. Wortmannin inhibited insulin-induced phosphorylation/activation of PDE3B and formation of macromolecular complexes [17]. Thus, although in virtually all of our studies phosphorylation/activation of PDE3B is associated with its recruitment into macromolecular complexes, we cannot be certain whether phosphorylation is required for recruitment, or whether recruitment of PDE3B into (or its association with) macromolecular complexes is required for effective phosphorylation of PDE3B. Future studies with phosphorylation-site mutants in recombinant PDE3B, expressed in adipocytes, may shed light on the overall relationship, if any, between signalling and complex formation, as well as the role of phosphorylation of PDE3B in its activation and/or recruitment into HMWCs.
Taken together, these studies provide an important example, in a single cell, for subcellular localization of PDE3B, cav-1 and other signalling molecules, and their roles in the differential regulation of membrane-associated PDE3B in different subcellular compartments, via PKA and PKB signalling pathways. Different PDE4D variants, i.e. PDE4D3 and PDE4D8, have been reported to co-immunoprecipitate with PKA–AKAP (PKA-anchoring protein) complexes in different cells, i.e. cardiac and vascular myocytes respectively [37]. The present study also demonstrates the importance of membrane integrity in the formation/maintenance of insulin- and CL-induced macromolecular assemblies that contain PDE3B, and suggests a chaperone or scaffolding role of cav-1 in cholesterol-rich lipid domains that may be necessary for stabilization and phosphorylation/activation of PDE3B within these macromolecular signalling complexes.
Supplementary Material
Acknowledgments
FUNDING
Part of this work was supported by the NHLBI (National Heart, Lung and Blood Institute) Intramural Research Programme [project number HL002540-15]. E.D. was supported, in part, by the Swedish Medical Research Council [grant number 3362].
Abbreviations used
- Ad
adenoviral
- β3-AR
β3-adrenergic receptor
- BCA
bicinchoninic acid
- BiP
immunoglobulin heavy-chain-binding protein
- cav-1
caveolin-1
- CL
- CREB
cAMP-response-element-binding protein
- CT
C-terminal
- DMEM
Dulbecco’s modified Eagle’s medium
- ER
endoplasmic reticulum
- FBS
fetal bovine serum
- GM130
Golgi matrix protein 130
- HMWC
high-molecular-mass macromolecular complex
- HMWC-CL
HMWC induced by CL
- HMWC-ins
HMWC induced by insulin
- HSL
hormone-sensitive lipase
- HSP-90
heat-shock protein 90
- IBMX
isobutylmethylxanthine
- IRS
insulin receptor substrate
- IGF
insulin-like growth factor
- KD
knockdown
- KRH
Krebs–Ringer-Hepes medium
- NT
N-terminal
- PDE
phosphodiesterase
- PI3K
phosphoinositide 3-kinase
- PKA
cAMP-dependent protein kinase
- PKA-RII
PKA-regulatory subunit
- PKB
protein kinase B
- PM
plasma membrane
- PP2A
protein phosphatase 2A
- pPKB
phosphorylated PKB
- qRT-PCR
quantitative real-time PCR
- siRNA
small interfering RNA
Footnotes
AUTHOR CONTRIBUTION
Faiyaz Ahmad, Peter Strålfors, Eva Degerman and Vincent Manganiello designed the research; Faiyaz Ahmad, Rebecka Lindh, Yan Tang, Iida Ruishalme, Anita Öst and Bobby Sahachartsiri carried out the research; Faiyaz Ahmad, Peter Strålfors, Eva Degerman and Vincent Manganiello analysed the data; Faiyaz Ahmad, Peter Strålfors, Eva Degerman and Vincent Manganiello wrote the manuscript.
References
- 1.Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem. 2007;76:481–511. doi: 10.1146/annurev.biochem.76.060305.150444. [DOI] [PubMed] [Google Scholar]
- 2.Shakur Y, Holst LS, Landstrom TR, Movsesian M, Degerman E, Manganiello V. Regulation and function of the cyclic nucleotide phosphodiesterase (PDE3) gene family. Prog Nucleic Acid Res Mol Biol. 2001;66:241–277. doi: 10.1016/s0079-6603(00)66031-2. [DOI] [PubMed] [Google Scholar]
- 3.Degerman E, Rahn LT, Stenson HL, Goransson O, Harndahl L, Ahmad F, Choi Y-H, Masciarelli S, Liu H, Manganiello V. Role for phosphodiesterase 3B in regulation of lipolysis and insulin secretion. In: LeRoith D, Taylor SI, Olefsky JM, editors. Diabetes Mellitus: a Fundamental and Clinical Text. 3. Lippincott Raven; Philadelphia: 2004. pp. 373–383. [Google Scholar]
- 4.Choi YH, Park S, Hockman S, Zmuda-Trzebiatowska E, Svennelid F, Haluzik M, Gavrilova O, Ahmad F, Pepin L, Napolitano M, et al. Alterations in regulation of energy homeostasis in cyclic nucleotide phosphodiesterase 3B-null mice. J Clin Invest. 2006;116:3240–3251. doi: 10.1172/JCI24867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harndahl L, Jing XJ, Ivarsson R, Degerman E, Ahren B, Manganiello VC, Renstrom E, Holst LS. Important role of phosphodiesterase 3B for the stimulatory action of cAMP on pancreatic β-cell exocytosis and release of insulin. J Biol Chem. 2002;277:37446–37455. doi: 10.1074/jbc.M205401200. [DOI] [PubMed] [Google Scholar]
- 6.Walz HA, Harndahl L, Wierup N, Zmuda-Trzebiatowska E, Svennelid F, Manganiello VC, Ploug T, Sundler F, Degerman E, Ahren B, Holst LS. Early and rapid development of insulin resistance, islet dysfunction and glucose intolerance after high-fat feeding in mice overexpressing phosphodiesterase 3B. J Endocrinol. 2006;189:629–641. doi: 10.1677/joe.1.06522. [DOI] [PubMed] [Google Scholar]
- 7.Wijkander J, Landstrom TR, Manganiello V, Belfrage P, Degerman E. Insulin-induced phosphorylation and activation of phosphodiesterase 3B in rat adipocytes: possible role for protein kinase B but not mitogen-activated protein kinase or p70 S6 kinase. Endocrinology. 1998;139:219–227. doi: 10.1210/endo.139.1.5693. [DOI] [PubMed] [Google Scholar]
- 8.Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 9.Kitamura T, Kitamura Y, Kuroda S, Hino Y, Ando M, Kotani K, Konishi H, Matsuzaki H, Kikkawa U, Ogawa W, Kasuga M. Insulin-induced phosphorylation and activation of cyclic nucleotide phosphodiesterase 3B by the serine-threonine kinase Akt. Mol Cell Biol. 1999;19:6286–6296. doi: 10.1128/mcb.19.9.6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rahn T, Ridderstrale M, Tornqvist H, Manganiello V, Fredrikson G, Belfrage P, Degerman E. Essential role of phosphatidylinositol 3-kinase in insulin-induced activation and phosphorylation of the cGMP-inhibited cAMP phosphodiesterase in rat adipocytes. Studies using the selective inhibitor wortmannin. FEBS Lett. 1994;350:314–318. doi: 10.1016/0014-5793(94)00797-7. [DOI] [PubMed] [Google Scholar]
- 11.Ahmad F, Gao G, Wang LM, Landstrom TR, Degerman E, Pierce JH, Manganiello VC. IL-3 and IL-4 activate cyclic nucleotide phosphodiesterases 3 (PDE3) and 4 (PDE4) by different mechanisms in FDCP2 myeloid cells. J Immunol. 1999;162:4864–4875. [PubMed] [Google Scholar]
- 12.Ahmad F, Cong LN, Stenson Holst L, Wang LM, Rahn LT, Pierce JH, Quon MJ, Degerman E, Manganiello VC. Cyclic nucleotide phosphodiesterase 3B is a downstream target of protein kinase B and may be involved in regulation of effects of protein kinase B on thymidine incorporation in FDCP2 cells. J Immunol. 2000;164:4678–4688. doi: 10.4049/jimmunol.164.9.4678. [DOI] [PubMed] [Google Scholar]
- 13.Zmuda-Trzebiatowska E, Oknianska A, Manganiello V, Degerman E. Role of PDE3B in insulin-induced glucose uptake, GLUT-4 translocation and lipogenesis in primary rat adipocytes. Cell Signalling. 2006;18:382–390. doi: 10.1016/j.cellsig.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 14.Shakur Y, Takeda K, Kenan Y, Yu ZX, Rena G, Brandt D, Houslay MD, Degerman E, Ferrans VJ, Manganiello VC. Membrane localization of cyclic nucleotide phosphodiesterase 3 (PDE3). Two N-terminal domains are required for the efficient targeting to, and association of, PDE3 with endoplasmic reticulum. J Biol Chem. 2000;275:38749–38761. doi: 10.1074/jbc.M001734200. [DOI] [PubMed] [Google Scholar]
- 15.Nilsson R, Ahmad F, Sward K, Andersson U, Weston M, Manganiello V, Degerman E. Plasma membrane cyclic nucleotide phosphodiesterase 3B (PDE3B) is associated with caveolae in primary adipocytes. Cell Signalling. 2006;18:1713–1721. doi: 10.1016/j.cellsig.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 16.Razani B, Combs TP, Wang XB, Frank PG, Park DS, Russell RG, Li M, Tang B, Jelicks LA, Scherer PE, Lisanti MP. Caveolin-1-deficient mice are lean, resistant to diet-induced obesity, and show hypertriglyceridemia with adipocyte abnormalities. J Biol Chem. 2002;277:8635–8647. doi: 10.1074/jbc.M110970200. [DOI] [PubMed] [Google Scholar]
- 17.Ahmad F, Lindh R, Tang Y, Weston M, Degerman E, Manganiello VC. Insulin-induced formation of macromolecular complexes involved in activation of cyclic nucleotide phosphodiesterase 3B (PDE3B) and its interaction with PKB. Biochem J. 2007;404:257–268. doi: 10.1042/BJ20060960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stralfors P, Honnor RC. Insulin-induced dephosphorylation of hormone-sensitive lipase. Correlation with lipolysis and cAMP-dependent protein kinase activity. Eur J Biochem. 1989;182:379–385. doi: 10.1111/j.1432-1033.1989.tb14842.x. [DOI] [PubMed] [Google Scholar]
- 19.Danielsson A, Ost A, Lystedt E, Kjolhede P, Gustavsson J, Nystrom FH, Stralfors P. Insulin resistance in human adipocytes occurs downstream of IRS1 after surgical cell isolation but at the level of phosphorylation of IRS1 in type 2 diabetes. FEBS J. 2005;272:141–151. doi: 10.1111/j.1432-1033.2004.04396.x. [DOI] [PubMed] [Google Scholar]
- 20.Sudo T, Tachibana K, Toga K, Tochizawa S, Inoue Y, Kimura Y, Hidaka H. Potent effects of novel anti-platelet aggregatory cilostamide analogues on recombinant cyclic nucleotide phosphodiesterase isozyme activity. Biochem Pharmacol. 2000;59:347–356. doi: 10.1016/s0006-2952(99)00346-9. [DOI] [PubMed] [Google Scholar]
- 21.Manganiello V, Vaughan M. An effect of insulin on cyclic adenosine 3′,5′-monophosphate phosphodiesterase activity in fat cells. J Biol Chem. 1973;248:7164–7170. [PubMed] [Google Scholar]
- 22.Liu P, Rudick M, Anderson RG. Multiple functions of caveolin-1. J Biol Chem. 2002;277:41295–41298. doi: 10.1074/jbc.R200020200. [DOI] [PubMed] [Google Scholar]
- 23.Gonzalez-Munoz E, Lopez-Iglesias C, Calvo M, Palacin M, Zorzano A, Camps M. Caveolin-1 loss of function accelerates glucose transporter 4 and insulin receptor degradation in 3T3-L1 adipocytes. Endocrinology. 2009;150:3493–3502. doi: 10.1210/en.2008-1520. [DOI] [PubMed] [Google Scholar]
- 24.Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- 25.Cohen AW, Razani B, Schubert W, Williams TM, Wang XB, Iyengar P, Brasaemle DL, Scherer PE, Lisanti MP. Role of caveolin-1 in the modulation of lipolysis and lipid droplet formation. Diabetes. 2004;53:1261–1270. doi: 10.2337/diabetes.53.5.1261. [DOI] [PubMed] [Google Scholar]
- 26.Ortegren U, Aboulaich N, Ost A, Stralfors P. A new role for caveolae as metabolic platforms. Trends Endocrinol Metab. 2007;18:344–349. doi: 10.1016/j.tem.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 27.Quinlan CL, Costa AD, Costa CL, Pierre SV, Dos SP, Garlid KD. Conditioning the heart induces formation of signalosomes that interact with mitochondria to open mitoKATP channels. Am J Physiol Heart Circ Physiol. 2008;295:H953–H961. doi: 10.1152/ajpheart.00520.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamamoto M, Toya Y, Schwencke C, Lisanti MP, Myers MG, Jr, Ishikawa Y. Caveolin is an activator of insulin receptor signaling. J Biol Chem. 1998;273:26962–26968. doi: 10.1074/jbc.273.41.26962. [DOI] [PubMed] [Google Scholar]
- 29.Karlsson M, Thorn H, Danielsson A, Stenkula KG, Ost A, Gustavsson J, Nystrom FH, Stralfors P. Colocalization of insulin receptor and insulin receptor substrate-1 to caveolae in primary human adipocytes. Cholesterol depletion blocks insulin signalling for metabolic and mitogenic control. Eur J Biochem. 2004;271:2471–2479. doi: 10.1111/j.1432-1033.2004.04177.x. [DOI] [PubMed] [Google Scholar]
- 30.Ost A, Ortegren U, Gustavsson J, Nystrom FH, Stralfors P. Triacylglycerol is synthesized in a specific subclass of caveolae in primary adipocytes. J Biol Chem. 2005;280:5–8. doi: 10.1074/jbc.C400429200. [DOI] [PubMed] [Google Scholar]
- 31.Ortegren U, Yin L, Ost A, Karlsson H, Nystrom FH, Stralfors P. Separation and characterization of caveolae subclasses in the plasma membrane of primary adipocytes: segregation of specific proteins and functions. FEBS J. 2006;273:3381–3392. doi: 10.1111/j.1742-4658.2006.05345.x. [DOI] [PubMed] [Google Scholar]
- 32.Madore N, Smith KL, Graham CH, Jen A, Brady K, Hall S, Morris R. Functionally different GPI proteins are organized in different domains on the neuronal surface. EMBO J. 1999;18:6917–6926. doi: 10.1093/emboj/18.24.6917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang W, Hemler ME. Caveolin-1 regulates matrix metalloproteinases-1 induction and CD147/EMMPRIN cell surface clustering. J Biol Chem. 2004;279:11112–11118. doi: 10.1074/jbc.M312947200. [DOI] [PubMed] [Google Scholar]
- 34.Kazui A, Ono M, Handa K, Hakomori S. Glycosylation affects translocation of integrin, Src, and caveolin into or out of GEM. Biochem Biophys Res Commun. 2000;273:159–163. doi: 10.1006/bbrc.2000.2903. [DOI] [PubMed] [Google Scholar]
- 35.Sztalryd C, Xu G, Dorward H, Tansey JT, Contreras JA, Kimmel AR, Londos C. Perilipin A is essential for the translocation of hormone-sensitive lipase during lipolytic activation. J Cell Biol. 2003;161:1093–1103. doi: 10.1083/jcb.200210169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lindh R, Ahmad F, Resjo S, James P, Yang JS, Fales HM, Manganiello V, Degerman E. Multisite phosphorylation of adipocyte and hepatocyte phosphodiesterase 3B. Biochim Biophys Acta. 2007;1773:584–592. doi: 10.1016/j.bbamcr.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 37.Raymond DR, Carter RL, Ward CA, Maurice DH. Distinct phosphodiesterase-4D variants integrate into protein kinase A-based signaling complexes in cardiac and vascular myocytes. Am J Physiol Heart Circ Physiol. 2009;296:H263–H271. doi: 10.1152/ajpheart.00425.2008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.